
Characterization of Properties and Kinetic Analysis of Unsaturated Polyester Resin Synthesized from PET Alcoholysis Waste


Abstract
1. Introduction
2. Materials and Methods
2.1. Raw Materials
2.2. Preparation of Oligomers and UPRs
2.3. Test Methods
3. Results and Discussion
3.1. Characterization of UPRs
3.1.1. Distribution of Molecular Weight
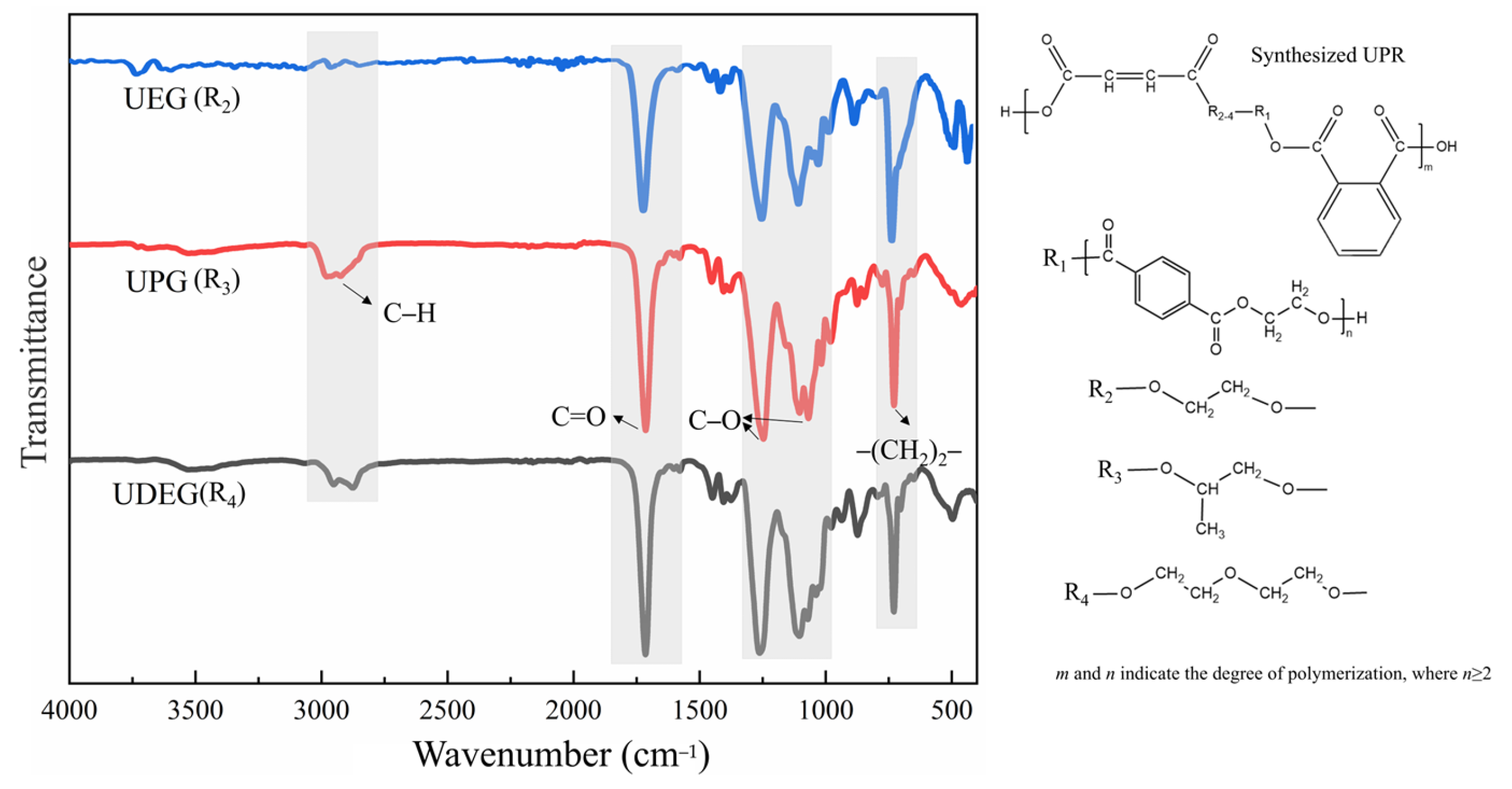
3.1.2. Analysis of Functional Groups
3.1.3. Thermal Stability
3.2. Mechanical Properties of UPR
3.2.1. Results of Tensile Test
3.2.2. Results of DMA
3.3. Kinetic Analysis of UPRs
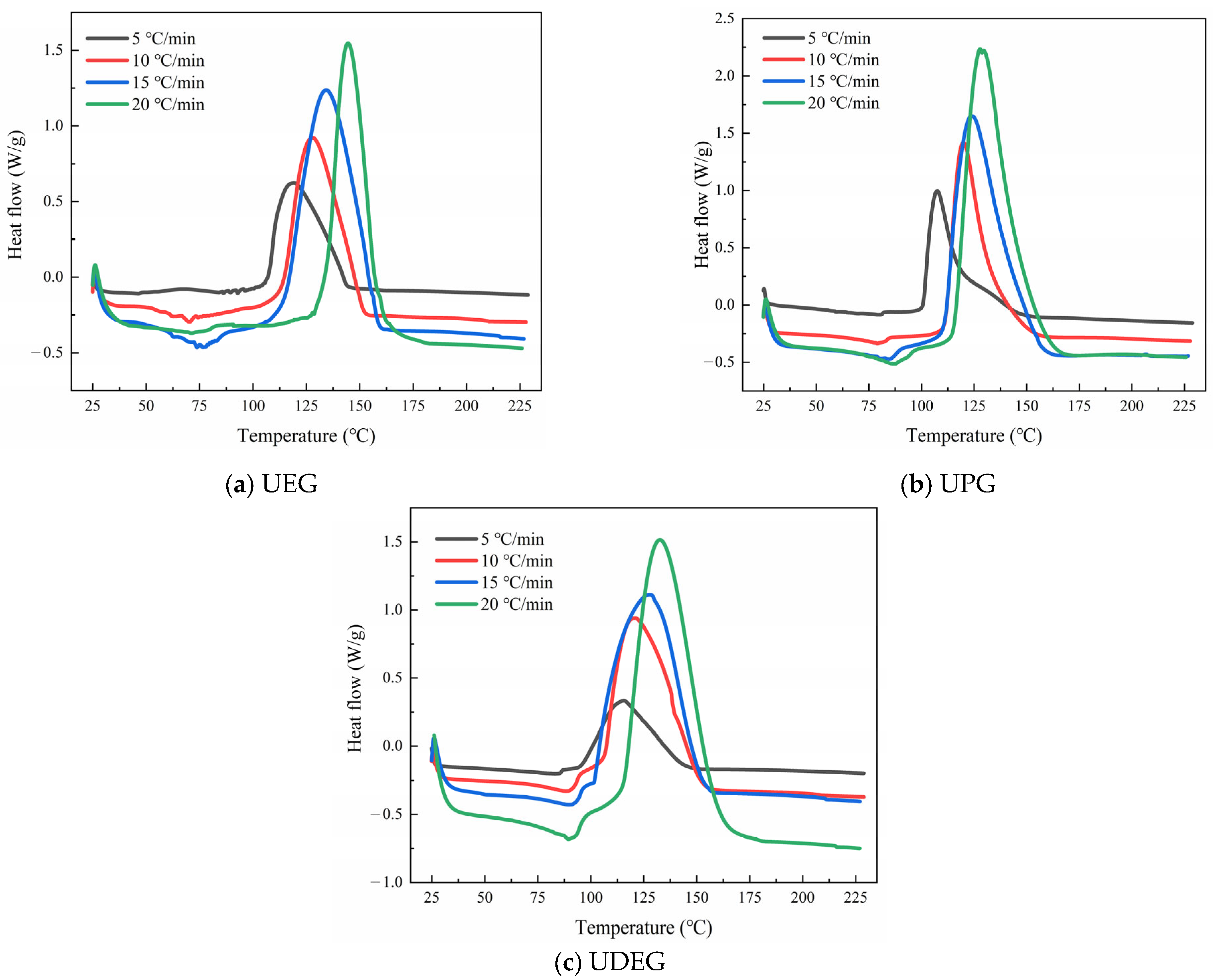
3.3.1. Analysis of Curing Process
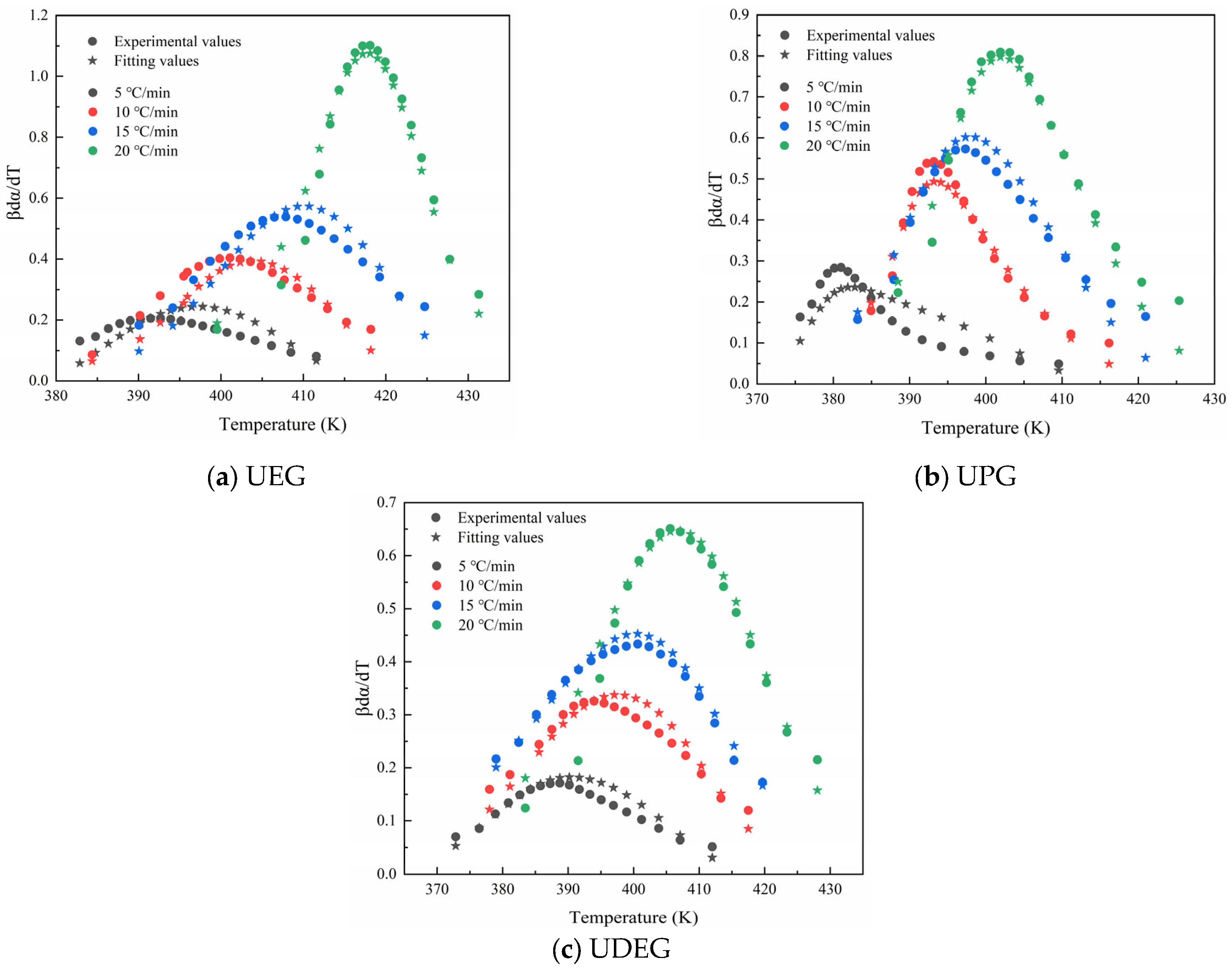
3.3.2. Calculation of Kinetic Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ubeda, S.; Aznar, M.; Nerín, C. Determination of oligomers in virgin and recycled polyethylene terephthalate (PET) samples by UPLC-MS-QTOF. Anal. Bioanal. Chem. 2018, 410, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Koshti, R.; Mehta, L.; Samarth, N. Biological recycling of polyethylene terephthalate: A mini-review. J. Polym. Environ. 2018, 26, 3520–3529. [Google Scholar]
- Kirshanov, K.; Toms, R.; Melnikov, P.; Gervald, A. Unsaturated Polyester Resin Nanocomposites Based on Post-Consumer Polyethylene Terephthalate. Polymers 2022, 14, 1602. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Li, Z.; Zhang, B.; Tang, H.; Yuan, X.; Xz, T. Study on Alcoholysis of Waste PET and Its Application in Wood Modification. Chem. Biochem. Eng. Q. 2024, 38, 71–81. [Google Scholar] [CrossRef]
- Enache, A.C.; Grecu, I.; Samoila, P. Polyethylene Terephthalate (PET) Recycled by Catalytic Glycolysis: A Bridge toward Circular Economy Principles. Materials 2024, 17, 2991. [Google Scholar] [CrossRef]
- Cao, F.; Wang, L.; Zheng, R.; Guo, L.; Chen, Y.; Qian, X. Research and progress of chemical depolymerization of waste PET and high-value application of its depolymerization products. RSC Adv. 2022, 12, 31564–31576. [Google Scholar]
- Zhang, M.; Ye, W.; Liao, Z. Preparation, characterization and properties of flame retardant unsaturated polyester resin based on r-PET. J. Polym. Environ. 2021, 30, 1984–1994. [Google Scholar] [CrossRef]
- Rorrer, N.A.; Nicholson, S.; Carpenter, A.; Biddy, M.J.; Grundl, N.J.; Beckham, G.T. Combining reclaimed PET with bio-based monomers enables plastics upcycling. Joule 2019, 3, 1006–1027. [Google Scholar]
- Karayannidis, G.P.; Nikolaidis, A.K.; Sideridou, I.D.; Bikiaris, D.N.; Achilias, D.S. Chemical recycling of PET by glycolysis: Polymerization and characterization of the dimethacrylated glycolysate. Macromol. Mater. Eng. 2006, 291, 1338–1347. [Google Scholar]
- Mersha, D.A.; Gesese, T.N.; Sendekie, Z.B.; Admase, A.T.; Bezie, A.J. Operating conditions, products and sustainable recycling routes of aminolysis of polyethylene terephthalate (PET)–a review. Polym. Bull. 2024, 81, 11563–11579. [Google Scholar] [CrossRef]
- Liu, B.; Lu, X.M.; Ju, Z.Y.; Sun, P.; Xin, J.Y.; Yao, X.Q.; Zhou, Q.; Zhang, S.J. Ultrafast homogeneous glycolysis of waste polyethylene terephthalate via a dissolution-degradation strategy. Ind. Eng. Chem. Res. 2018, 57, 16239–16245. [Google Scholar] [CrossRef]
- Lozano-Escarcega, R.J.; Sanchez-Anguiano, M.G.; Serrano, T.; Chen, J.Y.; Gomez, I. Synthesis of unsaturated polyester resin from waste cellulose and polyethylene terephthalate. Polym. Bull. 2019, 76, 4157–4188. [Google Scholar] [CrossRef]
- Raheem, A.B.; Noor, Z.Z.; Hassan, A.; Abd, H.M.K.; Samsudin, S.A.; Sabeen, A.H. Current developments in chemical recycling of post-consumer polyethylene terephthalate wastes for new materials production: A review. J. Clean. Prod. 2019, 225, 1052–1064. [Google Scholar] [CrossRef]
- Bórquez-Mendivil, A.; Hurtado-Macías, A.; Leal-Pérez, J.E.; Flores-Valenzuela, J.; Vargas-Ortíz, R.Á.; Cabrera-Covarrubias, F.G.; Almaral-Sánchez, J.L. Hybrid coatings of SiO2–recycled PET unsaturated polyester resin by Sol-Gel process. Polymers 2022, 14, 3280. [Google Scholar] [CrossRef]
- Wilson García, N.A.; Almaral Sánchez, J.L.; Vargas Ortiz, R.Á.; Hurtado Macías, A.; Flores Ramírez, N.; Aguilar Palazuelos, E.; Alvarado Beltrán, C.G. Physical and mechanical properties of unsaturated polyester resin matrix from recycled PET (based PG) with corn straw fiber. J. Appl. Polym. Sci. 2021, 138, 51305. [Google Scholar] [CrossRef]
- Ramirez-Palma, G.; Alcocer-Marquez, L.F.; Alcantar-Gonzalez, F.S.; Turati-Ramirez de Arellano, P.; Cruz-Gomez, M.J. Unsaturated polyester resins from glycolised waste PET: Synthesis and characterisation. Rev. Mex. Ing. Quim. 2017, 16, 1003–1010. [Google Scholar]
- Zhou, X.; Wang, C.; Fang, C.; Yu, R.; Li, Y.; Lei, W. Structure and thermal properties of various alcoholysis products from waste poly (ethylene terephthalate). Waste Manag. 2019, 85, 164–174. [Google Scholar] [CrossRef]
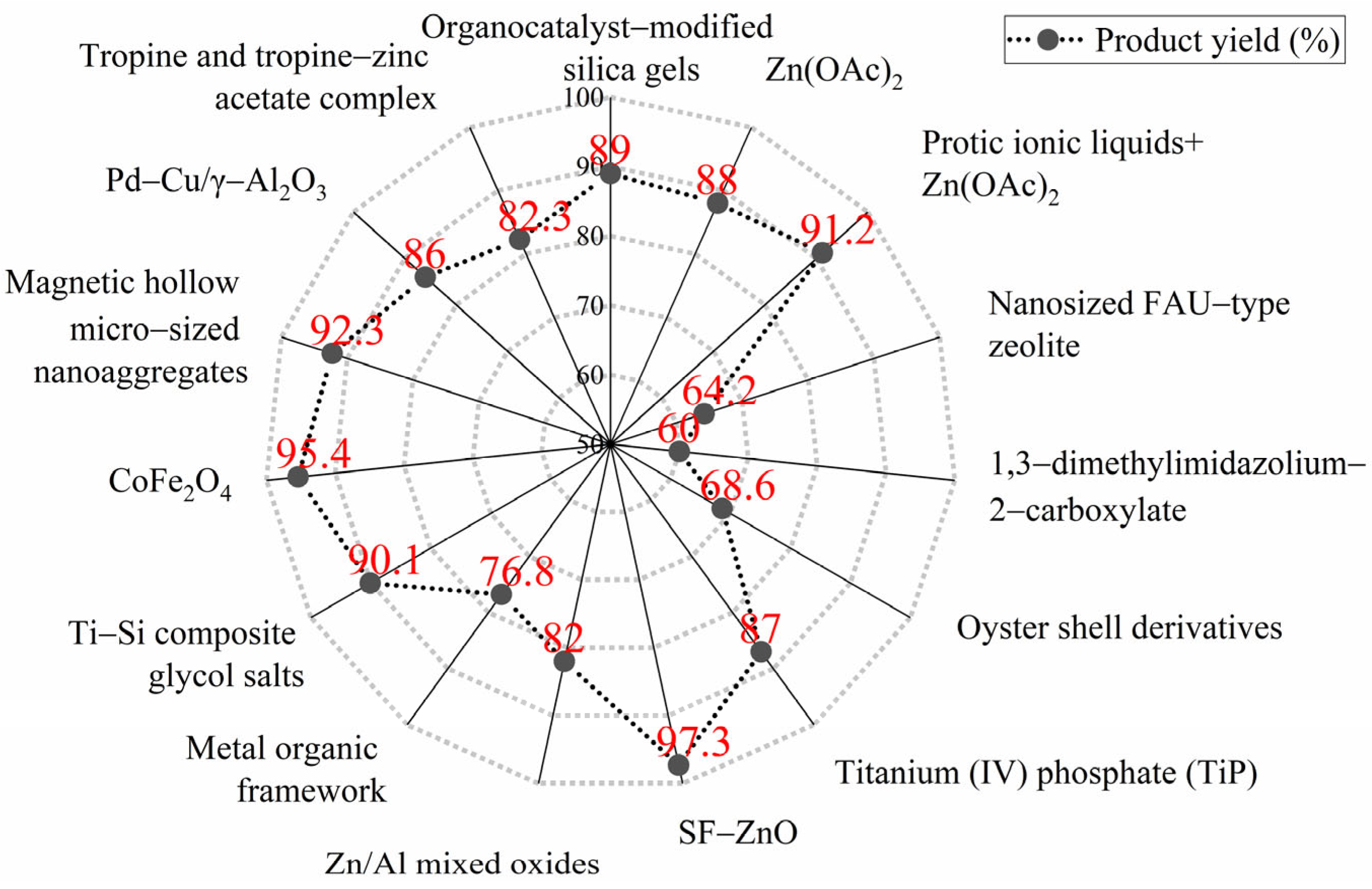
- Fehér, Z.; Kiss, J.; Kisszékelyi, P.; Molnár, J.; Huszthy, P.; Kárpáti, L.; Kupai, J. Optimisation of PET glycolysis by applying recyclable heterogeneous organocatalysts. Green Chem. 2022, 24, 8447–8459. [Google Scholar] [CrossRef]
- Aguado, A.; Becerra, L.; Martínez, L. Glycolysis optimisation of different complex PET waste with recovery and reuse of ethylene glycol. Chem. Pap. 2023, 77, 3293–3303. [Google Scholar] [CrossRef]
- Yang, G.; Wu, H.; Huang, K.; Ma, Y.; Chen, Q.; Chen, Y.; Lin, S.; Guo, H.; Li, Z. The Recyclable Dual-Functional Zeolite Nanocrystals Promoting the High Efficiency Glycolysis of PET. J. Polym. Environ. 2024, 32, 5071–5085. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, T.; Zhou, L.; Zhang, P.; Wang, Z.; Chen, X. Synergistic catalysis of ionic liquids and metal salts for facile PET glycolysis. Eur. Polym. J. 2023, 201, 112578. [Google Scholar]
- Wang, L.; Nelson, G.A.; Toland, J.; Holbrey, J.D. Glycolysis of PET using 1, 3-dimethylimidazolium-2-carboxylate as an organocatalyst. ACS Sustain. Chem. Eng. 2020, 8, 13362–13368. [Google Scholar]
- Zahova, S.; Tsacheva, I.; Troev, K.; Mitova, V. Conventional and MW assisted PET glycolysis promoted by titanium based catalyst. Polym. Degrad. Stabil. 2023, 212, 110353. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, M.; Hwang, J.; Im, E.; Moon, G.D. Optimizing PET glycolysis with an oyster shell-derived catalyst using response surface methodology. Polymers 2022, 14, 656. [Google Scholar] [CrossRef]
- Ao, Z.; Deng, J.; He, W.; Liu, T.; Wang, J.; Yang, H.; Shen, Z.; Chen, J. Low-temperature one-step synthesis of surfactant-free ZnO nanoparticles for efficient glycolysis of PET. Chem. Eng. J. 2024, 494, 153037. [Google Scholar]
- Chen, F.; Zhou, Q.; Bu, R.; Yang, F.; Li, W. Kinetics of poly (ethylene terephthalate) fiber glycolysis in ethylene glycol. Fiber Polym. 2015, 16, 1213–1219. [Google Scholar] [CrossRef]
- Suo, Q.; Zi, J.; Bai, Z.; Qi, S. The glycolysis of poly (ethylene terephthalate) promoted by metal organic framework (MOF) catalysts. Catal. Lett. 2017, 147, 240–252. [Google Scholar]
- Yu, Y.; Shen, G.; Xu, T.J.; Wen, R.; Qiao, Y.C.; Cheng, R.C.; Huo, Y. Ti–Si composite glycol salts: Depolymerization and repolymerization studies of PET. RSC Adv. 2023, 13, 36337–36345. [Google Scholar] [CrossRef]
- Mohammadi, S.; Bouldo, M.G.; Enayati, M. FeCl3-Doped Cobalt Ferrite as an Efficient Magnetic Catalyst for PET Glycolysis Depolymerization. J. Polym. Environ. 2024, 32, 5738–5749. [Google Scholar]
- Yun, L.X.; Wei, Y.; Sun, Q.; Li, Y.T.; Zhang, B.; Zhang, H.T.; Shen, Z.; Wang, J.X. Magnetic hollow micro-sized nanoaggregates for synergistically accelerating PET glycolysis. Green Chem. 2023, 25, 6901–6913. [Google Scholar]
- Zhou, L.; Qin, E.; Huang, H.; Wang, Y.; Li, M. PET Glycolysis to BHET Efficiently Catalyzed by Stable and Recyclable Pd-Cu/γ-Al2O3. Molecules 2024, 29, 4305. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Li, R.; Chen, Y.; Wang, J.; Song, H. New effective catalysts for glycolysis of polyethylene terephthalate waste: Tropine and tropine-zinc acetate complex. J. Mol. Liq. 2021, 334, 116419. [Google Scholar]
- Senra, E.M.; Silva, A.L.; Pacheco, E.B. A review of waterborne polyurethane coatings and adhesives with polyester polyol from poly (ethylene terephthalate) waste. J. Polym. Environ. 2023, 31, 3719–3739. [Google Scholar]
- Naguib, H.M.; Zhang, X.H. Advanced recycled polyester based on PET and oleic acid. Polym. Test. 2018, 69, 450–455. [Google Scholar]
- Zhang, Z.; Zhang, H.; Ma, J.; Wang, N. Multi-objective optimization, shrinkage and fracture properties of unsaturated polyester resin modified concrete for bridge deck pavement based on system theory. Constr. Build. Mater. 2024, 443, 137694. [Google Scholar]
- GB/T 2567-2021; Test Methods for Properties of Resin Casting body. China Standard Press: Beijing, China, 2021.
- ASTM D7028-07; Standard Test Method for Glass Transition Temperature (DMA Tg) of Polymer MatrixComposites by Dynamic Mechanical Analysis (DMA). ASTM international: West Conshohocken, PA, USA, 2007.
- GB/T 19466.1-2004; Plastics--Differential Scanning Calorimetry (DSC)—Part 1: General Principles. China Standard Press: Beijing, China, 2004.
- Bai, Z.; Song, L.; Hu, Y.; Gong, X.; Yuen, R. Investigation on flame retardancy, combustion and pyrolysis behavior of flame retarded unsaturated polyester resin with a star-shaped phosphorus-containing compound. J. Anal. Appl. Pyrol. 2014, 105, 317–326. [Google Scholar]
- Guo, Q.; Cheng, H.; Zhang, H.; Zhong, Z.; Chen, S. Improving flame retardancy and mechanical properties of halogen-free unsaturated polyester resin with diethylene glycol as comonomer. J. Therm. Anal. Calorim 2019, 135, 2171–2181. [Google Scholar]
- Feng, L.; Li, R.; Yang, H.; Chen, S.; Yang, W. The hyperbranched polyester reinforced unsaturated polyester resin. Polymers 2022, 14, 1127. [Google Scholar] [CrossRef]
- Tan, C.S.; Chan, M.Y.; Koay, S.C. Preliminary Study of the Mechanical Properties of Hybrid Fibres Reinforced Unsaturated Polyester Composites. J. Phys. Sci. 2021, 32, 45–59. [Google Scholar]
- Salasinska, K.; Celiński, M.; Barczewski, M.; Leszczyński, M.K.; Borucka, M.; Kozikowski, P. Fire behavior of flame retarded unsaturated polyester resin with high nitrogen content additives. Polym. Test. 2020, 84, 106379. [Google Scholar]
- Case, S.L.; O’Brien, E.P.; Ward, T.C. Cure profiles, crosslink density, residual stresses, and adhesion in a model epoxy. Polymer 2005, 46, 10831–10840. [Google Scholar]
- Nakka, J.S.; Jansen, K.M.B.; Ernst, L.J.; Jager, W.F. Effect of the epoxy resin chemistry on the viscoelasticity of its cured product. J. Appl. Polym. Sci. 2008, 108, 1414–1420. [Google Scholar]
- Pistor, V.; Ornaghi, F.G.; Ornaghi, H.L.; Zattera, A.J. Dynamic mechanical characterization of epoxy/epoxycyclohexyl–POSS nanocomposites. Mater. Sci. Eng. A 2012, 532, 339–345. [Google Scholar]
- Zheng, M.; Li, X.Y.; Guan, R.Q.; Liu, Y.M.; Zhao, Y.J.; Tai, Y.L. Ionic liquid-catalyzed synthesis of Diethylene Glycol. Adv. Mater. Res. 2012, 549, 287–291. [Google Scholar]
- Hong, C.; Wang, X.; Pan, Z.; Zhang, Y. Curing thermodynamics and kinetics of unsaturated polyester resin with different chain length of saturated aliphatic binary carboxylic acid. J. Therm. Anal. Calorim. 2015, 122, 427–436. [Google Scholar]
- Jia, H.; Zhu, Y.; Yan, C.; Chen, M.; Chen, G.; Liu, D. Reliability evaluation of a new method developed for DSC curing kinetic model of epoxy resin. J. Appl. Polym. Sci. 2023, 140, e53986. [Google Scholar]
- Chencheni, A.; Belkhiri, S.; Tarchoun, A.F.; Abdelaziz, A.; Bessa, W.; Boucheffa, Y.; Trache, D. Unravelling the thermal behavior and kinetics of unsaturated polyester resin supplemented with organo-nanoclay. RSC Adv 2024, 14, 517–528. [Google Scholar]
- Chencheni, A.; Belkhiri, S.; Tarchoun, A.F.; Abdelaziz, A.; Boucheffa, Y.; Trache, D. Insights into the curing and thermal behavior of orthophtalic unsaturated polyester resin with organically modified montmorillonite nanoclay. React. Kinet. Mech. Catal. 2024, 1–23. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn | Mw | Mz | PDI | |
|---|---|---|---|---|
| UEG | 1065 | 2480 | 5557 | 2.328 |
| UPG | 2345 | 5970 | 11,588 | 2.545 |
| UDEG | 1799 | 6593 | 15,006 | 3.664 |
| Sample | Er (90 °C) (MPa) | Tg (°C) | ve (mol/cm3) |
|---|---|---|---|
| UEG | 3.554 | 68 | 0.392 |
| UPG | 0.902 | 53 | 0.100 |
| UDEG | 1.153 | 35 | 0.127 |
| Sample | 5 °C/min | 10 °C/min | 15 °C/min | 20 °C/min | ||||
|---|---|---|---|---|---|---|---|---|
| Tp (K) | Pw (K) | Tp (K) | Pw (K) | Tp (K) | Pw (K) | Tp (K) | Pw (K) | |
| UEG | 392.34 | 28.69 | 401.20 | 33.21 | 407.30 | 34.66 | 417.69 | 31.74 |
| UPG | 384.72 | 33.91 | 394.04 | 32.81 | 400.33 | 37.78 | 405.01 | 36.89 |
| UDEG | 381.78 | 39.15 | 393.12 | 39.51 | 397.47 | 40.67 | 401.86 | 44.56 |
| lnA | E | m | n | |
|---|---|---|---|---|
| UEG | 26.150 | 81.339 KJ/mol | 0.588 | 1.204 |
| UPG | 25.512 | 81.630 KJ/mol | 0.416 | 1.340 |
| UDEG | 26.457 | 87.610 KJ/mol | 0.222 | 1.232 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Zhang, H.; Liu, J.; Wei, T. Characterization of Properties and Kinetic Analysis of Unsaturated Polyester Resin Synthesized from PET Alcoholysis Waste. Polymers 2025, 17, 820. https://doi.org/10.3390/polym17060820
Wang R, Zhang H, Liu J, Wei T. Characterization of Properties and Kinetic Analysis of Unsaturated Polyester Resin Synthesized from PET Alcoholysis Waste. Polymers. 2025; 17(6):820. https://doi.org/10.3390/polym17060820
Chicago/Turabian StyleWang, Ruixiang, Hongliang Zhang, Jingshuang Liu, and Tongjun Wei. 2025. "Characterization of Properties and Kinetic Analysis of Unsaturated Polyester Resin Synthesized from PET Alcoholysis Waste" Polymers 17, no. 6: 820. https://doi.org/10.3390/polym17060820
APA StyleWang, R., Zhang, H., Liu, J., & Wei, T. (2025). Characterization of Properties and Kinetic Analysis of Unsaturated Polyester Resin Synthesized from PET Alcoholysis Waste. Polymers, 17(6), 820. https://doi.org/10.3390/polym17060820