
Exploring the Thermal Degradation of Bakelite: Non-Isothermal Kinetic Modeling, Thermodynamic Insights, and Evolved Gas Analysis via Integrated In Situ TGA/MS and TGA/FT-IR Techniques


Abstract
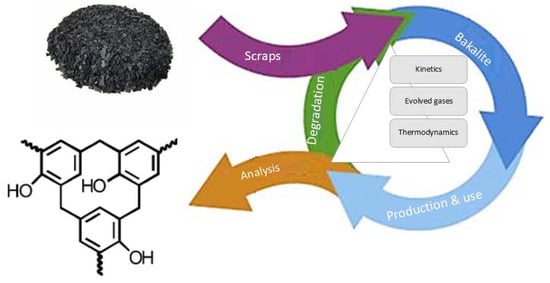
1. Introduction
2. Materials and Methods
2.1. Material Preparation and Characteristics
2.2. Thermoanalytical Measurements
2.3. Kinetic Analysis
2.4. Thermodynamic Analysis
3. Results and Discussion
3.1. Elemental Composition of the Material and Analysis of TG/dTG Thermograms
3.2. Kinetic Analysis Results
3.3. Thermodynamic Analysis
3.4. Evolved Gas Analysis Using Simultaneous In Situ TGA/FT-IR and TGA-MS Analyses
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, B.D.; Kadla, J.F. Thermal degradation kinetics of resole phenol-formaldehyde resin/multi-walled carbon nanotube/cellulose nanocomposite. Thermochim. Acta 2012, 540, 107–115. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, H.; Jiang, N. Study on the pyrolysis of phenol-formaldehyde (PF) resin and modified PF resin. Thermochim. Acta 2009, 496, 136–142. [Google Scholar] [CrossRef]
- Chen, P.; Li, G.; Shao, J.; Bai, B.; Hu, J.; Han, X.; Zhou, A.; Wang, Q.; Chen, F. Preparation of Nitrogen-Rich Tar by Co-Pyrolysis and Analysis of Nitrogen-Containing Compounds in Pyrolysis Products. Appl. Sci. 2025, 15, 6284. [Google Scholar] [CrossRef]
- Gola, A.; Knysak, T.; Mucha, I.; Musiał, W. Synthesis, thermogravimetric analysis, and kinetic study of poly-N-isopropylacrylamide with varied initiator content. Polymers 2023, 15, 2427. [Google Scholar] [CrossRef] [PubMed]
- Tworek, M.; Makarewicz, E.; Shyychuk, I.; Kowalik, J.; Zalewska, A.; Tomaszewska, J. Studies of thermal decomposition of poly (vinyl chloride) composition with cadmium yellow and stearic acid. Int. J. Polym. Anal. Charact. 2024, 29, 172–188. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, J.; Wu, S.; Yuan, Z.; Hu, Z.; Wu, R.; Liu, Q. The pyrolysis mechanism of phenol formaldehyde resin. Polym. Degrad. Stab. 2012, 97, 1527–1533. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, J.; Wu, S.; Wang, B.; Wang, Z. Pyrolysis kinetics of phenol–formaldehyde resin by non-isothermal thermogravimetry. Carbon 2010, 48, 352–358. [Google Scholar] [CrossRef]
- Jaques, N.G.; Nicácio, P.H.; Wellen, R.M. Non-isothermal curing kinetics of isosorbide based on epoxy with anhydrides. J. Polym. Environ. 2024, 32, 2365–2379. [Google Scholar] [CrossRef]
- Shang, F.; Zhang, J.; Zhu, T.; Guo, Y.; Fan, Y.; Tao, S.; Liu, R.; Ding, Y. Combined kinetic analysis of thermal degradation characteristics and reaction mechanism of thin intumescent fire-retardant coating of steel structure. Int. J. Polym. Anal. Charact. 2024, 29, 635–646. [Google Scholar] [CrossRef]
- Doğan, F. Thermal decomposition behavior of oligo (4-hydroxyquinoline). Polym. Eng. Sci. 2014, 54, 992–1002. [Google Scholar] [CrossRef]
- Yin, F.; Fan, F.; Yuan, X.; Wu, H.; Chang, T.; Lei, J.; Liang, D. Pyrolysis of raw and film PP/LDPE mixtures by step heating process. J. Polym. Res. 2023, 30, 247. [Google Scholar] [CrossRef]
- Li, J.; Lu, W.; Yang, D.; Jia, Y.; Su, H.; Deng, J.; Gong, Z.; Zhao, Y. A Novel Rare Earth Enhanced Epoxy Composites: Mechanical Properties, Thermal Stability and Curing Kinetics. Polym. Sci. Ser. B 2024, 66, 201–212. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, L.; Liu, S.; Wang, T.; Xue, P.; Wu, M.; Yun, J. Study on the Combustion Behavior and Kinetic Characteristics of Semi-Coke from Oil Shale. Appl. Sci. 2025, 15, 5797. [Google Scholar] [CrossRef]
- Yildirim, Y.; Saltan, F.; Şirin, K.; Küçük, V.A. Thermal decomposition kinetics and mechanical analysis of boron carbide-reinforced polymer nanocomposites. Polym. Eng. Sci. 2025, 65, 2308–2322. [Google Scholar] [CrossRef]
- Perez, B.A.; Krishna, J.J.; Toraman, H.E. Insights into co-pyrolysis of polyethylene terephthalate and polyamide 6 mixture through experiments, kinetic modeling and machine learning. Chem. Eng. J. 2023, 468, 143637. [Google Scholar] [CrossRef]
- Al-Mulla, A.; Mathew, J.; Al-Omairi, L.; Bhattacharya, S. Thermal decomposition kinetics of tricomponent polyester/polycarbonate systems. Polym. Eng. Sci. 2011, 51, 2335–2344. [Google Scholar] [CrossRef]
- Alpaslan, T.M.; Özsin, G. A Comparative Study on the Carbonization of Chitin and Chitosan: Thermo-Kinetics, Thermodynamics and Artificial Neural Network Modeling. Appl. Sci. 2025, 15, 6141. [Google Scholar] [CrossRef]
- Jen, T.H.; Wang, K.K.; Chen, S.A. Effect of thermal stability on performance of β-phase poly (9, 9-di-n-octylfluorene) in deep blue electroluminescence. Polymer 2012, 53, 5850–5855. [Google Scholar] [CrossRef]
- Wu, S.E.; Sharma, S.; Chen, H.L.; Chen, S.A.; Komarov, P.V.; Ivanov, V.A.; Khokhlov, A.R. Single conjugated polymer with four stepwise HOMO levels for effective hole injection across large barrier 1.4 eV to core–shell quantum dot layer for electroluminescence in inverted QLED. Adv. Opt. Mater. 2022, 10, 2102508. [Google Scholar] [CrossRef]
- Horikawa, T.; Ogawa, K.; Mizuno, K.; Hayashi, J.I.; Muroyama, K. Preparation and characterization of the carbonized material of phenol–formaldehyde resin with addition of various organic substances. Carbon 2003, 41, 465–472. [Google Scholar] [CrossRef]
- Xu, L.; Zhong, Q.Q.; Dong, Q.; Zhang, L.Y.; Fang, Z. Co-production of phenolic oil and CaO/char deoxidation catalyst via catalytic fast pyrolysis of phenol-formaldehyde resin with Ca(OH)2. J. Anal. Appl. Pyrolysis 2019, 142, 104663. [Google Scholar] [CrossRef]
- Kasar, P.; Ahmaruzzaman, M. Studies on Catalytic Co-Pyrolysis of Bakelite and Refineries Residual Fuel Oil Using ZSM-5 Catalyst to Produce Lighter Fuel Oil. Available online: http://nopr.niscpr.res.in/handle/123456789/48788 (accessed on 4 June 2025).
- Mahapatra, P.M.; Aech, S.; Dash, S.R.; Tripathy, U.P.; Mishra, P.C.; Panda, A.K. Co-pyrolysis of discarded bakelite with polystyrene to fuel: Thermokinetic, thermodynamic, synergetic analysis and batch pyrolysis studies. J. Energy Inst. 2023, 110, 101336. [Google Scholar] [CrossRef]
- Mahapatra, P.M.; Panda, A.K.; Ahmed, S.; Kumar, S. Pyrolysis kinetics and thermodynamics of discarded Bakelite. Korean J. Chem. Eng. 2023, 40, 1380–1388. [Google Scholar] [CrossRef]
- Mahapatra, P.M.; Pradhan, D.; Kumar, S.; Panda, A.K. Isothermal pyrolysis of discarded bakelite: Kinetics analysis and batch pyrolysis studies. Circ. Econ. 2024, 3, 100102. [Google Scholar] [CrossRef]
- Hall, W.J.; Williams, P.T. Analysis of products from the pyrolysis of plastics recovered from the commercial scale recycling of waste electrical and electronic equipment. J. Anal. Appl. Pyrolysis 2007, 79, 375–386. [Google Scholar] [CrossRef]
- Yang, X.; Sun, L.; Xiang, J.; Hu, S.; Su, S. Pyrolysis and dehalogenation of plastics from waste electrical and electronic equipment (WEEE): A review. Waste Manag. 2013, 33, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Alshrah, M.; Adeyemi, I.; Janajreh, I. Kinetic study on thermal degradation of crosslinked polyethylene cable waste. J. Polym. Res. 2022, 29, 289. [Google Scholar] [CrossRef]
- Liu, W.J.; Tian, K.; Jiang, H.; Zhang, X.S.; Yang, G.X. Preparation of liquid chemical feedstocks by co-pyrolysis of electronic waste and biomass without formation of polybrominated dibenzo-p-dioxins. Bioresour. Technol. 2013, 128, 1–7. [Google Scholar] [CrossRef]
- AlAbbad, M.; Gautam, R.; Romero, E.G.; Saxena, S.; Barradah, E.; Chatakonda, O.; Kloosterman, J.W.; Middaugh, J.; D’Agostini, M.D.; Sarathy, S.M. TG-DSC and TG-FTIR analysis of heavy fuel oil and vacuum residual oil pyrolysis and combustion: Characterization, kinetics, and evolved gas analysis. J. Therm. Anal. Calorim. 2023, 148, 1875–1898. [Google Scholar] [CrossRef]
- Saha, S.; Chowdhury, A.; Kumbhakarna, N. Effect of copper chromite on ammonium perchlorate decomposition-A TGA-FTIR-MS and FE-SEM study. J. Therm. Anal. Calorim. 2024, 149, 9401–9412. [Google Scholar] [CrossRef]
- Muhammed Raji, A.; Manescau, B.; Chetehouna, K.; Ogabi, R. Overview of thermal and analytical characterization techniques for biofuels and its blends. J. Therm. Anal. Calorim. 2025, 150, 3007–3036. [Google Scholar] [CrossRef]
- Lei, Y.; Huo, J. An Alkenyl Polyethylene Glycol Binder: Room Temperature Curing Properties via Nitrile Oxide, Thermodynamics and Kinetics Study. Polym. Sci. Ser. B 2024, 66, 531–543. [Google Scholar] [CrossRef]
- Zheng, B.; Li, Y.; Xie, Q.; Gu, Y.; Wang, C.; Chen, G.; Li, H. The exploration of pyrolysis characteristics, kinetics and products of cotton via TG, FTIR, GC/MS and shuffled complex evolution. J. Therm. Anal. Calorim. 2024, 149, 14677–14686. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, J.; Kim, S.S.; Park, Y.K. Pyrolysis characteristics and kinetics of oak trees using thermogravimetric analyzer and micro-tubing reactor. Bioresour. Technol. 2009, 100, 400–405. [Google Scholar] [CrossRef]
- Kim, Y.M.; Pyo, S.; Hakimian, H.; Yoo, K.S.; Rhee, G.H.; Park, Y.K. Kinetic analysis for the catalytic pyrolysis of polypropylene over low-cost mineral catalysts. Sustainability 2021, 13, 13386. [Google Scholar] [CrossRef]
- Doyle, C.D. Estimating isothermal life from thermogravimetric data. J. Appl. Polym. Sci. 1962, 6, 639–642. [Google Scholar] [CrossRef]
- Agrawal, R.K.; Sivasubramanian, M.S. Integral approximations for nonisothermal kinetics. AIChE J. 1987, 33, 1215–1217. [Google Scholar] [CrossRef]
- Senum, G.I.; Yang, R.T. Rational approximations of the integral of the Arrhenius function. J. Therm. Anal. 1977, 11, 445–447. [Google Scholar] [CrossRef]
- Yousef, S.; Eimontas, J.; Striūgas, N.; Subadra, S.P.; Abdelnaby, M.A. Thermal degradation and pyrolysis kinetic behaviour of glass fibre-reinforced thermoplastic resin by TG-FTIR, Py-GC/MS, linear and nonlinear isoconversional models. J. Mater. Res. Technol. 2021, 15, 5894–5908. [Google Scholar] [CrossRef]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. J. Polym. Sci. Part C 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Ozawa, T. A new method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. General treatment of the thermogravimetry of polymers. J. Res. Natl. Bur. Stand. A Phys. Chem. 1966, 70, 487–523. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Akahira, T.J.; Sunose, T. Method of determining activation deterioration constant of electrical insulating materials. Res. Rep. Chiba Inst. Technol. (Sci. Technol.) 1971, 16, 22–31. [Google Scholar]
- Starink, M.J. A new method for the derivation of activation energies from experiments performed at constant heating rate. Thermochim. Acta 1996, 288, 97–104. [Google Scholar] [CrossRef]
- Tang, W.; Liu, Y.; Zhang, H.; Wang, C. New approximate formula for Arrhenius temperature integral. Thermochim. Acta 2003, 408, 39–43. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Boonchom, B. Kinetics and thermodynamic properties of the thermal decomposition of manganese dihydrogenphosphate dihydrate. J. Chem. Eng. Data 2008, 53, 1137–1141. [Google Scholar] [CrossRef]
- Latoui, R.; Bouzid, D.; Belhani, M.A.; Boyron, O. Enhancing photopolymerization and modeling kinetic degradation in dental composites. J. Polym. Res. 2024, 31, 95. [Google Scholar] [CrossRef]
- Kim, B.S.; Kim, Y.M.; Lee, H.W.; Jae, J.; Kim, D.H.; Jung, S.C.; Watanabe, C.; Park, Y.K. Catalytic copyrolysis of cellulose and thermoplastics over HZSM-5 and HY. ACS Sustain. Chem. Eng. 2016, 4, 2532–2540. [Google Scholar] [CrossRef]
- Li, J.; Qiao, Y.; Zong, P.; Wang, C.; Tian, Y.; Qin, S. Thermogravimetric analysis and isoconversional kinetic study of biomass pyrolysis derived from land, coastal zone, and marine. Energy Fuels 2019, 33, 4330–4340. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z.; Lu, Y.; Yang, L.; Xu, T.; Wu, H.; Zhang, J.; He, L. Apparent activation energy and characteristic temperatures of thermal decomposition research of microwave prepared melamine foam. J. Polym. Res. 2023, 30, 163. [Google Scholar] [CrossRef]
- Trick, K.A.; Saliba, T.E. Mechanisms of the pyrolysis of phenolic resin in a carbon/phenolic composite. Carbon 1995, 33, 1509–1515. [Google Scholar] [CrossRef]
- Trick, K.A.; Saliba, T.E.; Sandhu, S.S. A kinetic model of the pyrolysis of phenolic resin in a carbon/phenolic composite. Carbon 1997, 35, 1315–1322. [Google Scholar] [CrossRef]
- Wong, H.W.; Peck, J.; Bonomi, R.E.; Assif, J.; Panerai, F.; Reinisch, G.; Lachaud, J.; Mansour, N.N. Quantitative determination of species production from phenol-formaldehyde resin pyrolysis. Polym. Degrad. Stab. 2015, 113, 44–55. [Google Scholar] [CrossRef]
- Ouchi, K. Infra-red study of structural changes during the pyrolysis of a phenol-formaldehyde resin. Carbon 1966, 4, 209–214. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Xiao, S.; Liu, H. A novel thermal degradation mechanism of phenol–formaldehyde type resins. Thermochim. Acta 2008, 474, 101–105. [Google Scholar] [CrossRef]
- Costa, L.; Di Montelera, L.R.; Camino, G.; Weil, E.D.; Pearce, E.M. Structure-charring relationship in phenol-formaldehyde type resins. Polym. Degrad. Stab. 1997, 58, 41–53. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, N.; Feng, M.W. Thermal degradation characteristics of phenol–formaldehyde resins derived from beetle infested pine barks. Thermochim. Acta 2013, 556, 50–55. [Google Scholar] [CrossRef]
- Özsin, G.; Dermenci, K.B.; Turan, S. Thermokinetic and thermodynamics of Pechini derived Li7−3xAlxLa3Zr2O12 xerogel decomposition under oxidative conditions. J. Therm. Anal. Calorim. 2021, 143, 2441–2451. [Google Scholar] [CrossRef]
- Özsin, G.; Alpaslan Takan, M.; Takan, A.; Pütün, A.E. A combined phenomenological artificial neural network approach for determination of pyrolysis and combustion kinetics of polyvinyl chloride. Int. J. Energy Res. 2022, 46, 18214–18231. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Pande, R.; Baraiya, N.A. Novel approach by combining model-free and model-fitting technique for kinetic and thermodynamic analysis of bamboo, bagasse, and rice husk. J. Therm. Anal. Calorim. 2025, 149, 5679–5694. [Google Scholar] [CrossRef]
- Shan, T.; Wang, K.; Bian, H.; Wang, C.; Tian, X. Study on kinetics of spent FCC catalyst applied to waste plastics. Polym. Eng. Sci. 2023, 63, 2502–2512. [Google Scholar] [CrossRef]
- Albin Zaid, Z.A.; Otaru, A.J. Thermal decomposition of date seed/polypropylene homopolymer: Machine learning CDNN, kinetics, and thermodynamics. Polymers 2025, 17, 307. [Google Scholar] [CrossRef]
- Tariq, R.; Inayat, A.; Shahbaz, M.; Zeb, H.; Ghenai, C.; Al-Ansari, T.; Kim, J. Kinetic and thermodynamic evaluation of pyrolysis of jeans waste via Coats-Redfern method. Korean J. Chem. Eng. 2023, 40, 1512–1520. [Google Scholar] [CrossRef]
- Kumar, P.; Mohanty, K. Insight into the pyrolysis of Melocanna baccifera biomass: Pyrolysis behavior, kinetics, and thermodynamic parameters analysis based on iso-conversional methods. ACS Omega 2025, 10, 4210–4222. [Google Scholar] [CrossRef] [PubMed]
- Özsin, G.; Apaydın-Varol, E.; Kılıç, M.; Pütün, A.E.; Pütün, E. Pyrolysis of petroleum sludge under non-isothermal conditions: Thermal decomposition behavior, kinetics, thermodynamics, and evolved gas analysis. Fuel 2021, 304, 120980. [Google Scholar] [CrossRef]
- Prayoga, M.Z.; Putra, H.P.; Adelia, N.; Luktyansyah, I.M.; Ifanda, I.; Prismantoko, A.; Darmawan, A.; Hartono, J.; Wirawan, S.S.; Aziz, M.; et al. Co-combustion performance of oil palm biomass with coal: Thermodynamics and kinetics analyses. J. Therm. Anal. Calorim. 2024, 149, 5821–5837. [Google Scholar] [CrossRef]
- Escalante, J.; Chen, W.H.; Tabatabaei, M.; Hoang, A.T.; Kwon, E.E.; Lin, K.Y.; Saravanakumar, A. Pyrolysis of lignocellulosic, algal, plastic, and other biomass wastes for biofuel production and circular bioeconomy: A review of thermogravimetric analysis (TGA) approach. Renew. Sustain. Energy Rev. 2022, 167, 112914. [Google Scholar] [CrossRef]
- Kim, Y.M.; Han, T.U.; Hwang, B.; Lee, Y.; Watanabe, A.; Teramae, N.; Kim, S.S.; Park, Y.K.; Kim, S. New approach for the kinetic analysis of cellulose using EGA-MS. Polym. Test. 2017, 60, 53–59. [Google Scholar] [CrossRef]
- Özsin, G.; Pütün, A.E. An investigation on pyrolysis of textile wastes: Kinetics, thermodynamics, in-situ monitoring of evolved gasses and analysis of the char residue. J. Environ. Chem. Eng. 2022, 10, 107748. [Google Scholar] [CrossRef]
- Ballice, L. Investigating the pyrolysis properties of cellulose and lignin isolated from different Turkish biomass using TG-FTIR. Korean J. Chem. Eng. 2024, 41, 1058–1067. [Google Scholar] [CrossRef]
- Vaught, L.O.; Meyer, J.L.; El Arwadi, O.; Mandal, T.; Amiri, A.; Naraghi, M.; Polycarpou, A.A. Complex cure kinetics of self-healing copolyester vitrimers via isothermal thermogravimetric analysis. Polym. Test. 2025, 123, 108724. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Friedman: | . |
| FWO: | . |
| KAS: | . |
| Starink: | . |
| Tang: | . |
| C (%) | 75.4 |
| H (%) | 5.5 |
| N (%) | 2.2 |
| * O (%) | 16.9 |
| H/C | 0.87 |
| O/C | 0.17 |
| β (°C/min) | Ti (°C) | Tp (°C) | Tf (°C) | Rp (%/min·mg) |
|---|---|---|---|---|
| 5 | 219.4 | 343.7 | 849.2 | 0.14 |
| 10 | 222.0 | 351.8 | 855.6 | 0.16 |
| 20 | 260.5 | 366.4 | 858.0 | 0.47 |
| 40 | 262.6 | 379.2 | 862.2 | 0.87 |
| α | A (s−1) | Kinetic Compensation Plot |
|---|---|---|
| 0.1 | 2.73 × 107 |  |
| 0.2 | 2.22 × 108 | |
| 0.3 | 2.76 × 107 | |
| 0.4 | 1.37 × 104 | |
| 0.5 | 2.72 × 107 | |
| 0.6 | 3.70 × 1010 | |
| 0.7 | 8.00 × 1011 | |
| 0.8 | 3.01 × 1015 | |
| 0.9 | 3.74 × 1023 | |
| Average | 4.15 × 1022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özsin, G. Exploring the Thermal Degradation of Bakelite: Non-Isothermal Kinetic Modeling, Thermodynamic Insights, and Evolved Gas Analysis via Integrated In Situ TGA/MS and TGA/FT-IR Techniques. Polymers 2025, 17, 2197. https://doi.org/10.3390/polym17162197
Özsin G. Exploring the Thermal Degradation of Bakelite: Non-Isothermal Kinetic Modeling, Thermodynamic Insights, and Evolved Gas Analysis via Integrated In Situ TGA/MS and TGA/FT-IR Techniques. Polymers. 2025; 17(16):2197. https://doi.org/10.3390/polym17162197
Chicago/Turabian StyleÖzsin, Gamzenur. 2025. "Exploring the Thermal Degradation of Bakelite: Non-Isothermal Kinetic Modeling, Thermodynamic Insights, and Evolved Gas Analysis via Integrated In Situ TGA/MS and TGA/FT-IR Techniques" Polymers 17, no. 16: 2197. https://doi.org/10.3390/polym17162197
APA StyleÖzsin, G. (2025). Exploring the Thermal Degradation of Bakelite: Non-Isothermal Kinetic Modeling, Thermodynamic Insights, and Evolved Gas Analysis via Integrated In Situ TGA/MS and TGA/FT-IR Techniques. Polymers, 17(16), 2197. https://doi.org/10.3390/polym17162197