
Enhanced Melt Memory Effects in Poly(butylene succinate) Through Incorporation of Extended-Chain Crystals



Abstract

1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of PBS/Urea Complex
2.3. Extraction of Extended-Chain Crystals
2.4. Preparation of PBS/ECCs Composites
2.5. Characterization
- Non-isothermal Crystallization Analysis: Samples were cooled to 40 °C at a constant rate of 10 °C/min to investigate the crystallization behavior, followed by reheating to 160 °C at the same rate to examine the melting characteristics. The crystallization temperature (Tc), melting temperature (Tm), and their corresponding enthalpies (ΔHc, ΔHm) were determined from the first heating and cooling curves, respectively.
- Isothermal Crystallization Analysis: Samples were rapidly quenched (50 °C/min) to predetermined crystallization temperatures and maintained isothermally until complete crystallization. The evolution of crystallization was monitored by recording the exothermic heat flow as a function of time.
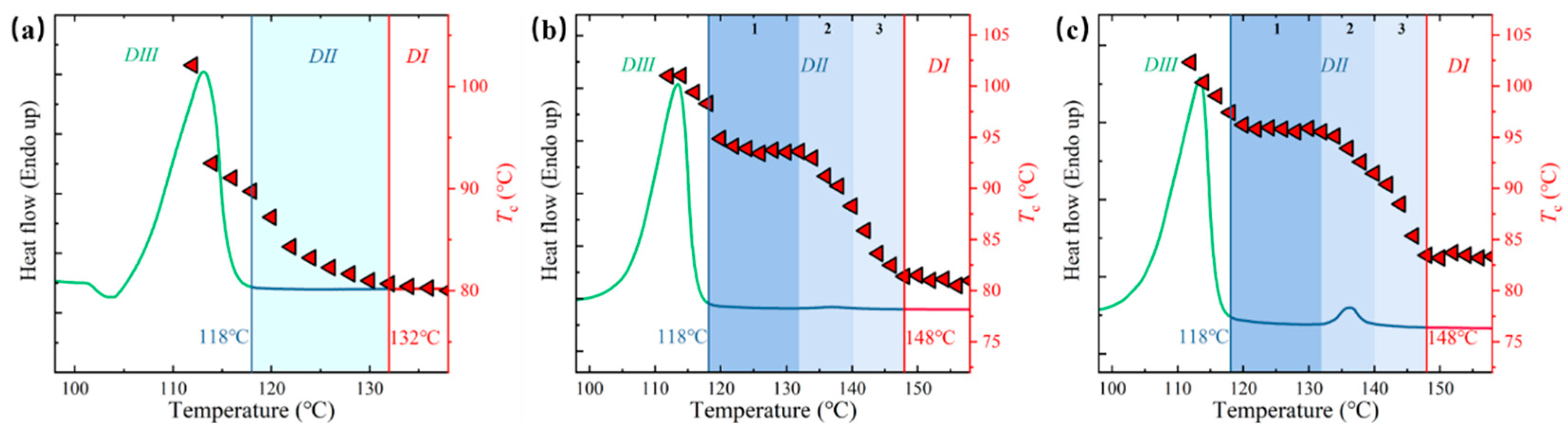
- Self-nucleation (SN) investigation: The self-nucleation behavior was investigated using a modified thermal protocol specifically designed to account for the unique structural characteristics of ECCs. Unlike conventional semi-crystalline polymers, ECCs undergo irreversible structural changes upon complete melting, necessitating a carefully tailored three-step thermal procedure (Figure S1 in Supporting Information): (i) The initial step involved controlled heating to a predetermined self-nucleation temperature (Ts) at a rate of 10 °C/min, followed by an isothermal hold period of 5 min. The Ts values were systematically varied to probe different self-nucleation domains. (ii) Subsequently, samples underwent controlled cooling from Ts to 40 °C at a constant rate of 10 °C/min. This cooling stage was designed to observe the nucleation efficiency of any remaining self-nuclei or memory effects through the analysis of crystallization temperatures and peak shapes. (iii) The final stage consisted of reheating to 160 °C at 10 °C/min to evaluate the melting behavior of crystals formed during the cooling process. This methodology facilitates quantitative assessment of nucleation efficiency through crystallization temperature shifts and exotherm characteristics. The thermograms revealed three distinct self-nucleation domains characterized by their crystallization behavior: [36,38] Domain I exhibiting complete melting with no correlation between Tc and Ts; Domain II displaying prominent self-nucleation effects at intermediate Ts; and Domain III manifesting incomplete melting at lower Ts values.
3. Results and Discussion
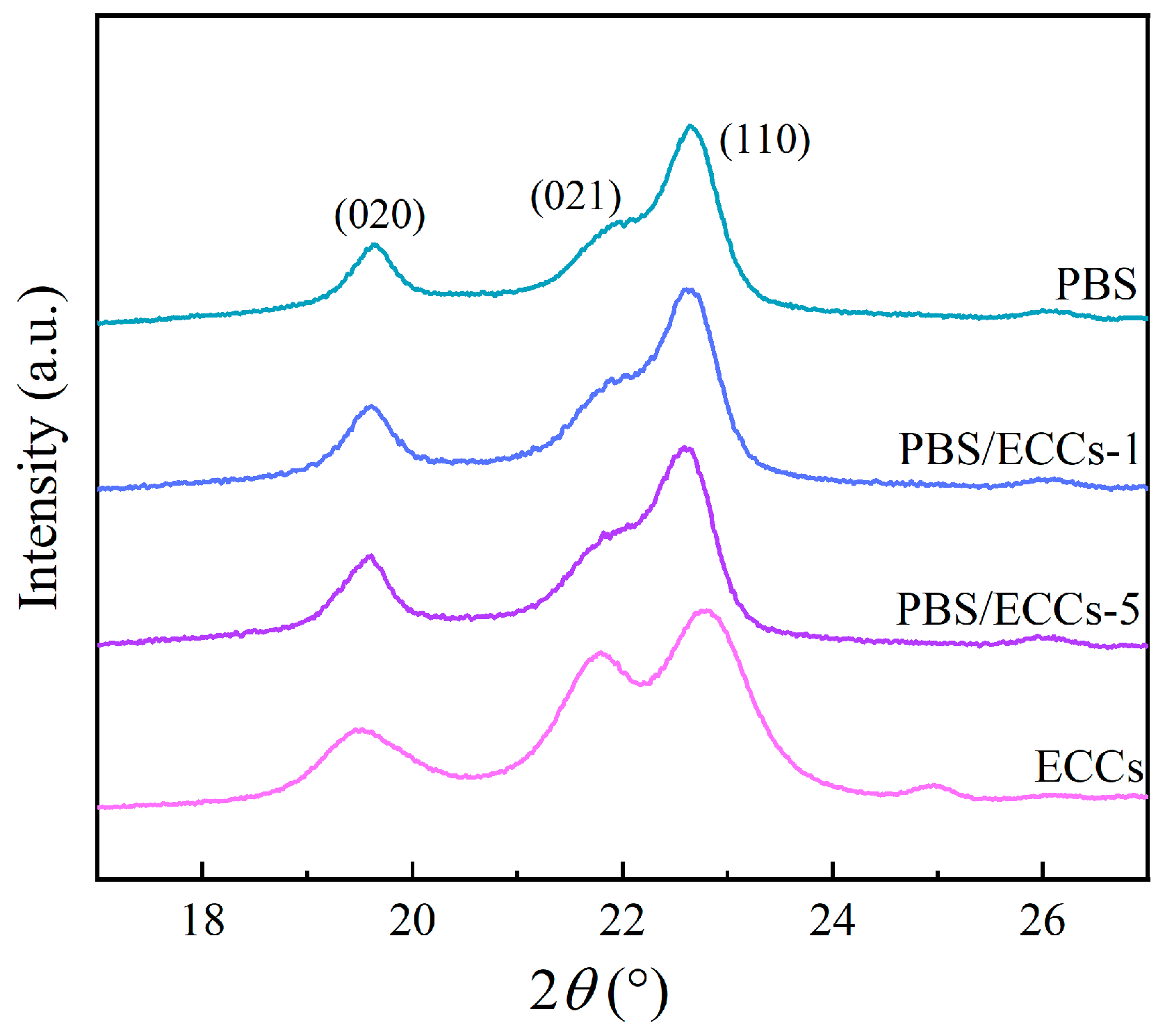
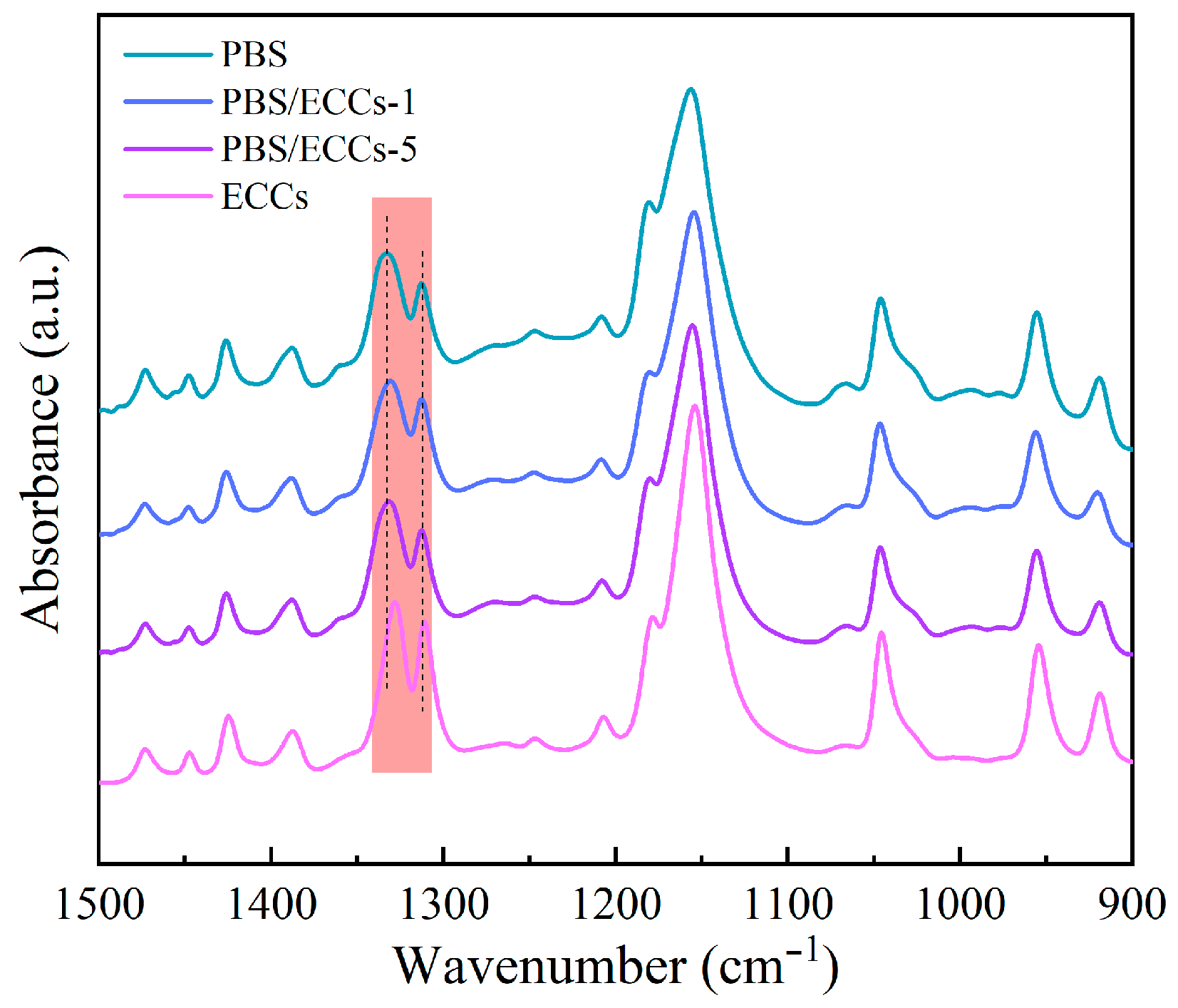
3.1. Structural Characterization
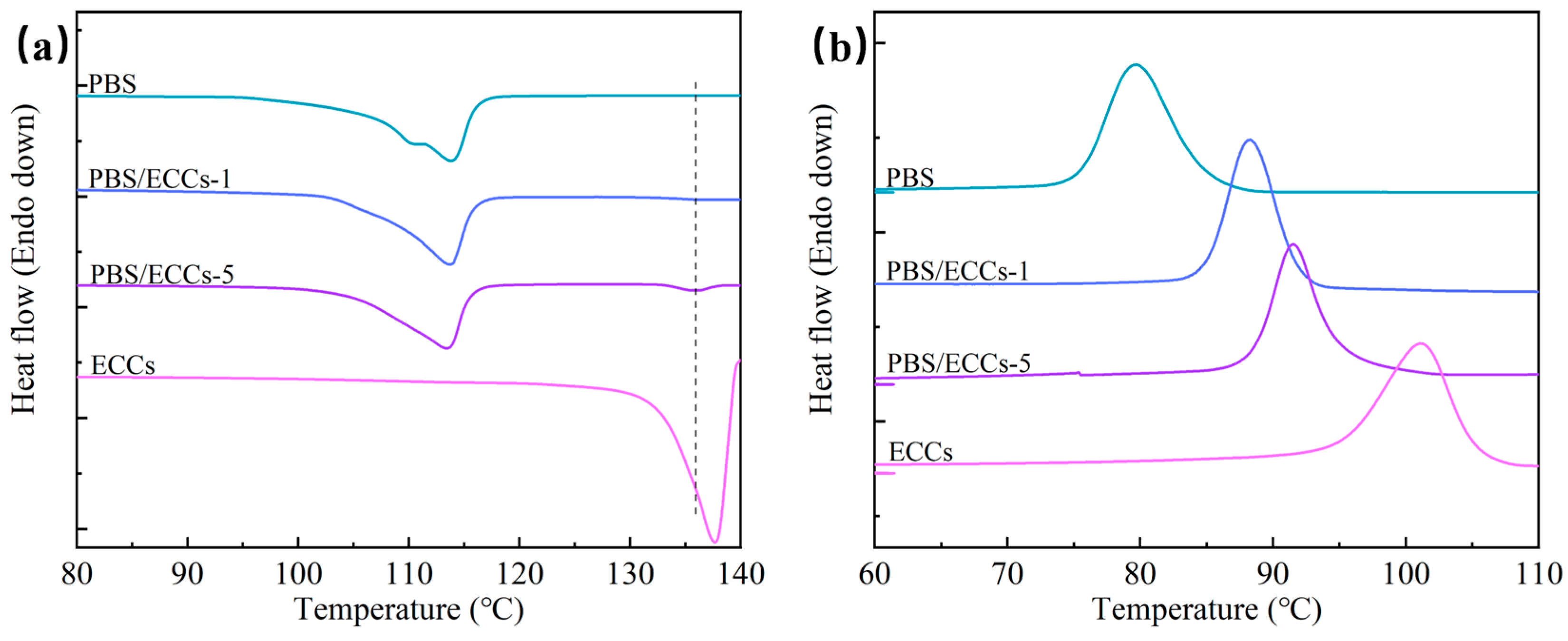
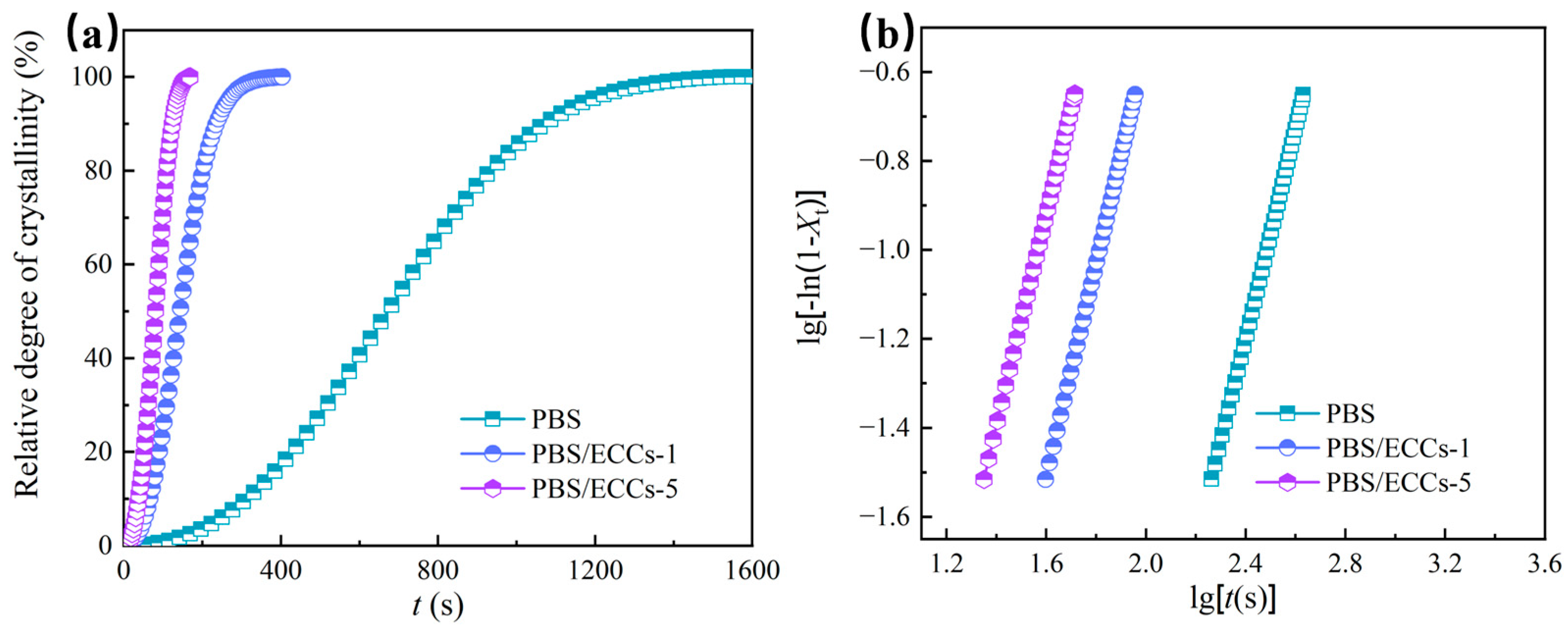
3.2. Crystallization Behavior and Thermal Properties
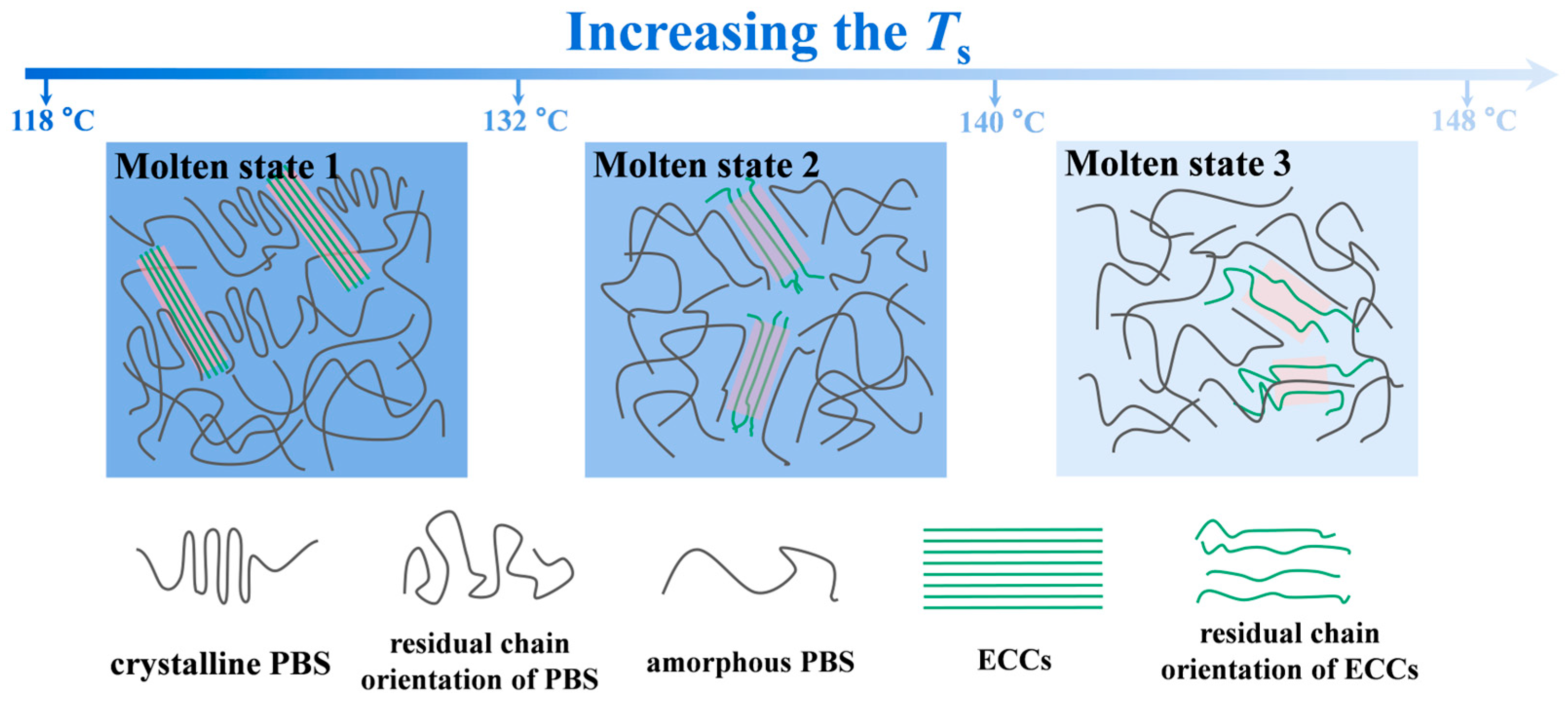
3.3. Temperature-Dependent Melt Memory Effects
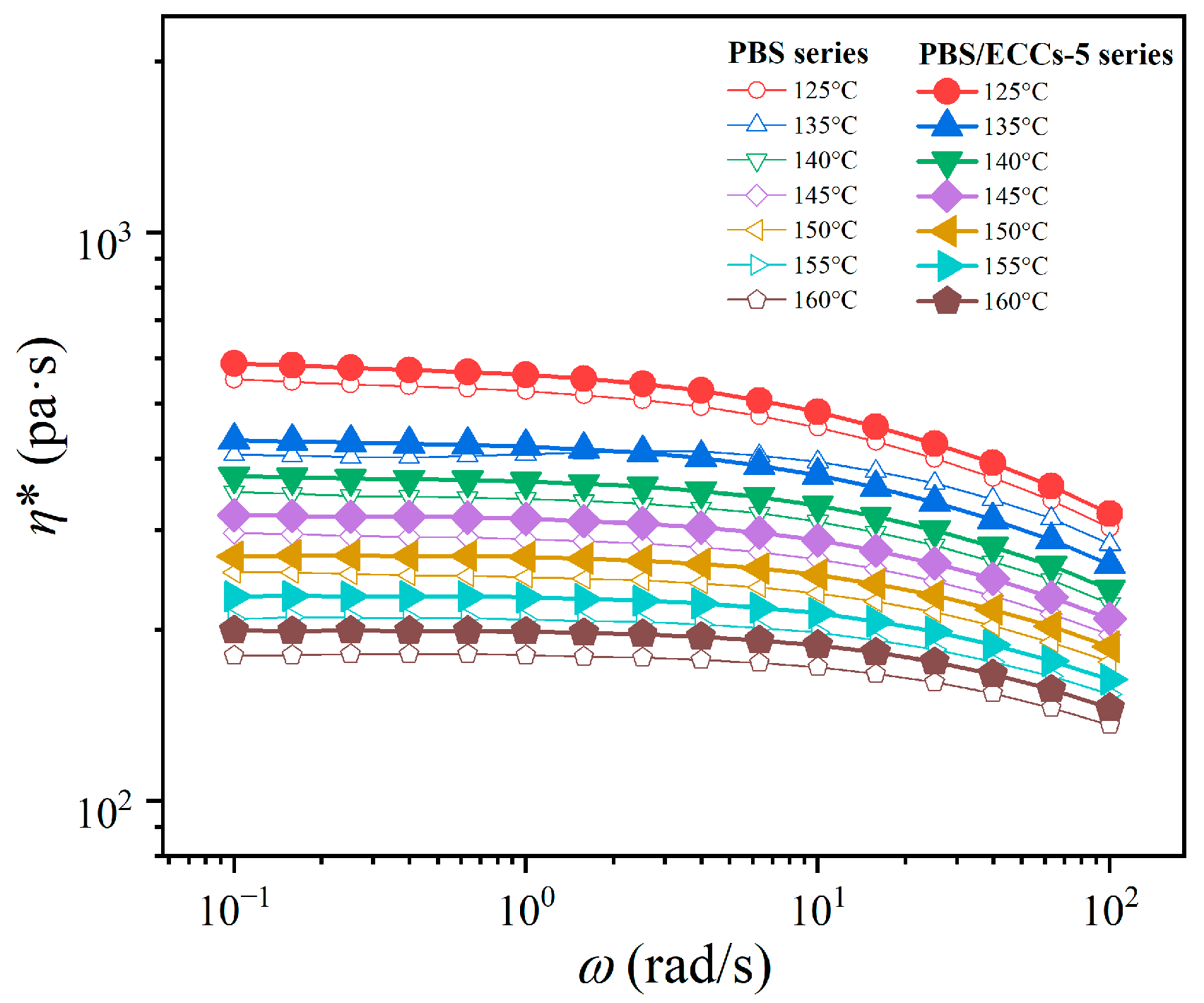
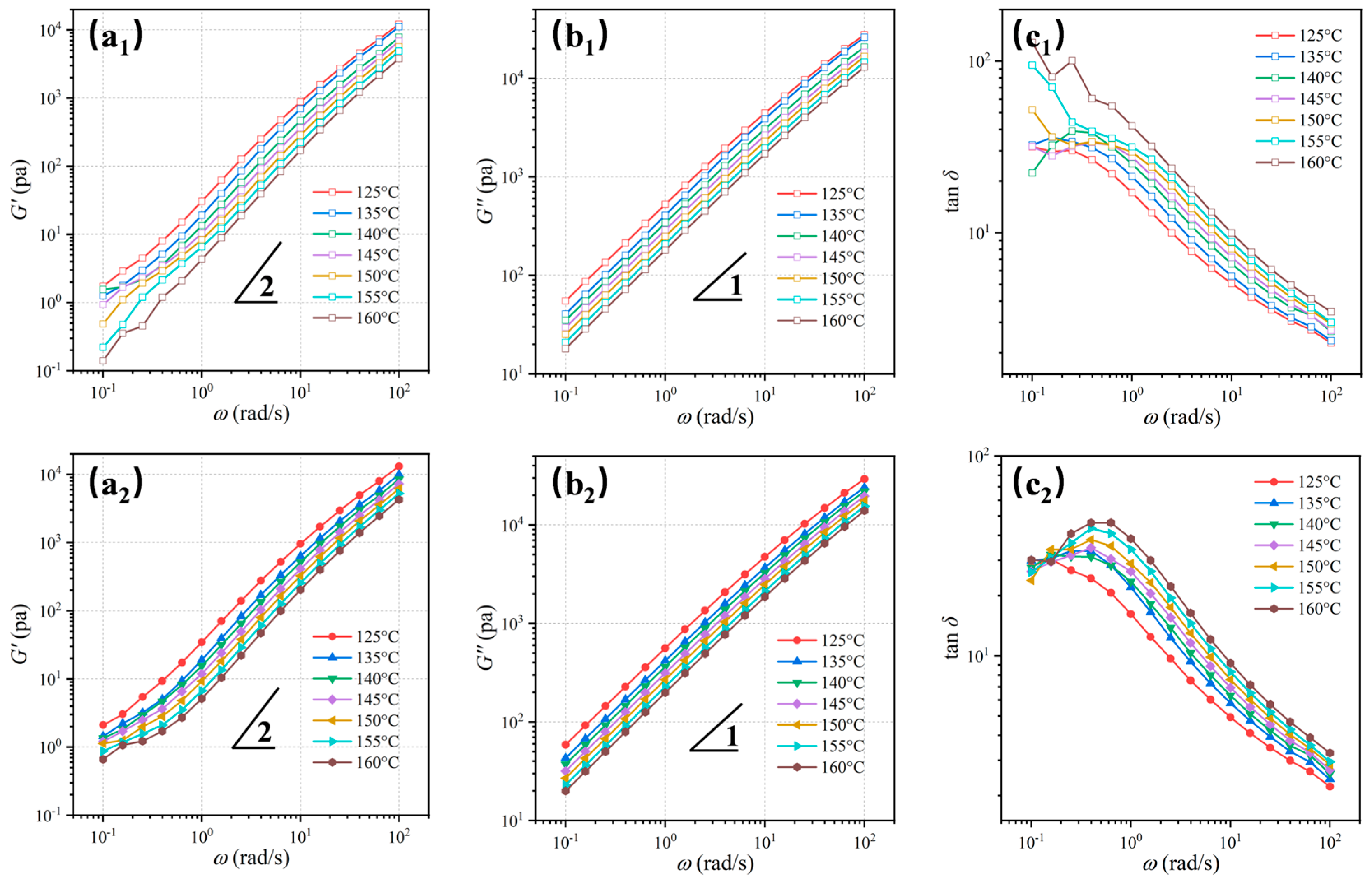
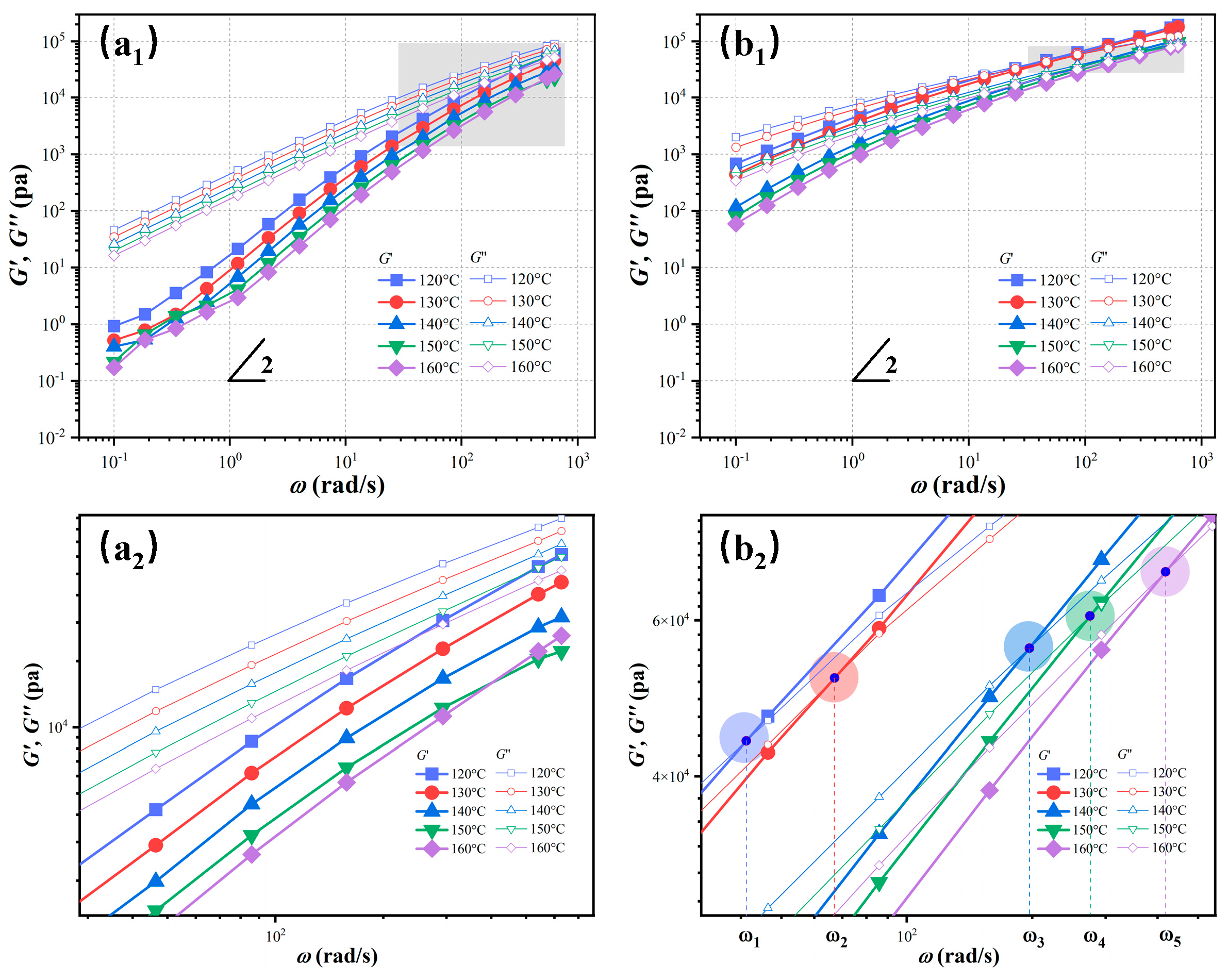
3.4. Rheological Analysis of Melt Structure Evolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, J.; Guo, B.H. Poly(butylene succinate) and its copolymers: Research, development and industrialization. Biotechnol. J. 2010, 5, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.S.; Okamoto, M. Biodegradable polylactide and its nanocomposites: Opening a new dimension for plastics and composites. Macromol. Rapid Commun. 2003, 24, 815–840. [Google Scholar] [CrossRef]
- Pérez-Camargo, R.A.; Liu, G.; Cavallo, D.; Wang, D.; Müller, A.J. Effect of the Crystallization Conditions on the Exclusion/Inclusion Balance in Biodegradable Poly(butylene succinate-ran-butylene adipate) Copolymers. Biomacromolecules 2020, 21, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Fabbri, M.; Lotti, N.; Gamberini, R.; Rimini, B.; Munari, A. Poly(butylene succinate)-based polyesters for biomedical applications: A review. Eur. Polym. J. 2016, 75, 431–460. [Google Scholar] [CrossRef]
- Barletta, M.; Aversa, C.; Ayyoob, M.; Gisario, A.; Hamad, K.; Mehrpouya, M.; Vahabi, H. Poly(butylene succinate)(PBS): Materials, processing, and industrial applications. Prog. Polym. Sci. 2022, 132, 101579. [Google Scholar] [CrossRef]
- Li, Y.; Huang, G.; Chen, C.; Wei, X.W.; Dong, X.; Zhao, W.; Ye, H.M. Poly(butylene succinate-co-butylene acetylenedicarboxylate): Copolyester with Novel Nucleation Behavior. Polymers 2021, 13, 365. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Achilias, D.S.; Bikiaris, D.N. Crystallization kinetics of biodegradable poly(butylene succinate) under isothermal and non-isothermal conditions. Macromol. Chem. Phys. 2007, 208, 1250–1264. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Kurokawa, H.; Doi, Y. Solid-state microstructures, thermal properties, and crystallization of biodegradable poly(butylene succinate) (PBS) and its copolyesters. Biomacromolecules 2001, 2, 605–613. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, P. Crystallization of biodegradable and biobased polyesters: Polymorphism, cocrystallization, and structure-property relationship. Prog. Polym. Sci. 2020, 109, 101291. [Google Scholar] [CrossRef]
- Fujimaki, T. Processability and properties of aliphatic polyesters, ‘BIONOLLE’, synthesized by polycondensation reaction. Polym. Degrad. Stab. 1998, 59, 209–214. [Google Scholar] [CrossRef]
- Tan, L.; Chen, Y.; Zhou, W.; Ye, S.; Wei, J. Novel approach toward poly(butylene succinate)/single-walled carbon nanotubes nanocomposites with interfacial-induced crystallization behaviors and mechanical strength. Polymer 2011, 52, 3587–3596. [Google Scholar] [CrossRef]
- Bian, J.; Han, L.; Wang, X.; Wen, X.; Han, C.; Wang, S.; Dong, L. Nonisothermal crystallization behavior and mechanical properties of poly(butylene succinate)/silica nanocomposites. J. Appl. Polym. Sci. 2010, 116, 902–912. [Google Scholar] [CrossRef]
- Jin, T.-X.; Liu, C.; Zhou, M.; Chai, S.-g.; Chen, F.; Fu, Q. Crystallization, mechanical performance and hydrolytic degradation of poly(butylene succinate)/graphene oxide nanocomposites obtained via in situ polymerization. Compos. Part A Appl. Sci. Manuf. 2015, 68, 193–201. [Google Scholar] [CrossRef]
- Shih, Y.-F.; Chen, L.; Jeng, R. Preparation and properties of biodegradable PBS/multi-walled carbon nanotube nanocomposites. Polymer 2008, 49, 4602–4611. [Google Scholar] [CrossRef]
- Li, J.; Luo, X.; Lin, X. Preparation and characterization of hollow glass microsphere reinforced poly(butylene succinate) composites. Mater. Des. 2013, 46, 902–909. [Google Scholar] [CrossRef]
- Kuilla, T.; Bhadra, S.; Yao, D.; Kim, N.H.; Bose, S.; Lee, J.H. Recent advances in graphene based polymer composites. Prog. Polym. Sci. 2010, 35, 1350–1375. [Google Scholar] [CrossRef]
- Wittmann, J.C.; Lotz, B. Epitaxial crystallization of polyethylene on organic substrates: A reappraisal of the mode of action of selected nucleating agents. J. Polym. Sci. Polym. Phys. Ed. 1981, 19, 1837–1851. [Google Scholar] [CrossRef]
- Bledsoe, J.C.; Crane, G.H.; Locklin, J.J. Beyond Lattice Matching: The Role of Hydrogen Bonding in Epitaxial Nucleation of Poly(hydroxyalkanoates) by Methylxanthines. ACS Appl. Polym. Mater. 2023, 5, 3858–3865. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, Y.; Zhang, C.; Dong, Z.; Wang, K.; Wang, D. Morphological Characteristics of β-Nucleating Agents Governing the Formation of the Crystalline Structure of Isotactic Polypropylene. Macromolecules 2021, 54, 6824–6834. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, W.; Deng, H.; Zhang, Q.; Fu, Q. Control of Crystal Morphology in Poly(l-lactide) by Adding Nucleating Agent. Macromolecules 2011, 44, 1233–1237. [Google Scholar] [CrossRef]
- Ye, H.-M.; Wang, R.-D.; Liu, J.; Xu, J.; Guo, B.-H. Isomorphism in poly(butylene succinate-co-butylene fumarate) and its application as polymeric nucleating agent for poly(butylene succinate). Macromolecules 2012, 45, 5667–5675. [Google Scholar] [CrossRef]
- Ye, H.-M.; Tang, Y.-R.; Xu, J.; Guo, B.-H. Role of poly(butylene fumarate) on crystallization behavior of poly(butylene succinate). Ind. Eng. Chem. Res. 2013, 52, 10682–10689. [Google Scholar] [CrossRef]
- Wei, X.-W.; Yang, L.-L.; Li, Y.; Meng, X.; Cai, L.-H.; Zhou, Q.; Ye, H.-M. Asymmetrical formation of isomorphism in the crystalline/crystalline blend of poly(butylene succinate) and poly(butylene fumarate). Polymer 2021, 235, 124282. [Google Scholar] [CrossRef]
- Zheng, Y.; Tian, G.; Xue, J.; Zhou, J.; Huo, H.; Li, L. Effects of isomorphic poly(butylene succinate-co-butylene fumarate) on the nucleation of poly(butylene succinate) and the formation of poly(butylene succinate) ring-banded spherulites. CrystEngComm 2018, 20, 1573–1587. [Google Scholar] [CrossRef]
- Varga, J.; Karger-Kocsis, J. Rules of supermolecular structure formation in sheared isotactic polypropylene melts. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 657–670. [Google Scholar] [CrossRef]
- Xu, J.; Ma, Y.; Hu, W.; Rehahn, M.; Reiter, G. Cloning polymer single crystals through self-seeding. Nat. Mater. 2009, 8, 348–353. [Google Scholar] [CrossRef]
- Ye, H.-M.; Chen, X.-T.; Li, H.-F.; Zhang, P.; Ma, W.; Li, B.; Xu, J. Industrializable and sustainable approach for preparing extended-chain crystals of biodegradable poly(butylene succinate) and their applications. Polymer 2019, 160, 93–98. [Google Scholar] [CrossRef]
- Ye, H.-M.; Chen, X.-T.; Liu, P.; Wu, S.-Y.; Jiang, Z.; Xiong, B.; Xu, J. Preparation of poly(butylene succinate) crystals with exceptionally high melting point and crystallinity from its inclusion complex. Macromolecules 2017, 50, 5425–5433. [Google Scholar] [CrossRef]
- Brochu, S.; Prud’homme, R.E.; Barakat, I.; Jerome, R. Stereocomplexation and Morphology of Polylactides. Macromolecules 1995, 28, 5230–5239. [Google Scholar] [CrossRef]
- Yamane, H.; Sasai, K. Effect of the addition of poly(D-lactic acid) on the thermal property of poly(L-lactic acid). Polymer 2003, 44, 2569–2575. [Google Scholar] [CrossRef]
- Rahman, N.; Kawai, T.; Matsuba, G.; Nishida, K.; Kanaya, T.; Watanabe, H.; Okamoto, H.; Kato, M.; Usuki, A.; Matsuda, M.; et al. Effect of Polylactide Stereocomplex on the Crystallization Behavior of Poly(L-lactic acid). Macromolecules 2009, 42, 4739–4745. [Google Scholar] [CrossRef]
- Wei, X.-F.; Bao, R.-Y.; Cao, Z.-Q.; Yang, W.; Xie, B.-H.; Yang, M.-B. Stereocomplex Crystallite Network in Asymmetric PLLA/PDLA Blends: Formation, Structure, and Confining Effect on the Crystallization Rate of Homocrystallites. Macromolecules 2014, 47, 1439–1448. [Google Scholar] [CrossRef]
- Schmidt, S.C.; Hillmyer, M.A. Polylactide stereocomplex crystallites as nucleating agents for isotactic polylactide. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 300–313. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wei, X.-W.; Wu, T.; Ye, H.-M. Crystallization manipulation of poly(butylene succinate) via hydrazide-based self-assembled hydrogen-bonding interactions. Polymer 2024, 298, 126933. [Google Scholar] [CrossRef]
- Reid, B.O.; Vadlamudi, M.; Mamun, A.; Janani, H.; Gao, H.; Hu, W.; Alamo, R.G. Strong memory effect of crystallization above the equilibrium melting point of random copolymers. Macromolecules 2013, 46, 6485–6497. [Google Scholar] [CrossRef]
- Sangroniz, L.; Cavallo, D.; Müller, A.J. Self-Nucleation Effects on Polymer Crystallization. Macromolecules 2020, 53, 4581–4604. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, R.-J.; Ma, G.-Q.; Ma, Z. Self-nucleation Effect of Crystallization with Various Nucleating Agents. Chin. J. Polym. Sci. 2023, 41, 1912–1920. [Google Scholar] [CrossRef]
- Fillon, B.; Wittmann, J.; Lotz, B.; Thierry, A. Self-nucleation and recrystallization of isotactic polypropylene (α phase) investigated by differential scanning calorimetry. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 1383–1393. [Google Scholar] [CrossRef]
- Solomon, O.; Ciutǎ, I. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Maghami, G.G.; Roberts, G.A. Evaluation of the viscometric constants for chitosan. Die Makromol. Chem. Macromol. Chem. Phys. 1988, 189, 195–200. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wei, X.-W.; Wu, T.; Meng, X.; Ye, H.-M. Manipulating the Co-crystallization Behavior in Isomorphic Copolymers by Introducing a Third Monomeric Unit. Macromolecules 2023, 56, 5080–5088. [Google Scholar] [CrossRef]
- Ihn, K.; Yoo, E.; Im, S. Structure and morphology of poly(tetramethylene succinate) crystals. Macromolecules 1995, 28, 2460–2464. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Kondo, H.; Igarashi, Y.; Noguchi, K.; Okuyama, K.; Washiyama, J. Crystal structures of α and β forms of poly(tetramethylene succinate). Polymer 2000, 41, 4719–4727. [Google Scholar] [CrossRef]
- Binsbergen, F. Heterogeneous nucleation in the crystallization of polyolefins: Part 1. Chemical and physical nature of nucleating agents. Polymer 1970, 11, 253–267. [Google Scholar] [CrossRef]
- Tang, Y.-R.; Lin, D.-W.; Gao, Y.; Xu, J.; Guo, B.-H. Prominent nucleating effect of finely dispersed hydroxyl-functional hexagonal boron nitride on biodegradable poly(butylene succinate). Ind. Eng. Chem. Res. 2014, 53, 4689–4696. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, Y.; Sato, H.; Tsuji, H.; Noda, I.; Yan, S.; Ozaki, Y. Crystal modifications and thermal behavior of poly(L-lactic acid) revealed by infrared spectroscopy. Macromolecules 2005, 38, 8012–8021. [Google Scholar] [CrossRef]
- Wei, M.; Shin, I.D.; Urban, B.; Tonelli, A.E. Partial miscibility in a nylon-6/nylon-66 blend coalesced from their common α-cyclodextrin inclusion complex. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 1369–1378. [Google Scholar] [CrossRef]
- Kongkhlang, T.; Tashiro, K.; Kotaki, M.; Chirachanchai, S. Electrospinning as a new technique to control the crystal morphology and molecular orientation of polyoxymethylene nanofibers. J. Am. Chem. Soc. 2008, 130, 15460–15466. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sakashita, M.; Hasegawa, M. Infrared and Raman spectra of macrocyclic poly(oxymethylene). Macromolecules 1991, 24, 4796–4800. [Google Scholar] [CrossRef]
- Sangroniz, L.; Alamo, R.G.; Cavallo, D.; Santamaría, A.; Müller, A.J.; Alegría, A. Differences between Isotropic and Self-Nucleated PCL Melts Detected by Dielectric Experiments. Macromolecules 2018, 51, 3663–3671. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, J.; Li, L. Multiple melting behavior of poly(butylene succinate). Eur. Polym. J. 2007, 43, 3163–3170. [Google Scholar] [CrossRef]
- Qiu, Z.; Komura, M.; Ikehara, T.; Nishi, T. DSC and TMDSC study of melting behaviour of poly(butylene succinate) and poly(ethylene succinate). Polymer 2003, 44, 7781–7785. [Google Scholar] [CrossRef]
- Yasuniwa, M.; Tsubakihara, S.; Satou, T.; Iura, K. Multiple melting behavior of poly(butylene succinate). II. Thermal analysis of isothermal crystallization and melting process. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2039–2047. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Arnal, M.L.; Albuerne, J.; Müller, A.J. DSC isothermal polymer crystallization kinetics measurements and the use of the Avrami equation to fit the data: Guidelines to avoid common problems. Polym. Test. 2007, 26, 222–231. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Tsuji, H.; Yamashita, Y. Highly accelerated stereocomplex crystallization by blending star-shaped 4-armed stereo diblock poly(lactide)s with poly(D-lactide) and poly(L-lactide) cores. Polymer 2014, 55, 6444–6450. [Google Scholar] [CrossRef]
- Li, Y.-D.; Fu, Q.-Q.; Wang, M.; Zeng, J.-B. Morphology, crystallization and rheological behavior in poly(butylene succinate)/cellulose nanocrystal nanocomposites fabricated by solution coagulation. Carbohydr. Polym. 2017, 164, 75–82. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Wang, Z.; Cavallo, D.; Müller, A.J.; Zhu, P.; Zhao, Y.; Dong, X.; Wang, D. The origin of memory effects in the crystallization of polyamides: Role of hydrogen bonding. Polymer 2020, 188, 122117. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Arnal, M.L.; Sanchez, J.J.; Müller, A.J. Effect of annealing time on the self-nucleation behavior of semicrystalline polymers. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 1738–1750. [Google Scholar] [CrossRef]
- Pérez, R.; Córdova, M.; López, J.; Hoskins, J.; Zhang, B.; Grayson, S.; Müller, A. Nucleation, crystallization, self-nucleation and thermal fractionation of cyclic and linear poly(ε-caprolactone) s. React. Funct. Polym. 2014, 80, 71–82. [Google Scholar] [CrossRef]
- Jing, Z.; Shi, X.; Zhang, G.; Li, J.; Li, J.; Zhou, L.; Zhang, H. Formation, structure and promoting crystallization capacity of stereocomplex crystallite network in the poly(lactide) blends based on linear PLLA and PDLA with different structures. Polymer 2016, 92, 210–221. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Bu, Z.; Li, B.-G.; Li, N.; Dai, J. Synthesis and Thermomechanical and Rheological Properties of Biodegradable Long-Chain Branched Poly(butylene succinate-co-butylene terephthalate) Copolyesters. Ind. Eng. Chem. Res. 2014, 53, 10380–10386. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, Y.; Wang, Z.; Sun, N.; Li, H. Enhancement of electrical conductivity by changing phase morphology for composites consisting of polylactide and poly(ε-caprolactone) filled with acid-oxidized multiwalled carbon nanotubes. ACS Appl. Mater. Interfaces 2011, 3, 4858–4864. [Google Scholar] [CrossRef]
- Liu, K.; Andablo-Reyes, E.; Patil, N.; Merino, D.H.; Ronca, S.; Rastogi, S. Influence of reduced graphene oxide on the rheological response and chain orientation on shear deformation of high density polyethylene. Polymer 2016, 87, 8–16. [Google Scholar] [CrossRef]
- Tsuji, H.; Hyon, S.H.; Ikada, Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. 3. Calorimetric studies on blend films cast from dilute solution. Macromolecules 1991, 24, 5651–5656. [Google Scholar] [CrossRef]
- Xu, Z.; Niu, Y.; Wang, Z.; Li, H.; Yang, L.; Qiu, J.; Wang, H. Enhanced nucleation rate of polylactide in composites assisted by surface acid oxidized carbon nanotubes of different aspect ratios. ACS Appl. Mater. Interfaces 2011, 3, 3744–3753. [Google Scholar] [CrossRef]
- Yamada, T.; Yamaguchi, M.; Kida, T. Evaluation of Rheological Properties and Generation of Cross Orientation of Lamellae and Amorphous Chains in a Thermoplastic Polyester Elastomer. ACS Appl. Polym. Mater. 2022, 4, 6322–6331. [Google Scholar] [CrossRef]
- Bai, J.; Wang, J.; Wang, W.; Fang, H.; Xu, Z.; Chen, X.; Wang, Z. Stereocomplex Crystallite-Assisted Shear-Induced Crystallization Kinetics at a High Temperature for Asymmetric Biodegradable PLLA/PDLA Blends. ACS Sustain. Chem. Eng. 2016, 4, 273–283. [Google Scholar] [CrossRef]
- Winter, H.H. Glass transition as the rheological inverse of gelation. Macromolecules 2013, 46, 2425–2432. [Google Scholar] [CrossRef]
- Winter, H.H.; Chambon, F. Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J. Rheol. 1986, 30, 367–382. [Google Scholar] [CrossRef]
- Elmoumni, A.; Winter, H.H.; Waddon, A.J.; Fruitwala, H. Correlation of material and processing time scales with structure development in isotactic polypropylene crystallization. Macromolecules 2003, 36, 6453–6461. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tc (°C) | ΔHc (J/g) | Tm (°C) | ΔHm (J/g) |
|---|---|---|---|---|
| PBS | 79.6 | 66.8 | 113.5 | 61.5 |
| PBS/ECCs-1 | 88.3 | 63.4 | 113.7 | 68.6 |
| PBS/ECCs-5 | 91.5 | 60.5 | 113.5/135.9 | 66.3/2.6 |
| ECCs | 101.1 | 79.7 | 137.6 | 90.0 |
| Samples | n | K (s−n) | t1/2 (s) | R2 |
|---|---|---|---|---|
| PBS | 2.4 | 1.47 × 10−7 | 672 | 0.99988 |
| PBS/ECCs-1 | 2.4 | 4.24 × 10−6 | 143 | 0.99995 |
| PBS/ECCs-5 | 2.4 | 1.95 × 10−5 | 81 | 0.99999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.-W.; Xu, J.; Chen, J.-Y.; Guo, B.-H.; Ye, H.-M. Enhanced Melt Memory Effects in Poly(butylene succinate) Through Incorporation of Extended-Chain Crystals. Polymers 2025, 17, 1086. https://doi.org/10.3390/polym17081086
Wei X-W, Xu J, Chen J-Y, Guo B-H, Ye H-M. Enhanced Melt Memory Effects in Poly(butylene succinate) Through Incorporation of Extended-Chain Crystals. Polymers. 2025; 17(8):1086. https://doi.org/10.3390/polym17081086
Chicago/Turabian StyleWei, Xue-Wei, Jun Xu, Jia-Yao Chen, Bao-Hua Guo, and Hai-Mu Ye. 2025. "Enhanced Melt Memory Effects in Poly(butylene succinate) Through Incorporation of Extended-Chain Crystals" Polymers 17, no. 8: 1086. https://doi.org/10.3390/polym17081086
APA StyleWei, X.-W., Xu, J., Chen, J.-Y., Guo, B.-H., & Ye, H.-M. (2025). Enhanced Melt Memory Effects in Poly(butylene succinate) Through Incorporation of Extended-Chain Crystals. Polymers, 17(8), 1086. https://doi.org/10.3390/polym17081086