1. Introduction
Wound healing is a complex process involving coordinated signaling and interactions between various cell types and the extracellular matrix to restore tissue structure and function. Full-thickness wounds are particularly challenging to repair and pose serious health risks [
1]. The increasing prevalence of non-healing wounds, driven by obesity, diabetes, and aging, places a significant burden on healthcare systems and the economy [
2,
3]. Traditional wound dressings are often inadequate for managing complex wounds, highlighting the need for advanced regenerative approaches. Mesenchymal stem cells (MSCs) have been widely explored for skin repair because of their regenerative properties and ability to enhance wound healing [
4]. MSCs can be derived from various sources, including bone marrow, adipose tissue, and umbilical cords. Among these, human umbilical cord MSCs (hUCMSCs) stand out due to their easy procurement, high proliferative capacity, and lack of ethical concerns [
5]. However, challenges such as tumor formation risks, immune rejection, and long-term clinical complexities hinder MSC-based therapies [
4]. Emerging research suggests that mesenchymal stem cells (MSCs) primarily exert their therapeutic effects through paracrine signaling, with extracellular vesicles (MSC-EVs) playing a critical role in mediating cell communication and signaling. These vesicles are key carriers of bioactive molecules that facilitate intercellular communication, influencing various biological processes such as tissue regeneration, immune modulation, and cellular repair mechanisms [
6]. MSC-EVs contain bioactive molecules, including mRNA, miRNA, proteins, and lipids, which regulate cellular processes and facilitate tissue repair [
7,
8,
9]. hUCMSC-derived small extracellular vesicles (hUCMSC-sEVs) have demonstrated potential in stimulating all phases of wound healing, including hemostasis, inflammation, proliferation, and remodeling [
10,
11]. However, their therapeutic efficacy alone may be insufficient for large or full-thickness wounds, which require both biochemical signaling and structural support [
12]. Chronic conditions such as aging and diabetes further complicate healing by impairing cellular responses [
13,
14]. Biomaterials such as hydrogels provide a three-dimensional structure that supports cell attachment, migration, and tissue formation. Hydrogels help maintain a favorable wound environment by retaining moisture and absorbing excess exudate while gradually degrading to facilitate tissue integration [
15]. This approach aligns with the principles of tissue engineering, which combine biomaterials, cells, and bioactive molecules to promote functional tissue regeneration [
16,
17]. Hydrogels also enable the controlled, localized release of MSC-EVs, preventing their rapid clearance and enhancing their regenerative potential [
12,
18]. This combination addresses both the structural and biochemical requirements for effective wound healing, particularly in chronic and large wounds. Gelatin hydrogels, in particular, have shown promise in combination with MSC-EVs [
10,
18,
19]. As a collagen-derived biopolymer, gelatin is widely used in wound care because of its natural degradability, minimal immune response, and resorbability [
20,
21,
22]. Additionally, it is cost-effective and can be modified to enhance its properties. However, its weak mechanical strength limits its application [
23]. Crosslinking is commonly employed to reinforce gelatin hydrogels, improving their thermal and mechanical stability [
24]. Although chemical crosslinkers like glutaraldehyde are effective, they pose cytotoxicity risks due to potential leaching during degradation [
25]. As a safer alternative, genipin (GNP), a naturally occurring crosslinker derived from Gardenia jasminoides, has gained attention for its ability to enhance gelatin’s properties without toxic effects [
26,
27]. This shift toward natural crosslinkers aligns with the growing interest in sustainable and biocompatible biomaterials for wound healing [
28]. Beyond biomaterials and MSC-EVs, advanced 3D printing technologies further enhance wound healing strategies. Three-dimensional bioprinting enables the fabrication of highly customized hydrogel scaffolds tailored to the wound size, shape, and specific patient needs [
29]. Techniques such as extrusion-based bioprinting, inkjet bioprinting, and stereolithography offer precise scaffold structuring, supporting cell encapsulation and tissue regeneration [
30]. Additionally, imaging techniques like computed tomography (CT) provide data for precise digital wound modeling, optimizing hydrogel application, and improving overall healing outcomes [
31]. By integrating 3D bioprinting with hydrogel-based MSC-EV delivery, researchers can develop highly efficient, patient-specific solutions for treating complex and chronic wounds. This research takes an integrated approach by combining advanced 3D bioprinting with a biocompatible gelatin–GNP hydrogel infused with hUCMSC-sEVs, aiming to fabricate precisely designed scaffolds that provide both structural support and a regenerative microenvironment for skin repair. Specifically, this study will investigate the physicochemical properties of the gelatin–GNP hydrogel and the biocompatibility of the gelatin–GNP hUCMSC-sEVs combination for potential future clinical applications. By addressing both the structural and biochemical needs of wound healing, this strategy offers an effective solution for treating large and chronic wounds.
2. Materials and Methods
2.1. Ethical Approval
This study received ethics approval from the Research Ethics Committee Universiti Kebangsaan Malaysia (approval code no.: JEP-2023-421). Written informed consent was obtained from all donors. All experimental procedures adhered to the relevant guidelines and regulations.
Figure 1 is a schematic illustration of the experimental design of this study.
2.2. Materials
Commercial-grade gelatin (GE) powder was procured from Nitta Gelatin Ltd. (Yao City, Osaka, Japan) and used as the primary biomaterial for the fabrication of the gelatin-based hydrogel. The natural crosslinking agent genipin was purchased from FUJIFILM Wako Pure Chemical Corporation (Chuo-ku, Osaka, Japan) and prepared in 70% ethanol (MERCK, Darmstadt, Germany) for hydrogel crosslinking. For the swelling ratio evaluation, phosphate-buffered saline (PBS) powder was obtained from Sigma-Aldrich (St. Louis, MO, USA) and reconstituted as a buffer medium. To assess enzymatic degradation, a 0.0006% (w/v) collagenase type I solution (Worthington Biochemical Corporation, Lakewood, NJ, USA) was used to simulate the in vitro enzymatic conditions. Distilled water was used as the solvent in various physicochemical characterization tests. Primary human umbilical cord mesenchymal stem cells (hUCMSCs) and their derived small extracellular vesicles (hUCMSC-sEVs) were used for in vitro biological assays. Primary human dermal fibroblasts (PHDFs) were used to evaluate the biocompatibility of the hydrogel systems. Cell viability was examined via qualitative live/dead staining using the LIVE/DEAD™ Viability/Cytotoxicity Kit (Invitrogen, Waltham, MA, USA), while quantitative analysis was performed using the MTT assay (Sigma-Aldrich, St. Louis, MO, USA) to determine the cell metabolic activity.
2.3. Hydrogel Preparation and 3D Bioprinting
To prepare the gelatin solution, different concentrations (8% and 10% w/v) were prepared by dissolving the gelatin powder (GE; Nitta-Gelatin Ltd., Yao City, Osaka Japan) in distilled water by continuous stirring at 37 °C using a magnetic stirrer set to 400 rpm for 30 min. After dissolution, the gelatin solution was incubated at 37 °C for an additional 30 min to ensure complete homogeneity. Meanwhile, a 0.3% (w/v) genipin (GNP) solution was prepared by dissolving GNP (Fujifilm Wako Pure Chemical, Chuo-ku, Osaka, Japan) in 10 mL of 70% ethanol (EtOH; Merck, Darmstadt, Germany). Polymerization times were assessed using the inverted tube test at 22–24 °C.
The hydrogel biomaterial ink was prepared in four groups based on the GE concentration and crosslinking state. To create the crosslinked GE–GNP hydrogel biomaterial ink, the GE and GNP solutions were mixed in a 3:1 ratio at 37 °C, resulting in a crosslinked solution (GECL). There were two crosslinked hydrogel bioink groups, 8% GECL (8% GE + 0.3% GNP) and 10% GECL (10% GE + 0.3% GNP), and two non-crosslinked bioink hydrogel (GNC) groups, 8% GNC (GE 8%, w/v) and 10% GNC (10%, w/v). The prepared hydrogel was loaded into a 10 mL syringe fitted with a nozzle of 400 μm diameter. The extrusion 3D bioprinting system Biogens XI (3D Gens, Shah Alam, Selangor, Malaysia) was used for the 3D bioprinting process. Autodesk Fusion AutoCAD version 9.20.0.0software (stl file format) was used to create the model. The designed structures were put into Simplify3D software (version 4.1). To maintain the biomaterial ink in a sol-gel state, the printing head temperature was controlled at 25 °C, allowing the ink to remain printable for the required duration. A CAD (Autodesk Fusion AutoCAD 2021 software) design was employed to generate a net structure with dimensions of L10.0 × W10.0 × H0.25 mm3 per layer. Using this design, a cubic scaffold consisting of 4 layers was fabricated. Before initiating the bioprinting process, the printing feed rate and pattern parameters were optimized to ensure uniform extrusion and a precise scaffold architecture.
2.4. Visual Assessment of the Gross Morphology
The 3D bioprinted hydrogels were visually assessed immediately after the polymerization process to evaluate their external morphology and uniformity. Using a digital camera, detailed images were captured to document the differences between the crosslinked and non-crosslinked samples.
2.5. Hydrogel Physical and Chemical Properties Assessment
2.5.1. Swelling Behavior
The swelling behavior of the hydrogels was investigated by submerging them in PBS at 37 °C for 6 h. Swelling ratios were calculated based on the weight difference before and after immersion, providing data on the hydrogel’s capacity to absorb fluids. The swelling ratio (SR) was calculated experimentally using Formula (1):
where W
2 is the swollen weight of the hydrogels, and W
1 is the dry weight of the hydrogels. The swelling ratio is the fractional increase in the weight of the hydrogel caused by water absorption.
2.5.2. Porosity Evaluation
The porosity of the freeze-dried hydrogels was evaluated using a combination of scanning electron microscopy (SEM) and the liquid displacement method.
SEM, operated at 15 kV, was employed to examine the surface morphology and internal structure of the hydrogels. SEM was used for the high-magnification imaging, providing detailed insights into the fibrous and microporous architecture.
In addition, the liquid displacement method was used to quantify the porosity. Ethanol served as the displacement medium, allowing for the measurement of the porosity percentage.
The lyophilized hydrogels were first weighed (M1) before being immersed in 99.5% ethanol for 24 h. Following immersion, excess ethanol was carefully removed using Whatman® No. 42 filter paper (Merck, Darmstadt, Germany), and the weight of the hydrogel (M2) was then recorded.
The porosity was calculated using Formula (2):
where p is the density of 99.5% EtOH, V is the volume of the hydrogel, and M1 and M2 are the initial and final weights of the lyophilized hydrogels, respectively.
2.5.3. Water Vapor Transmission Rate (WVTR)
This study was conducted on the basis of the American Society for Testing and Materials (ASTM) standard [
32,
33]. In brief, the hydrogels were placed on the opening of a glass vial that contained 10 mL of dH
2O. The samples were placed in a controlled environment (5% CO
2 at 37 °C). The water vapor transmission rate was recorded and calculated as shown in Formula (3) below:
where
Wi is the initial weight,
Wf is the final weight, and
A is the surface area of the glass vial.
2.5.4. Contact Angle and Wettability Analysis
The contact angle was measured to determine the surface wettability with distilled water droplets by dropping 10 µL of dH2O onto the surface of the hydrogel, and the angle was analyzed. The contact angle analysis performed using the ImageJ application (V1.5, Bethesda, MD, USA) revealed the hydrophilic or hydrophobic nature of the samples, which influences cell attachment and proliferation.
2.5.5. Enzymatic Biodegradation
The biodegradability of the hydrogels was analyzed by immersing the samples in 0.0006 mg/mL collagenase type I (Worthington-Biochemical Corporation, Lakewood, NJ, USA) in Dulbecco’s phosphate-buffered saline (DPBS) solution (Gibco, San Diego, CA, USA) at physiological temperature (37 °C) for 24 h. The weight loss of the hydrogels over time served as an indicator of enzymatic breakdown, simulating the in vivo microenvironment. The samples were rinsed, dried, and reweighed to calculate the percentage weight loss. This method provided insight into the hydrogel’s degradation profile, which is critical for determining its suitability as a biomaterial for regenerative applications. The percentage of weight loss was calculated as shown in Formula (4):
where W
1 is the initial weight, and W
2 is the final weight.
2.6. Chemical Analysis
2.6.1. Energy-Dispersive X-Ray
The elemental composition of the hydrogel surface was analyzed using Energy-Dispersive X-ray (EDX) using a Phenom Pro X SEM EDX microscope (Phenom, Eindhoven, The Netherlands).
2.6.2. Fourier Transform Infrared Spectrophotometry
Fourier Transform Infrared (FTIR) spectroscopy was employed to identify the functional groups within the hydrogels, confirming the chemical modifications introduced by crosslinking. Measurements were conducted using an FTIR spectrophotometer (PerkinElmer, Waltham, MA, USA) in the range of 4000–500 cm⁻1, with a resolution of 2 cm⁻1 per point at room temperature.
2.6.3. X-Ray Diffraction Study
X-ray diffraction (XRD) analysis of the sample was conducted at room temperature using an advanced X-ray diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) in θ–2θ scan mode. The diffraction patterns were recorded with CuKα radiation (λ = 1.542 Å) under operating conditions of 35 kV and 10 mA. The sample was scanned over a 2θ range from 10° to 70° in a constant scan mode. The diffractogram was further analyzed using the integrated software (Diffrac. Suite EVA, V4.0, Bruker, Coventry, UK) to identify specific peaks and interpret the results.
2.6.4. Mechanical Testing (Compression Test)
To assess the mechanical properties of the hydrogel biomaterials, specifically their elastic limit and compressive strength, a compression test was performed. This method involves placing each hydrogel sample on a flat surface and applying a defined load to evaluate the material’s response to stress. The test was designed to examine the behavior of the biomaterials under both constant and progressively applied loads, measuring the maximum stress they can withstand over time and providing insights into their resilience and structural integrity under mechanical forces.
The hydrogel samples, fabricated according to the optimized formulations, were polymerized and maintained at room temperature before testing. The mechanical properties were evaluated using a high-precision universal testing machine (Autograph AG-X, Shimadzu, Japan) at a crosshead speed of 5 mm/min. Cylindrical samples (15 mm in diameter, 10 mm in height) were employed to ensure the consistency and reproducibility of the measurements. The compressive strength was determined by calculating the stress values at the maximum load, offering a comprehensive understanding of the material’s mechanical behavior and suitability for load-bearing applications, such as tissue repair and implantable biomaterials.
2.7. Isolation and Culture of hUCMSCs
The umbilical cords (n = 3) were collected from healthy full-term births, of mothers aged 25–40 years at the Hospital Canselor Tuanku Muhriz, Universiti Kebangsaan Malaysia, Malaysia, during natural birth and elective cesarean sections. Mothers with conditions such as gestational diabetes or hypertension were excluded. Umbilical cords (6–10 cm) were disinfected, and vascular structures were removed before mincing the tissue into ~2 mm
2 pieces. The tissue was washed with DPBS (Gibco, Carlsbad, CA, USA), and digestion was performed using 0.6% collagenase type I (Worthington-Biochemical Corporation, Lakewood, NJ, USA) at 37 °C with shaking for 1 h. Cells were cultured in α-MEM (Gibco, Weilam Rhein, Germany) supplemented with 10% in-house human platelet lysate and 1% antibiotic-antimycotic (Gibco, Weilam Rhein, Germany), with media changes every 3 days until 70–80% confluency. Expanded cells were dissociated at passage 2 with 0.05% trypsin-EDTA (Gibco/BRL, Carlsbad, CA, USA). hUCMSCs were characterized in accordance with the criteria established by the International Society for Cell and Gene Therapy [
34]. Immunophenotyping of hUCMSCs was performed using flow cytometry (BD FACSVerse™, Franklin Lakes, NJ, USA) and the Human MSC Analysis Kit (BD Biosciences, Franklin Lakes, NJ, USA) to check for the presence of positive MSC markers (CD44, CD73, CD90, and CD105) and absence of negative markers (CD11b, CD19, CD34, CD45, and HLA-DR). Trilineage differentiation was induced using the StemPro™ (Osteogenesis/Chondrogenesis/Adipogenesis) Differentiation Kit (Gibco, Weilam Rhein, Germany) following the manufacturer’s protocol and confirmed by staining: Alizarin Red (Sigma-Aldrich, St. Louis, MO, USA) for osteogenesis, Oil Red O (Sigma-Aldrich, St. Louis, MO, USA) for adipogenesis, and Safranin O (Sigma-Aldrich, St. Louis, MO, USA) for chondrogenesis.
2.8. hUCMSC-sEV Isolation and Characterization
For hUCMSC-sEVs isolation, hUCMSCs from the three donors at passage 5 were pooled and cultured until they reached 80% confluency. Once confluency was achieved, the spent culture medium was carefully discarded, and the cells were washed three times with phosphate-buffered saline (PBS) to remove any residual serum or medium components. Subsequently, the cells were incubated in low-glucose Dulbecco’s Modified Eagle Medium (LG-DMEM; Sigma-Aldrich, St. Louis, MO, USA) without serum for 24 h to allow for hUCMSC-sEV production. The conditioned medium was then collected and filtered through a 0.22 µm filter membrane to eliminate cellular debris and other large particles.
2.8.1. Tangential Flow Filtration (TFF)
To further purify hUCMSC-sEVs, while minimizing the risk of contamination with protein aggregates, lipoproteins, or other unwanted impurities, the conditioned medium was centrifuged at 2000× g for 15 min followed by TFF. This method used the MINIMATE TFF Capsule fitted with a 100 kDa molecular weight cut-off (MWCO) OMEGA membrane (Pall, New York, NY, USA). Following the completion of the TFF process, the isolated hUCMSC-sEV were carefully resuspended in PBS, aliquoted to avoid repeated freeze–thaw cycles, and stored at −80 °C until further experiments or analyses were performed.
2.8.2. Characterization of hUCMSC-sEVs
The isolation and characterization of hUCMSC-sEVs adhered to the Minimal Information for Studies of Extracellular Vesicles 2023 (MISEV2023) guidelines to ensure reproducibility and reliability. Total protein was quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific™, Boston, MA, USA) according to the manufacturer’s protocol, ensuring standardized measurement procedures. The protein content served as a baseline for the downstream analyses and verification of sEV purity. Western blotting was employed to validate the presence of EV-specific markers, such as CD63 and TSG101, which are conventional markers of EV identity. Simultaneously, the absence of the cellular contaminant GP 96 (an endoplasmic reticulum marker) was evaluated. For intracellular marker analysis, hUCMSC-sEVs were lysed using a radioimmunoprecipitation assay (RIPA) buffer enriched with phenylmethyl sulfonyl fluoride (PMSF) and a protease inhibitor cocktail (Thermo Fisher Scientific) to maintain protein stability during extraction. The extracted proteins were then separated by 10–12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE). After electrophoresis, the proteins were transferred to nitrocellulose membranes for subsequent immunoblotting analysis, enabling the detection of specific intracellular markers. The membranes were incubated with primary antibodies targeting the desired markers, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies. Signal detection was performed using enhanced chemiluminescence (ECL) substrate, and protein bands were visualized and quantified using a chemiluminescent imaging system. Primary antibodies specific to CD63, TSG101, and GP 96 from Cell Signaling Technology, Danvers, IL, USA, were applied to the membranes, followed by incubation with HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, IL, USA). The protein bands were visualized using a chemiluminescent detection system, and images were captured using the Amersham Imager 600 (Cytiva, Marlborough, MA, USA), confirming the identity and purity of the isolated hUCMSC-sEVs.
Nanoparticle Tracking Analysis (NTA) was carried out to determine the size distribution and concentration of the hUCMSC-sEVs. For accurate measurements, the hUCMSC-sEVs samples were diluted 1:1000 in filtered phosphate-buffered saline (PBS) to achieve an optimal particle concentration range (20–200 particles per frame) suitable for the instrument’s sensitivity. Using a Nanosight NS300 instrument (Malvern Panalytical, Misterton, Doncaster, UK) equipped with a 488 nm laser, five 1 min videos were recorded for each sample at 25 °C. The videos were analyzed with the finite track length adjustment (FTLA) algorithm, which tracks the Brownian motion of particles in the suspension, yielding detailed information about the hUCMSC-sEVs size distribution and concentration. In addition, the structural integrity and morphology of the hUCMSC-sEVs were assessed using transmission electron microscopy (TEM). For this, the hUCMSC-sEVs were deposited onto formvar-coated copper grids, ensuring an even distribution, and were negatively stained with 2% phosphotungstic acid (PTA) to enhance the contrast for imaging. The grids were then examined under a LEO Libra-120 TEM.
2.9. Primary Human Dermal Fibroblast Isolation and Culture
Primary human dermal fibroblasts (PHDFs) were isolated from human skin samples collected as redundant tissue after surgery from three consenting patients. The skin samples were processed by cutting them into small (1–2 cm) pieces and cleaning them using DPBS. They were digested with 0.6% collagenase Type I (Worthington-Biochemical Corporation) for 4–6 h at 37 °C in a shaker incubator, followed by trypsinization with trypsin-EDTA for 10 min. The cell suspension was centrifuged and resuspended in F12: DMEM (Gibco/BRL) supplemented with 10% fetal bovine serum (FBS) (Gibco). Cells were seeded at 1 × 104 cells/cm2 in a 6-well polystyrene culture plate and incubated at 37 °C in 5% CO2. The medium was replenished every 2–3 days until the cells achieved 70–80% confluence, at which point differential trypsinization was performed. The fibroblasts were subsequently expanded in a 75 cm2 culture flask containing F12: DMEM with 10% FBS. For all experiments, PHDFs between passages 2 to 4 were used. A total of 50,000 cells in 20 µL of culture medium were seeded onto the top of the hydrogels prepared under sterile conditions in a biosafety cabinet.
2.10. hUCMSC-sEV Loaded Hydrogel Biocompatibility
To further functionalize the hydrogels, the purified hUCMSC-sEVs were incorporated into the 10% GECL hydrogel. The hUCMSC-sEVs, previously isolated and resuspended in 50 μL of PBS, were added to the 10% GECL solution at various concentrations (125, 100, 75, 50, and 25 μg/mL). The mixture was stirred at 4 °C to ensure a homogeneous distribution of hUCMSC-sEVs throughout the hydrogel matrix.
2.10.1. MTT Assay
This assay was conducted using hydrogel leachates from 10% GECL loaded with hUCMSC-sEVs at concentrations of 125, 100, 75, 50, and 25 µg/mL, according to the ISO 10993-12 protocol [
35]. A leachate (2 g/mL) was prepared, and PHDFs were seeded at a density of 5000 cells/cm
2 in a 48-well plate. After 24 h of incubation at 37 °C in 5% CO
2, 200 µL of hydrogel leachate at each concentration was added to the respective triplicate wells. The MTT assay (Sigma-Aldrich, St. Louis, MO, USA) was performed to determine cell viability after 48 h. The cells were rinsed with PBS, and 90 µL of pure medium with 10 µL of 5 mg/mL MTT solution was added, yielding a final concentration of 0.5 mg/mL. After 4 h at 37 °C, 87.5 µL of the solution was removed, and 100 µL of DMSO (Sigma-Aldrich, St. Louis, MO, USA) was added to dissolve the formazan. The absorbance was measured using a spectrophotometer (BioTek, Power Wave XS, Highland Park, IL, USA) to calculate cell viability (%) using Formula (5):
where Ab
t represents the absorbance of the treatment group, and Ab
c represents the control group.
2.10.2. LIVE/DEAD Assay
Cell viability was assessed using the LIVE/DEAD™ Cell (Invitrogen, Waltham, MA, USA). Various concentrations of hUCMSC-sEVs (125, 100, 75, 50, and 25 µg/mL) were incorporated with 10% GECL. PHDFs were seeded onto the hydrogels one day prior and then exposed to calcein-AM and EthD-1 in PBS for 30 min at 37 °C, following the manufacturer’s instructions. Cells were gently washed with PBS and observed under a fluorescence microscope (Nikon A1R-A1, Shinagawa-ku, Tokyo, Japan) at ×100 magnification. Live cells appeared green, while dead cells appeared red.
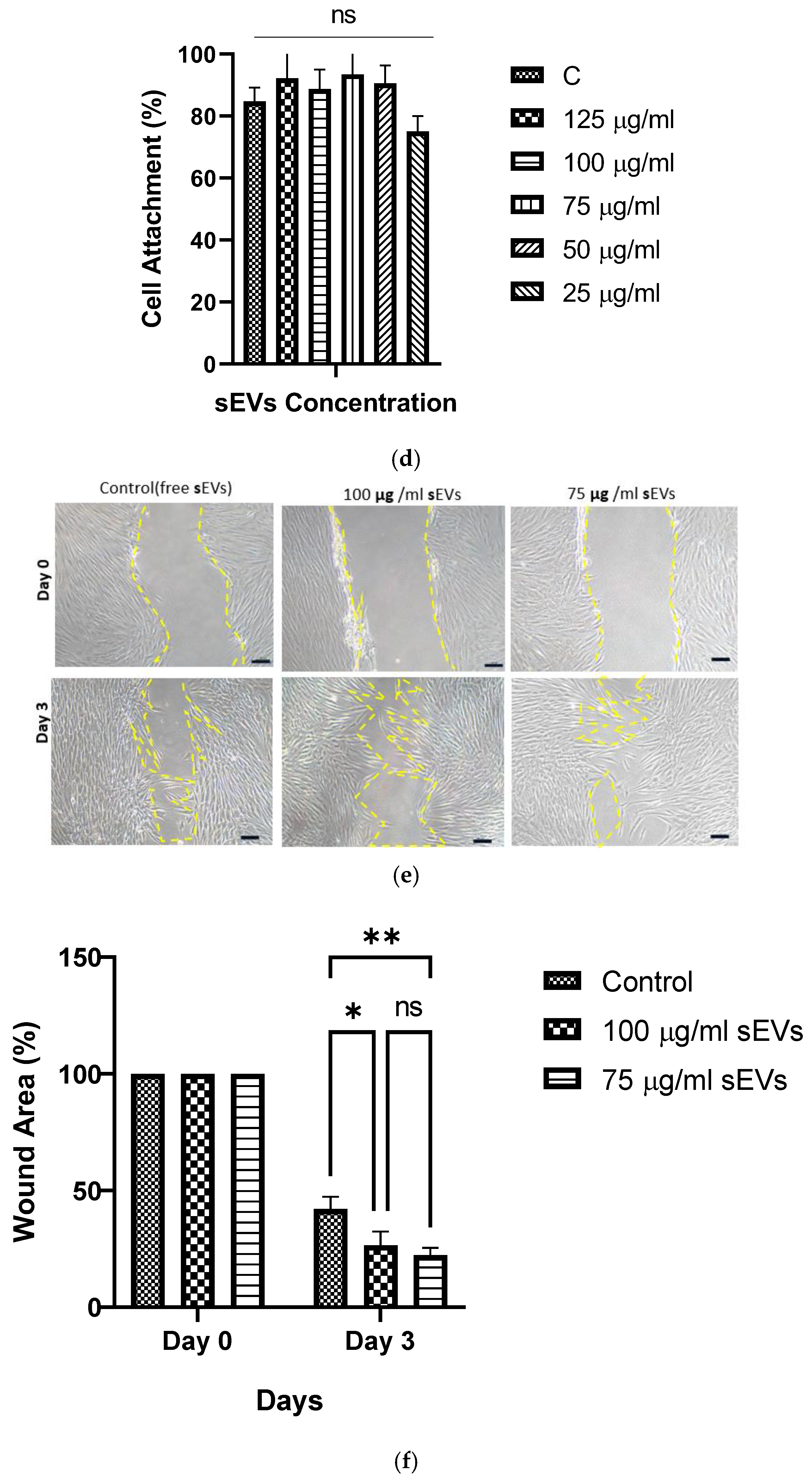
2.10.3. Cell Attachment Percentage
The cell attachment percentage was evaluated using trypan blue staining. After initial seeding and incubation, unattached cells were collected and visualized using a hemocytometer (Optik Labor, Görlitz, Germany). The cell attachment percentage was calculated using Formula (6):
where Ci Seeded Cell is the initial cell seeding, and Cd is the number of unattached cells in DPBS.
2.10.4. Scratch Wound Assay
The scratch assay was used to assess PHDF migration. To study the wound healing effect of 10% GECL leachate containing sEVs concentrations (75 and 100) µg/mL on PHDFs. The tip of a sterile pipette was used to scratch confluent HDF monolayers in the center of each well. After removing the culture medium, the cells were washed with DPBS (Sigma-Aldrich) and grown in the biomaterial leachate media containing sEVs at concentrations of 0, 75, and 100 µg/mL. The plate was then placed in an incubator, and images were taken every 12 h until the cells migrated to close the scratch, typically within 0 to 72 h. For each of the three biological samples (n = 3), three technical replicates were conducted. A Nikon A1R-A1 live-cell imaging microscope was used to obtain pictures at 1 h intervals to estimate the wound healing closure using Formula (7):
where A (0) is the initial wound area, and A(t) is the wound area at each time point.
The wound closure percentage was calculated using the ImageJ software.
2.11. Statistical Analysis
All experiments were conducted with three biological replicates and three technical replicates unless otherwise stated. Results are expressed as mean ± standard deviation (SD). Statistical comparisons were made using one-way or two-way Analysis of Variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons or an unpaired Student’s t-test for comparisons between two groups using GraphPad Prism 9.0 (GraphPad Software, Inc., San Diego, CA, USA). A p-value < 0.05 was considered statistically significant.
4. Discussion
The hUCMSC-EVs, known for their regenerative potential, deliver key bioactive signals that support tissue repair, reduce inflammation, stimulate angiogenesis, and enhance cell regeneration [
10]. However, for the complete regeneration of large wounds, a 3D scaffold is essential to support cell attachment, migration, and proper tissue formation. Hydrogels provide this structural support while maintaining hydration and allowing the controlled release of hUCMSC-EVs. This slows their degradation and clearance, extending their therapeutic effects [
12]. Additionally, advances in 3D printing technology allow for the design of personalized hydrogel scaffolds tailored to the unique shape and size of individual wounds, improving treatment precision and efficacy [
29]. The combination of hUCMSC-sEVs with bioengineered hydrogels offers a promising personalized approach for treating chronic and complex wounds.
Hence, this study aimed to develop and characterize 3D bioprinted gelatin–genipin (GNP)-crosslinked hydrogels loaded with hUCMSC-EVs for future skin wound healing applications. Initially, we formulated and optimized GNP-crosslinked gelatin hydrogels using four different combinations of gelatin concentrations, with and without GNP. The formulations included non-crosslinked gelatin hydrogels (GNC and 10% GNC, where gelatin concentration is expressed in w/v) and GNP-crosslinked hydrogels (8% GECL and 10% GECL, containing 0.3% GNP). Each hydrogel type was evaluated for its physicochemical properties to determine its potential for wound healing applications.
A key advantage of this hydrogel system is its sustainability and biocompatibility, as both gelatin and GNP are naturally derived eco-friendly biomaterials with well-documented therapeutic potential. Gelatin, sourced from various waste materials, not only provides a cost-effective and biodegradable alternative but also mimics the ECM, facilitating cell adhesion and tissue regeneration [
36,
37]. Likewise, GNP, a natural crosslinker, offers a green alternative to synthetic chemical crosslinker, enhancing the hydrogel’s mechanical properties while preserving high biocompatibility [
26]. These attributes make gelatin–GNP hydrogels a promising platform for wound healing applications, combining structural support with bioactive properties in a commercially viable and environmentally responsible manner.
The physicochemical properties of the hydrogels were evaluated to determine their suitability for both 3D bioprinting and potential wound healing applications. Among these, the polymerization time is a critical parameter influencing the practicality of the 3D printing process [
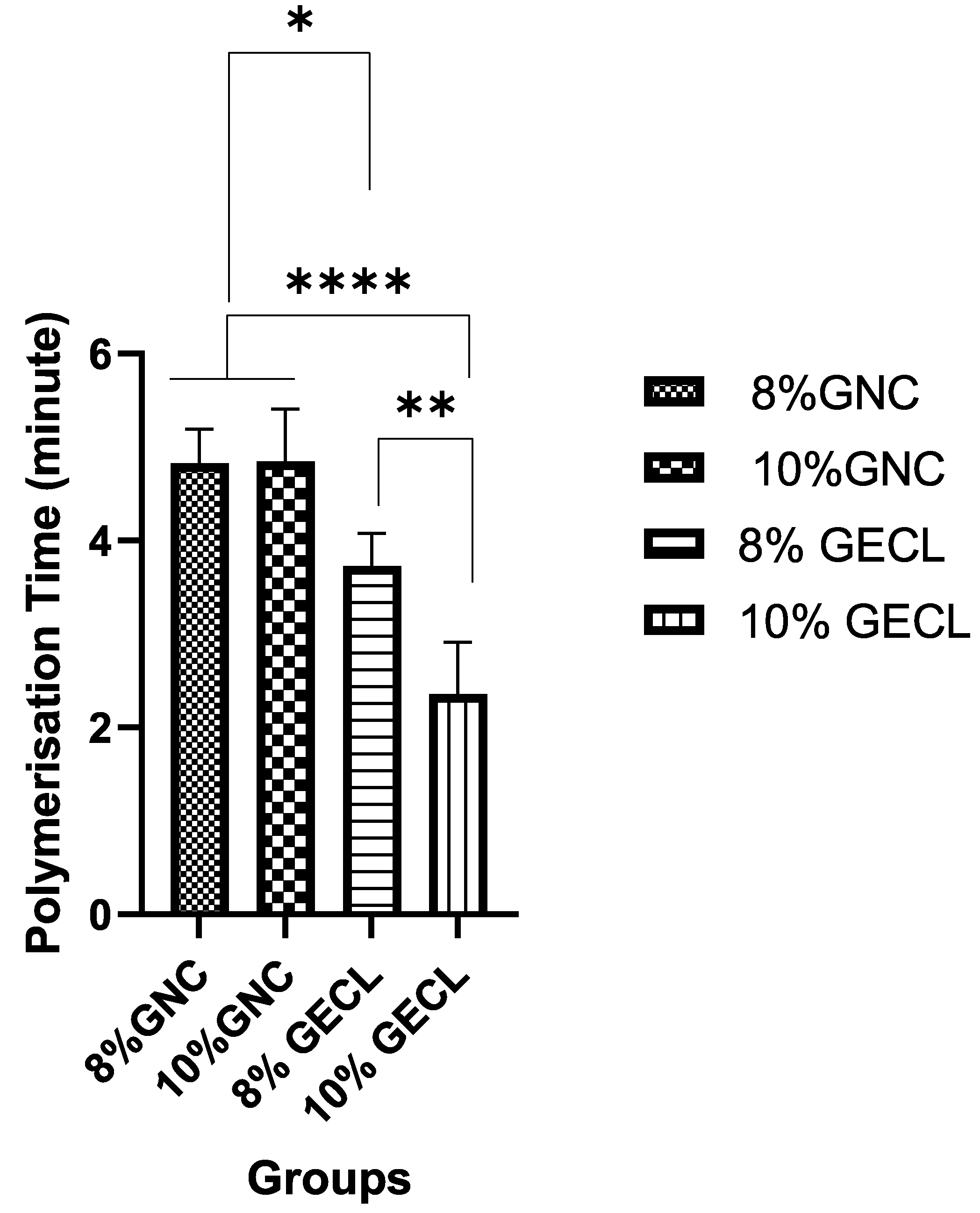
38]. In this study, polymerization occurred within two to five minutes at room temperature (22–24 °C), with the rate significantly improving as gelatin concentration increased. Notably, the hydrogel with 10% gelatin content (10% GECL) demonstrated the fastest polymerization, solidifying in approximately two minutes. This timeframe is considered optimal for 3D bioprinting, offering sufficient working time for precise deposition and structural fidelity, while ensuring rapid solidification post-printing. The fast gelation not only facilitates handling and positioning during clinical application but also supports the maintenance of construct integrity throughout the process.
Previous studies have identified the optimal gelation times for extrusion-based 3D bioprinting. For instance, a study using GNP hybrid gelatin–PVA hydrogels found three minutes to be ideal at 23 °C [
39]. In contrast, a similar study found that longer polymerization times of 4–5 min were less favorable, as they delayed solidification, compromising the efficiency of the process.
The bluish-green color change observed during the polymerization process in the GNP hydrogels indicated successful crosslinking. The hydrogels’ appearance revealed clear, translucent, non-crosslinked (GNC) gelatin hydrogels, whereas the crosslinked (GECL) hydrogels displayed a bluish-green hue. This color change is attributed to the reaction of GNPs with primary amines in the presence of oxygen, resulting in the formation of water-soluble blue pigments, as previously reported by Zawani et al. 2022 [
40]. Keeping the wound area hydrated is essential for optimal healing, as it prevents eschar formation and supports cell communication through signaling molecules. A moist environment enhances epithelial cell migration and collagen synthesis by fibroblasts and promotes the autolytic debridement of necrotic tissue [
41]. To assess the hydration ability of the hydrogel in maintaining a moist wound environment and exudate clearance, several key properties were tested, such as the swelling ratio, porosity, WVTR, and hydrophilicity.
The swelling ratio is a paramount parameter that quantifies a hydrogel’s ability to absorb and retain water, directly reflecting its capacity to maintain a hydrated environment—an essential factor in promoting tissue regeneration and wound healing. A high swelling ratio not only indicates superior absorption of wound exudates but also correlates with improved hydrogel performance as a moisture-retentive dressing. Furthermore, swelling behavior provides valuable insights into the structural integrity and crosslinking efficiency of the hydrogel as well as its potential for sustained drug delivery—both of which are pivotal to its therapeutic efficacy [
42]. In this study, all hydrogel formulations exhibited a swelling ratio exceeding 500%, affirming their suitability for wound healing. Notably, non-crosslinked hydrogels showed a higher swelling ratio, likely due to the absence of additional bonding restricting water uptake. Consistent with previous findings, the degree of crosslinking plays a crucial role in modulating swelling behavior, where a denser network may reduce water absorption while still maintaining adequate hydration to support cellular activities essential for tissue repair [
43]. However, the tested crosslinked hydrogels still maintained an optimal swelling ratio, striking a balance between excessive swelling, which could lead to hydrogel deformation, and controlled swelling, which ensures moisture retention and blood and body fluid absorption [
42].
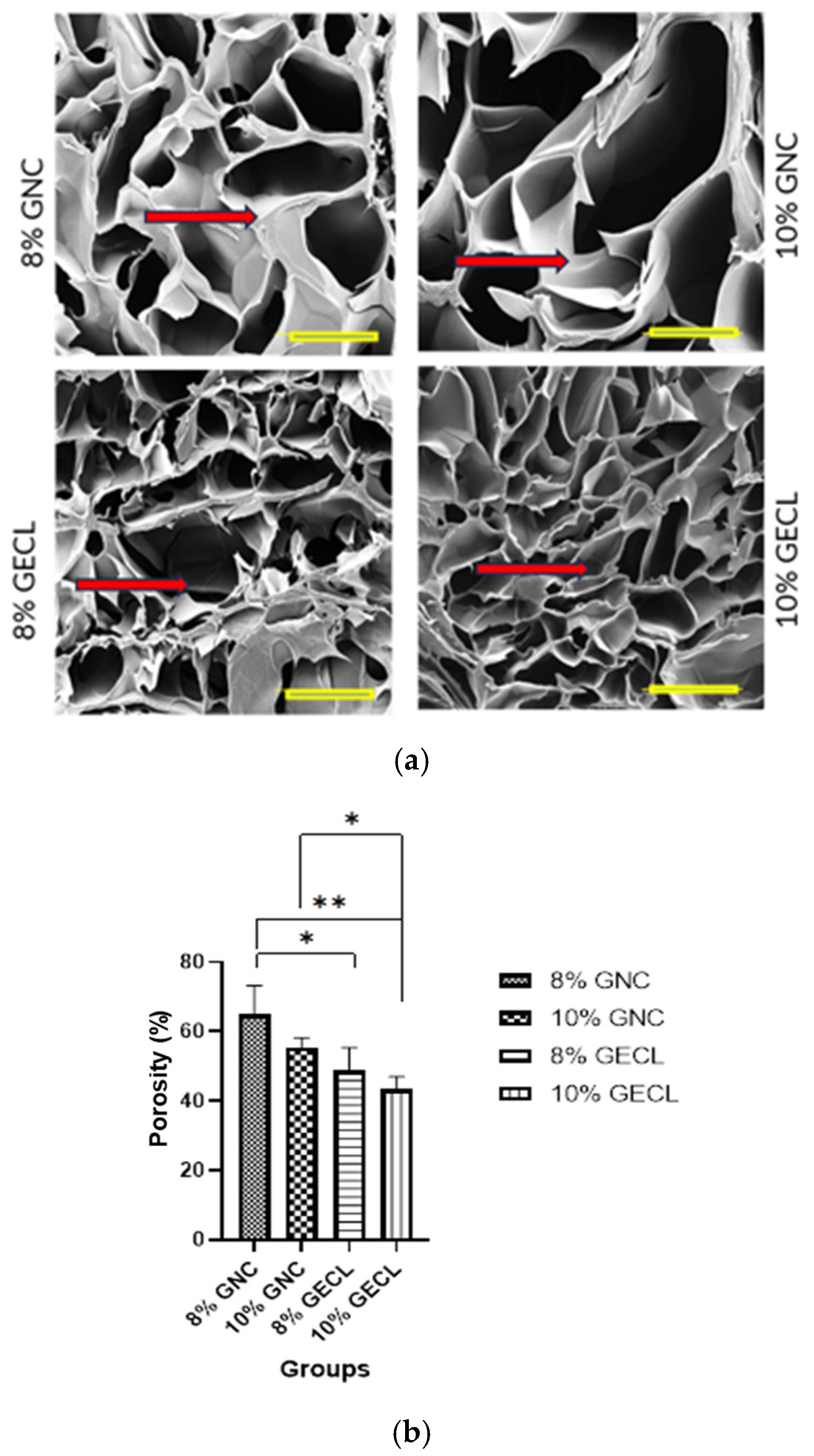
The variation in the swelling ratio was reflected in the porosity differences between the crosslinked and non-crosslinked hydrogels. As expected, crosslinking reduced the overall porosity [
40,
44]. The crosslinked hydrogels exhibited an interconnected pore network, which is vital for allowing efficient water absorption and retention within the hydrogel matrix. Beyond its influence on swelling behavior and water infiltration, this porous structure is also essential to support cell infiltration, facilitate the exchange of oxygen and nutrients, and ultimately, accelerate the healing process [
45]. This has been reported by another study that also showed reduced porosity due to GNP crosslinking; however, this reduction did not affect the viability of the cells cultured within the hydrogels [
46].
Given that porosity directly influences water permeability, it was essential to evaluate the water vapor transmission rate (WVTR), which represents the rate at which water vapor diffuses through the hydrogel matrix [
40]. This parameter is critical in assessing the hydrogel’s ability to maintain a moist wound environment—an important factor for effective wound healing. An optimal WVTR helps sustain the ideal moisture balance at the wound site, which is known to facilitate cell migration, enhance collagen synthesis, and minimize the risk of infection. Previous studies have reported that the ideal WVTR for skin biomimetic scaffolds ranges from 700 to 1200 g/m
2/h, providing a balance between moisture retention and the prevention of wound bed dehydration.
Surface wettability is another measure of hydrogel hydrophilicity, which is important to assess a hydrogel’s suitability for wound healing. A lower contact angle reflects higher hydrophilicity, indicating better cell attachment, moisture retention, and lesion exudate absorption, thus supporting effective healing [
47,
48]. Additionally, wettability influences the hydrogel’s adhesion capability, as higher wettability enhances adherence to wet biological tissues by reducing the resistance from physiological fluids [
48,
49]. All the hydrogels in this study demonstrated high hydrophilicity. While crosslinking modifications enhance wettability [
50], our findings revealed that GNP only trivially modified the inherently high wettability of the gelatin-based hydrogels. Overall, both the crosslinked and non-crosslinked hydrogels exhibited suitable hydrophilicity for a conducive environment for tissue regeneration.
In vitro biodegradation was evaluated to address the limitation of rapid gelatin degradation post-implantation. Crosslinked hydrogels (GECL) exhibited greater durability than gelatin-only hydrogels, meeting the requirement of at least 14 days for wound healing applications [
51]. The 10% GECL showed better stability compared with 8% GECL, apparently due to increased gelatin concentration. GNP crosslinking has been shown to enhance the stability of hydrogels by preventing enzymatic degradation [
52]. Rapid degradation could lead to premature scaffold loss before new skin tissue formation occurs [
52]. Furthermore, the crosslinker GNP has been reported to improve scaffold stability while providing antioxidant properties to support wound healing and sustain cell migration and differentiation from surrounding tissues [
26]. Given their enhanced durability and bioactive properties, crosslinked hydrogels (GECL) present a promising platform for hUCMSC-sEV incorporation and future wound healing applications.
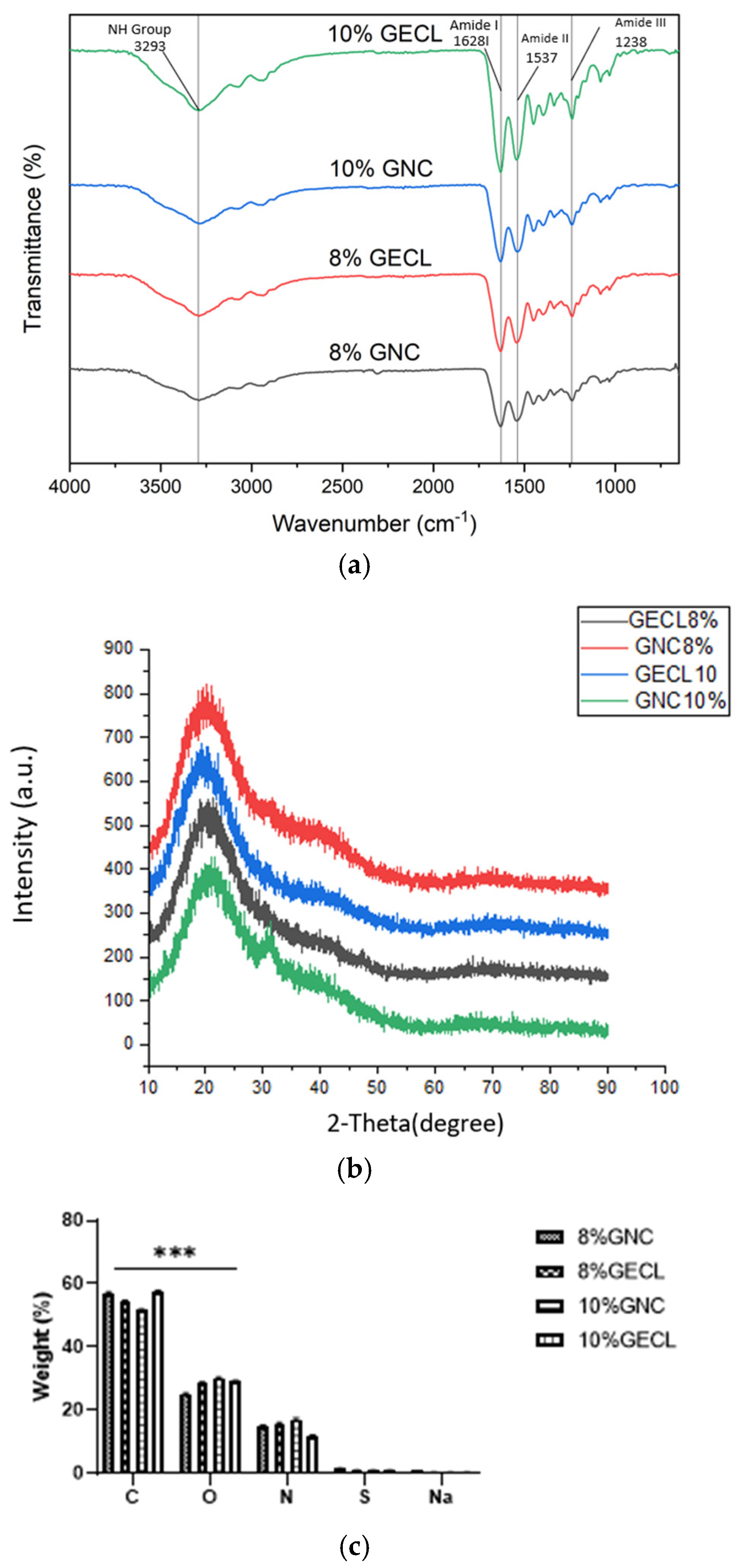
The chemical characterization of GECL and GNC via EDX revealed only minimal elemental changes following the addition of GNP. Gelatin, rich in amino acids and composed of carbon, oxygen, nitrogen, sulfur, and sodium, maintains key functional groups such as carboxylic, amide, ester, aromatic, and alkane [
53]. FTIR analysis showed no significant peak shifts between GECL and GNC; however, subtle differences in the intensity and shape of the amide I and II bands indicate that GNP crosslinking induces minor structural modifications in the gelatin matrix. This observation is consistent with earlier findings [
51] and another previous study by our research team reporting that genipin and PVA incorporation do not significantly alter the native amorphous structure of gelatin hydrogels [
54]. While FTIR provides important insight into functional group interactions, we acknowledge its limitations in resolving fine chemical changes. In future work, we aim to complement these results with more sensitive techniques.
Moreover, XRD results demonstrated that, while GNP incorporation marginally increased the degree of crystallinity, the hydrogels remained predominantly amorphous—an advantageous feature for biomedical applications [
55]. Excessive crystallinity in biomaterials has been associated with increased stiffness, which can hinder cellular adhesion and proliferation [
56]. The mechanical strength of biomaterials is critical for their ability to withstand pressure during implantation at wound sites [
57].
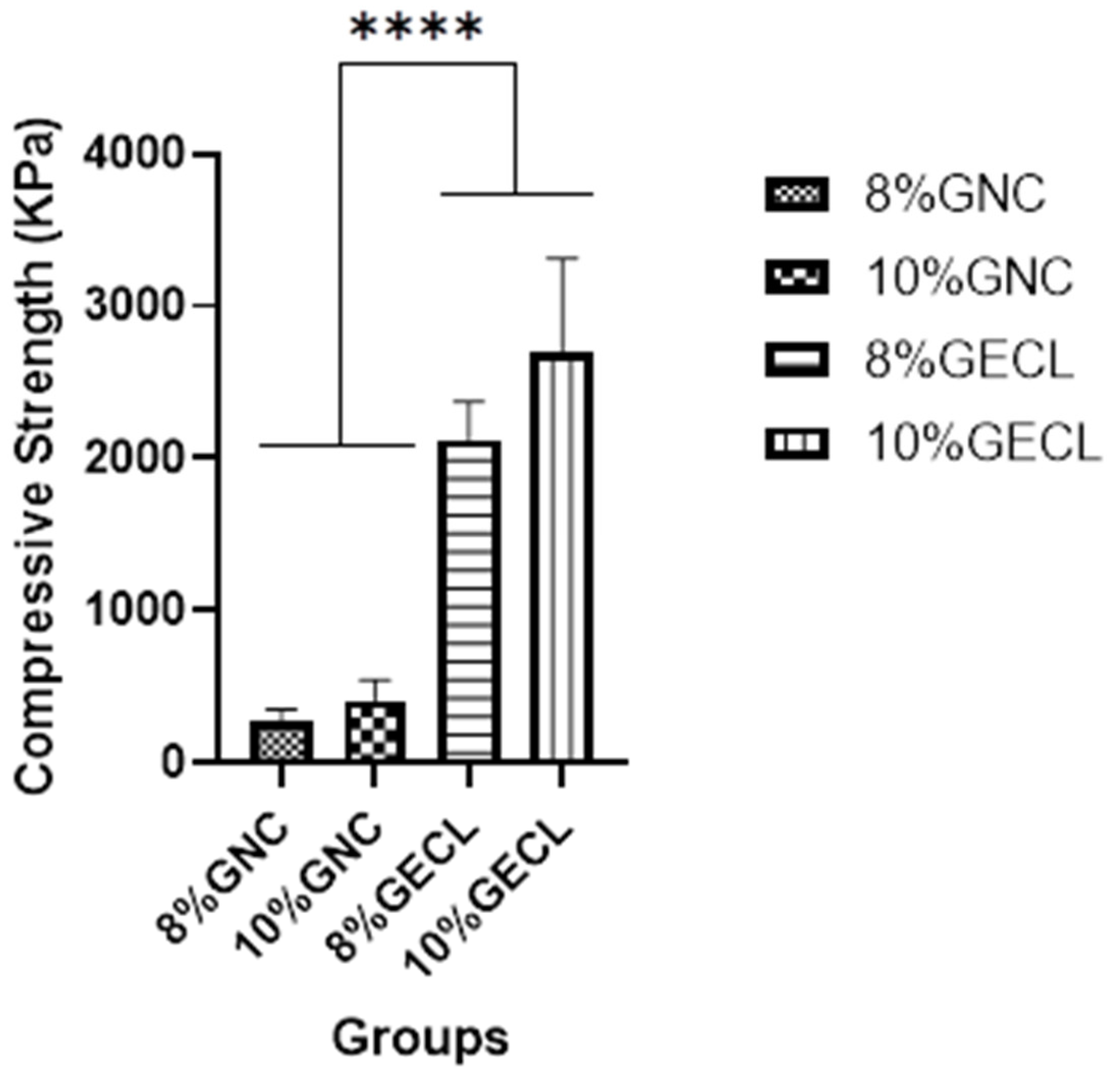
The mechanical properties of the hydrogels, particularly compressive strength, underscore their potential for biological applications. The GNP-crosslinked gelatin hydrogel exhibited enhanced mechanical stability, closely resembling the biomechanical characteristics of native skin, thereby making it well suited for skin-related applications. The incorporation of GECL contributed to a notable increase in compressive strength, reaching approximately 2 MPa. This enhancement is attributed to the stabilizing interactions between GECL and gelatin molecules, which reinforce the hydrogel matrix and improve its structural integrity. Comparable improvements have been observed in other crosslinked systems, such as chitosan composite hydrogels (CS-CGO), where compressive stress increased from 1.9 MPa to 4.2 MPa, further supporting the role of crosslinking in optimizing hydrogel performance [
58]. Overall, the improved mechanical properties position these hydrogels as promising candidates for dynamic and mechanically demanding applications, including wound healing and skin regeneration.
In summary, the incorporation of GNPs into the gelatin matrix induced only subtle structural modifications without significantly altering its overall chemical composition or amorphous nature. This balance is critical for wound healing applications, as the maintained amorphous structure facilitates optimal cell infiltration and nutrient diffusion, while the minor enhancements in crystallinity and crosslinking contribute to improved mechanical stability and controlled degradation [
58]. Collectively, these properties support a conducive environment for tissue regeneration and suggest that GECL hydrogels are well suited for use in wound healing therapies.
Functionalizing hydrogels with bioactive compounds has been explored to enhance their regenerative properties, such as angiogenesis promotion, immunomodulation, and cell proliferation stimulation [
59]. In this context, hUCMSC-sEVs have emerged as a promising tool for hydrogel functionalization because they inherently exhibit these therapeutic effects [
60]. The selection of hUCMSCs as an EV source offers several advantages. Their high proliferation capacity, immunomodulatory effects, and robust paracrine signaling contribute significantly to the therapeutic potential of the EVs they release. As key mediators that propagate the beneficial properties of their parent cells [
6], hUCMSC-sEVs are expected to deliver these effects to target tissues [
61]. Additionally, the non-invasive and ethically favorable process of obtaining hUCMSCs further supports their utility in both clinical and research applications [
62].
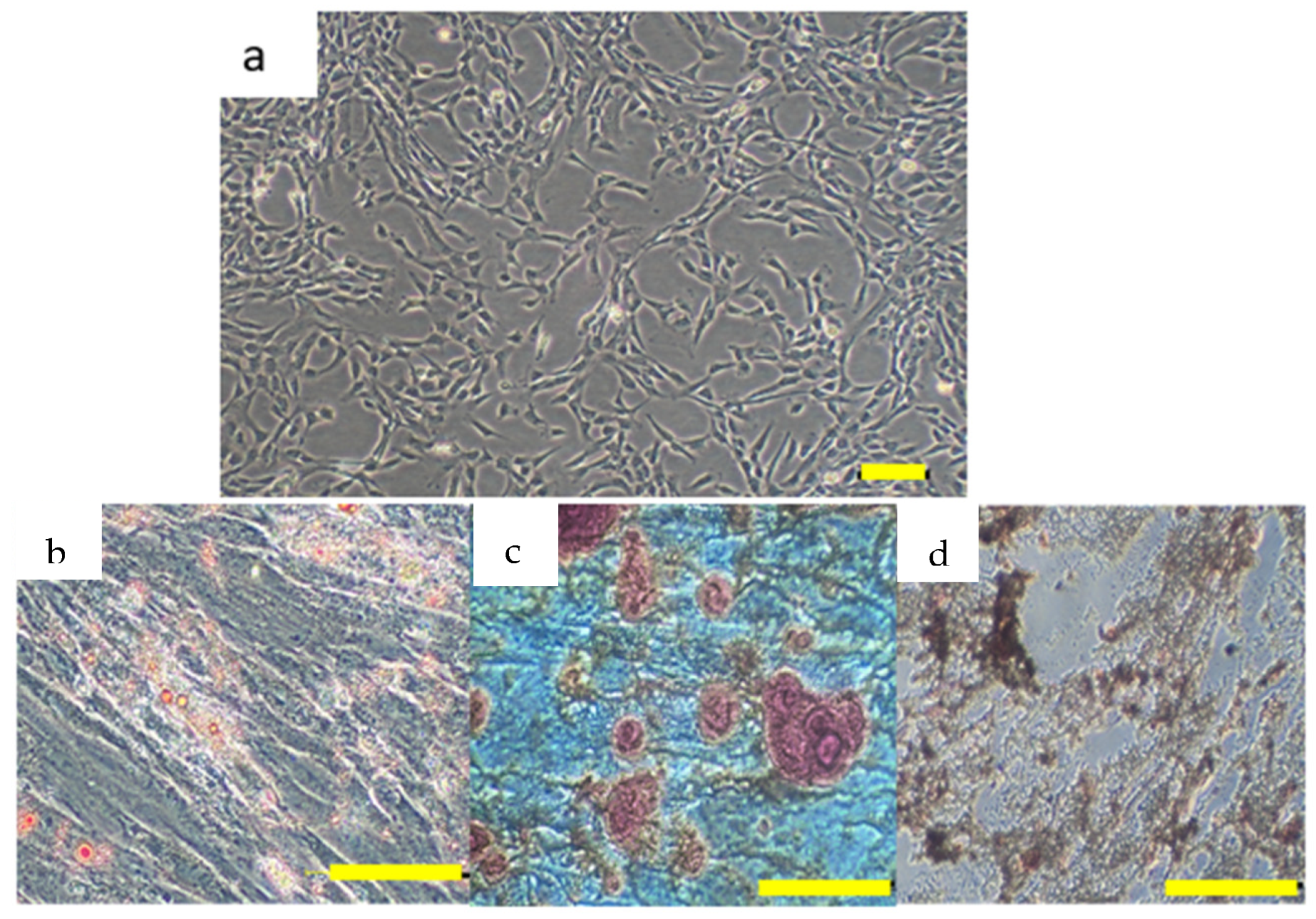
The characterization of the isolated hUCMSCs was performed according to the ISCT criteria, which are critical for establishing the MSC identity [
34]. The hUCMSCs exhibited the characteristic properties of MSCs, such as plastic adherence, spindle-shaped morphology, and the expression of the surface markers CD73, CD90, CD105, and CD44, while being negative for the hematopoietic markers CD34 and CD45 [
63]. These findings agree with previous studies that reported similar profiles for MSCs derived from various tissues, including bone marrow and adipose tissue, supporting the claim of the successful isolation of hUCMSCs [
64]. The multilineage differentiation potential of adipogenic, osteogenic, and chondrogenic lineages further corroborates the stemness of the isolated cells [
65]. Similarly, this comprehensive characterization aligns with our previous findings, confirming the identity of the cells [
66].
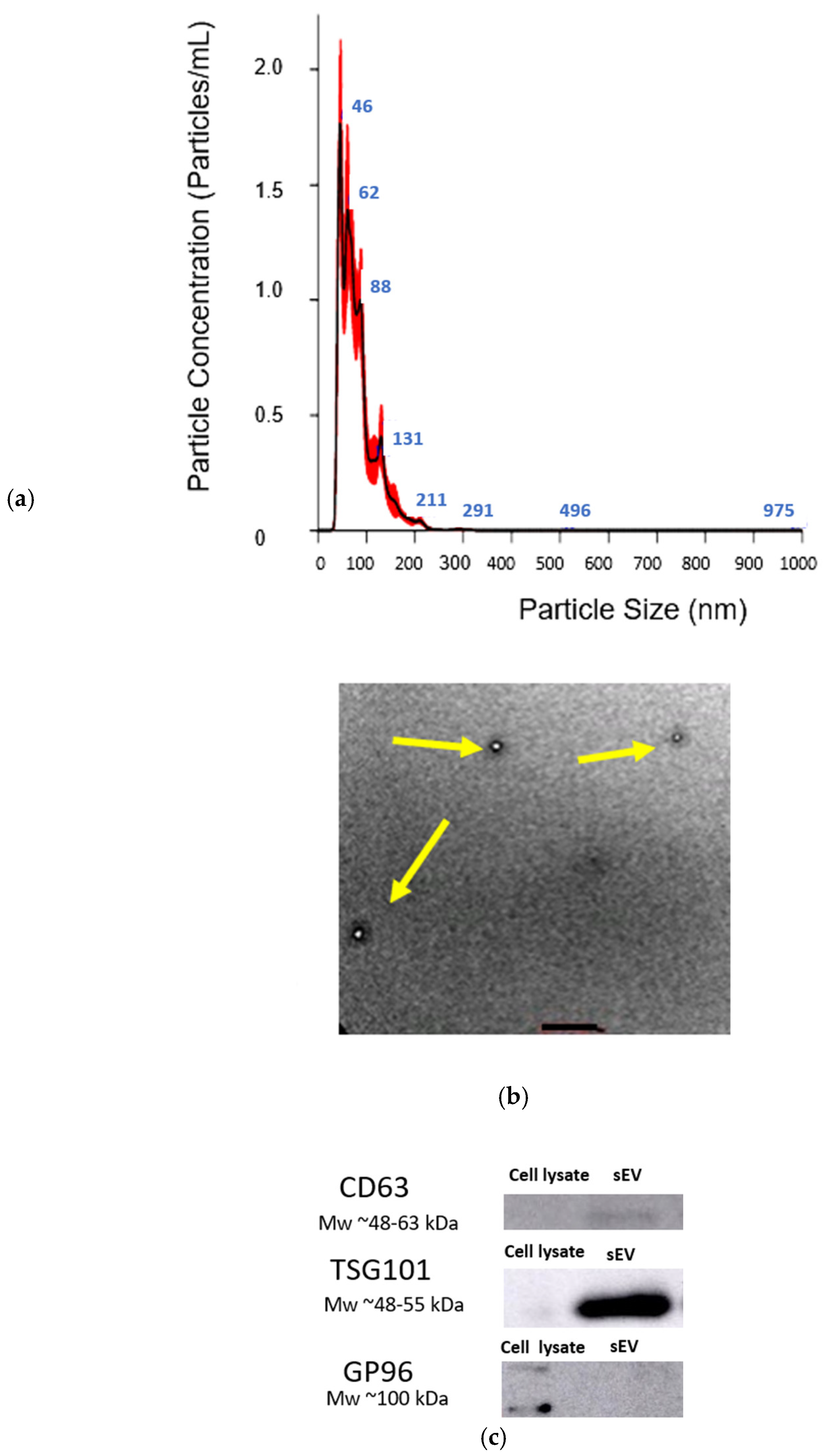
hUCMSC-sEVs were successfully isolated and characterized using TFF, a highly efficient and scalable technique for EV isolation, particularly suitable for processing large volumes of conditioned medium. Unlike conventional dead-end filtration, TFF directs the fluid tangentially across the membrane surface rather than perpendicularly, reducing the accumulation of retained material and minimizing membrane clogging. This tangential flow mechanism enhances the separation efficiency and ensures consistent performance.
The characterization of hUCMSC-sEVs confirmed their compliance with the criteria outlined in MISEV 2023 [
67]. The isolated hUCMSC-sEVs exhibited a round morphology and a size range of 50–200 nm and expressed the canonical EV markers CD63 and TSG101, validating their identity as sEVs [
68,
69]. The absence of the GP96 marker further indicated the purity of the preparation, confirming minimal cellular contamination [
70]. These findings establish the successful isolation and enrichment of hUCMSC-sEVs.
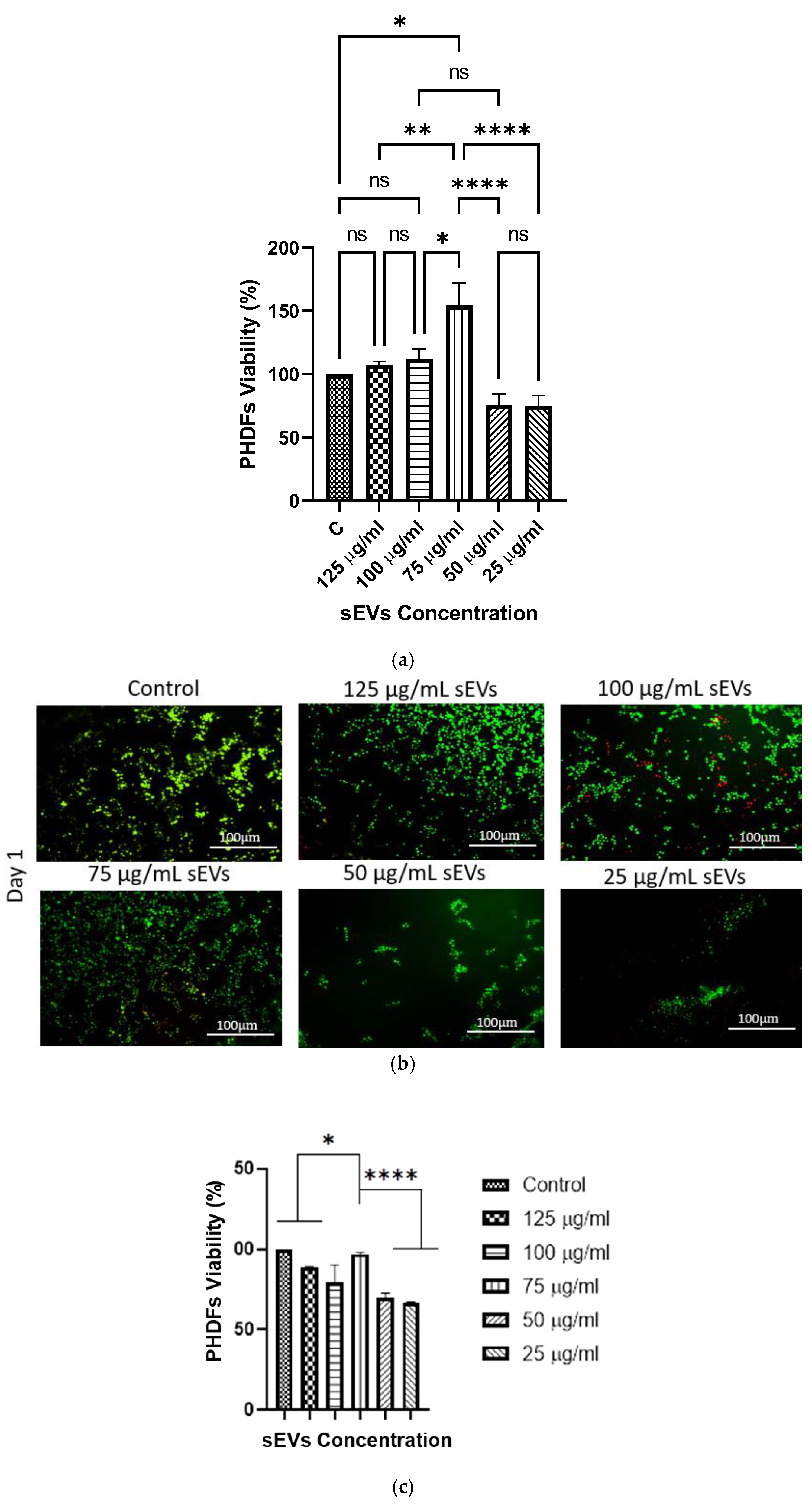
Based on the prior findings, the hydrogel formulation of 10% GECL was chosen for further cell biocompatibility testing. This selection was made due to its optimal polymerization time and slower biodegradation, along with its ideal swelling ratio, WVTR, porosity, and wettability. These characteristics collectively indicate its suitability for biomedical applications.
The biocompatibility of the 3D bioprinted hUCMSC-sEV-incorporated gelatin hydrogel 10% GECL was evaluated by assessing both PHDF viability and attachment. Compared to the hydrogels without hUCMSC-sEV, those incorporating hUCMSC-sEVs exhibited an additive effect on PHDFs viability. This finding indicates that the presence of hUCMSC-sEVs promotes fibroblast survival and proliferation without inducing cytotoxic effects, consistent with previously reported studies highlighting the pro-regenerative and cytocompatibility nature of sEV-based therapies [
10,
18]. Notably, the concentration of 75 μg/mL of hUCMSC-sEV resulted in significantly higher PHDF viability. Increasing the dose beyond 75 μg/mL led to a decline in PHDFs viability, highlighting the importance of optimizing hUCMSC-sEV concentration for optimal outcomes [
71]. Additionally, high PHDF attachment (80–90%) across all hUCMSC-sEVs concentrations further confirmed the excellent biocompatibility of the hydrogel platform. The hUCMSC-sEV-incorporated 10% GECL hydrogels exhibited high cell attachment, likely due to the combined influence of the hydrogel’s improved structure with crosslinking, which provides robust mechanical support for cell adhesion, and the bioactive properties of hUCMSC-sEV. These results underline the potential of 10% GECL hydrogels, particularly those incorporating 75 µg/mL hUCMSC-sEV, as biocompatible scaffolds for tissue engineering and wound healing. Meanwhile, the in vitro scratch wound healing assay demonstrated that small extracellular vesicles (sEVs) derived from human umbilical cord mesenchymal stem cells (hUCMSCs) significantly enhanced the migratory capacity of human dermal fibroblasts (HDFs), thereby accelerating wound closure. Treatment with both 75 µg/mL and 100 µg/mL concentrations of sEVs led to a marked reduction in wound area compared to the untreated control, with statistically significant differences observed by day 3. Notably, there was no significant difference between the two concentrations, suggesting that 75 µg/mL is sufficient to elicit an optimal therapeutic response within the evaluated timeframe. These findings underscore the robust regenerative potential of hUCMSC-sEVs in promoting fibroblast migration and cutaneous wound healing, reinforcing their promise as a therapeutic agent in skin tissue regeneration. Consistent with our findings, a previous study demonstrated that sEV treatment enhanced cellular migration toward the wound site, promoting partial gap closure compared to controls [
72]. However, further investigations are needed to assess long-term cell viability and functional outcomes, which will be crucial for fully establishing the therapeutic potential of these scaffolds in wound healing.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}