
Performance of Asphalt Materials Based on Molecular Dynamics Simulation: A Review


 , ,
, ,
Abstract
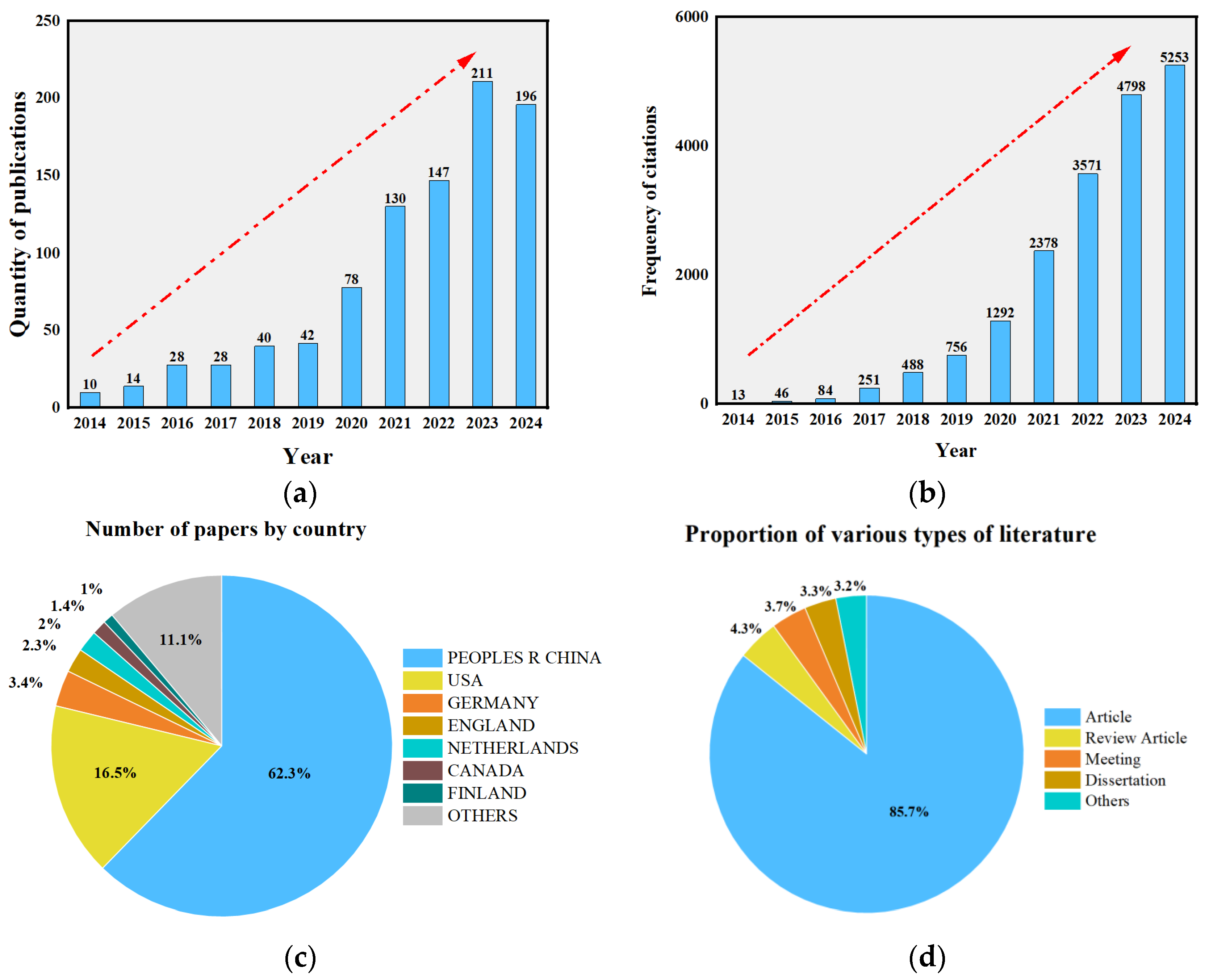
1. Introduction
2. Molecular Models of Asphalt, Modifiers, and Asphalt–Aggregate Interfaces
2.1. Generic Model Method
2.2. Molecular Assembly Method
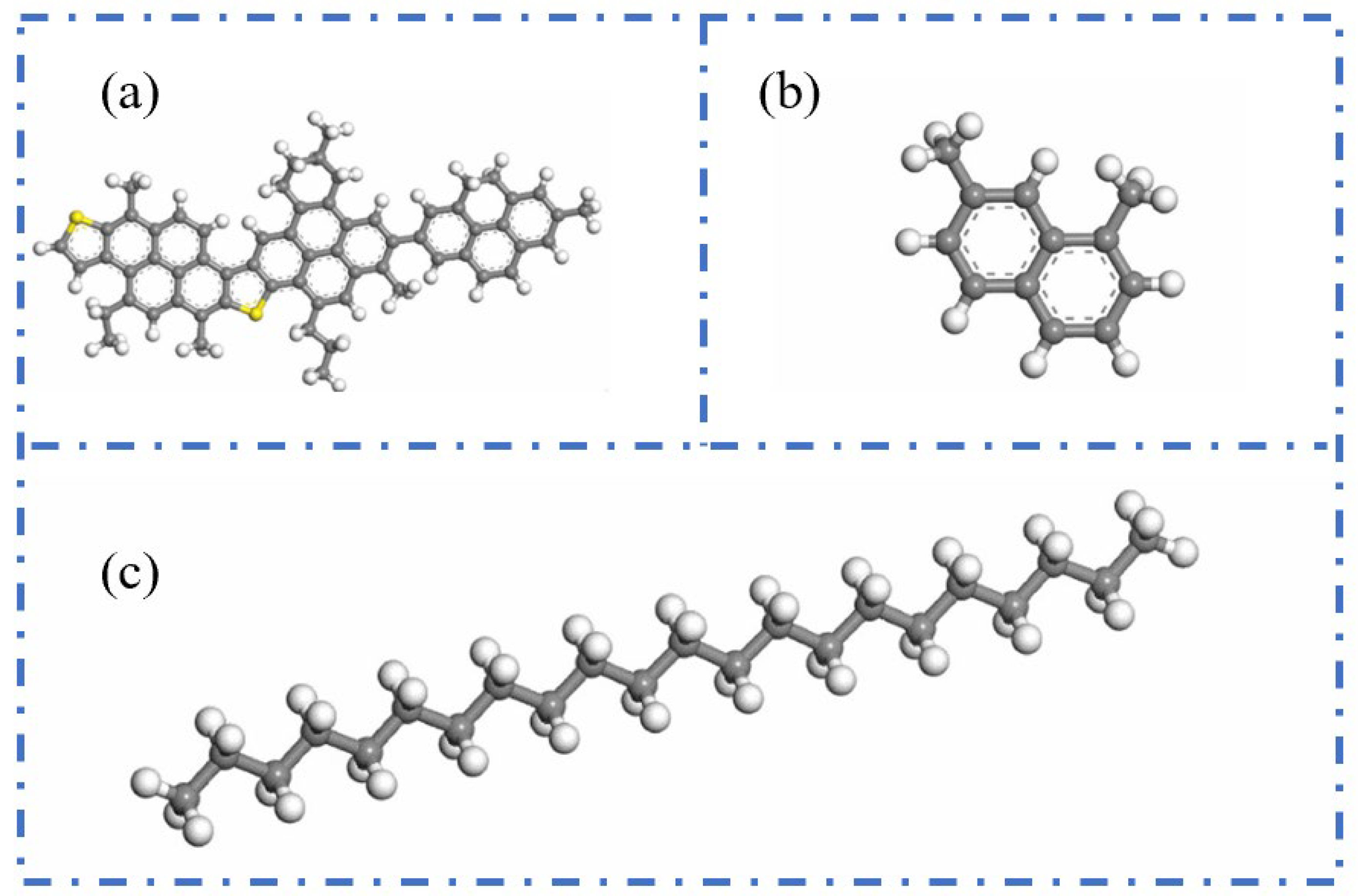
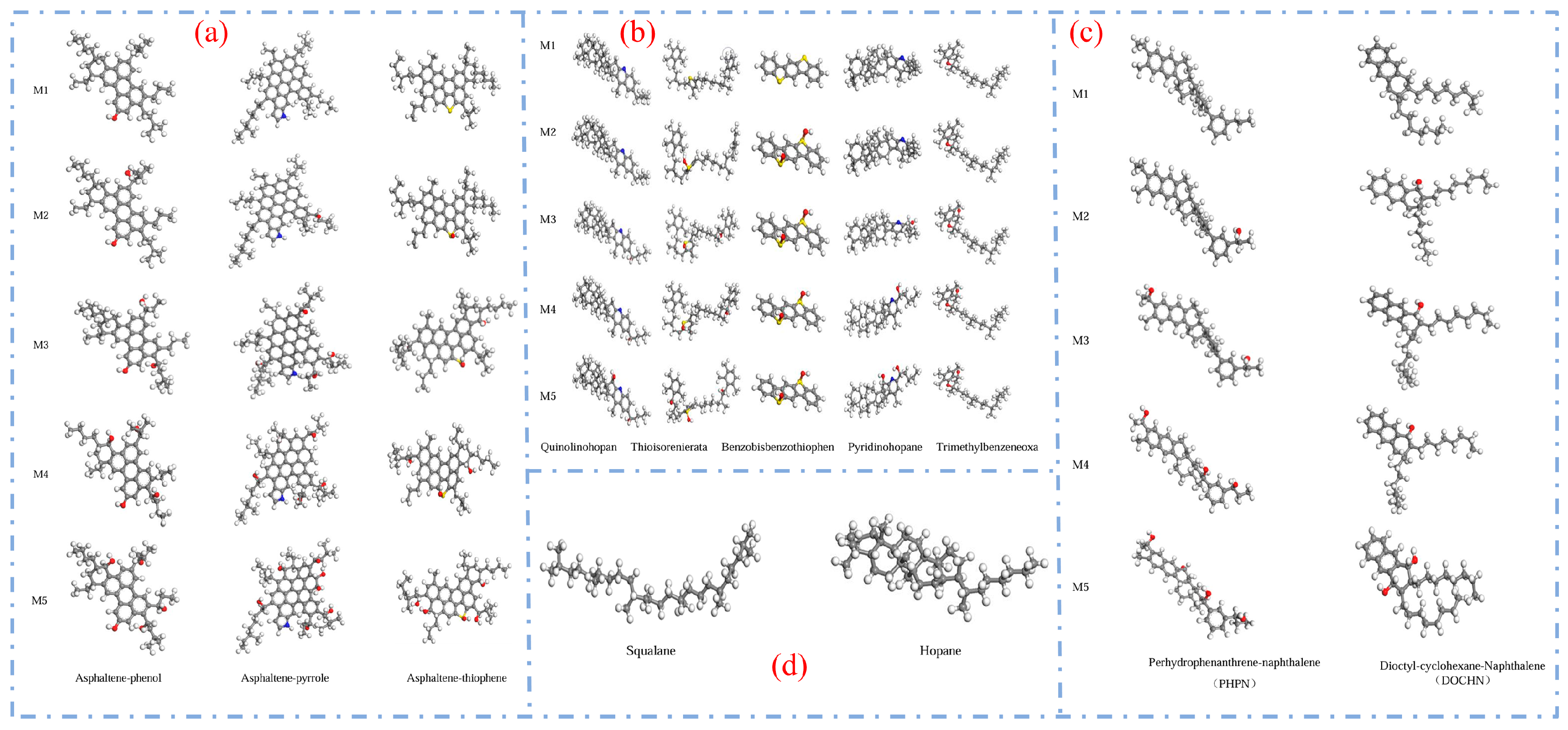
2.2.1. Three-Component Model
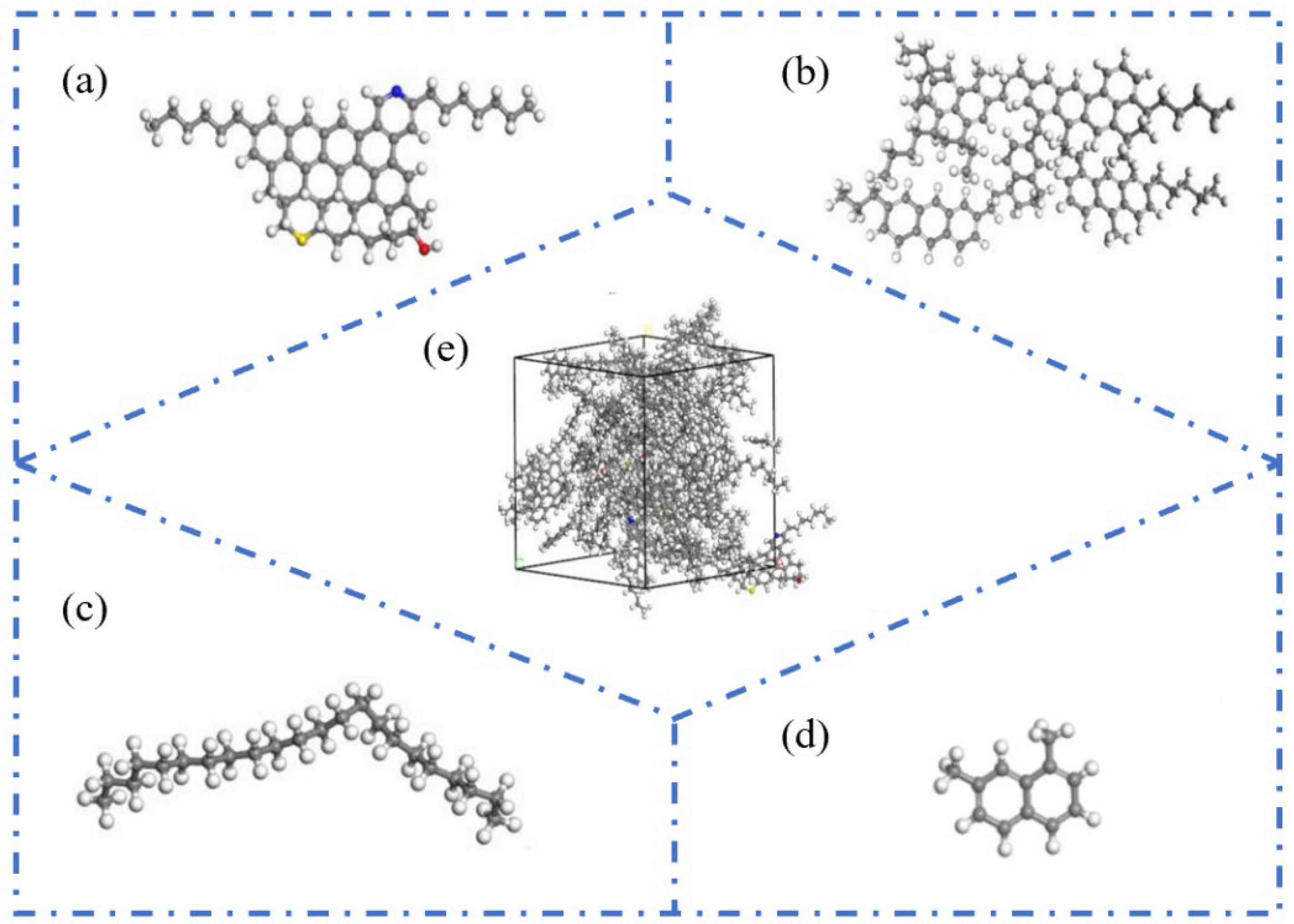
2.2.2. Four-Component Model
2.3. Common Modifier Models
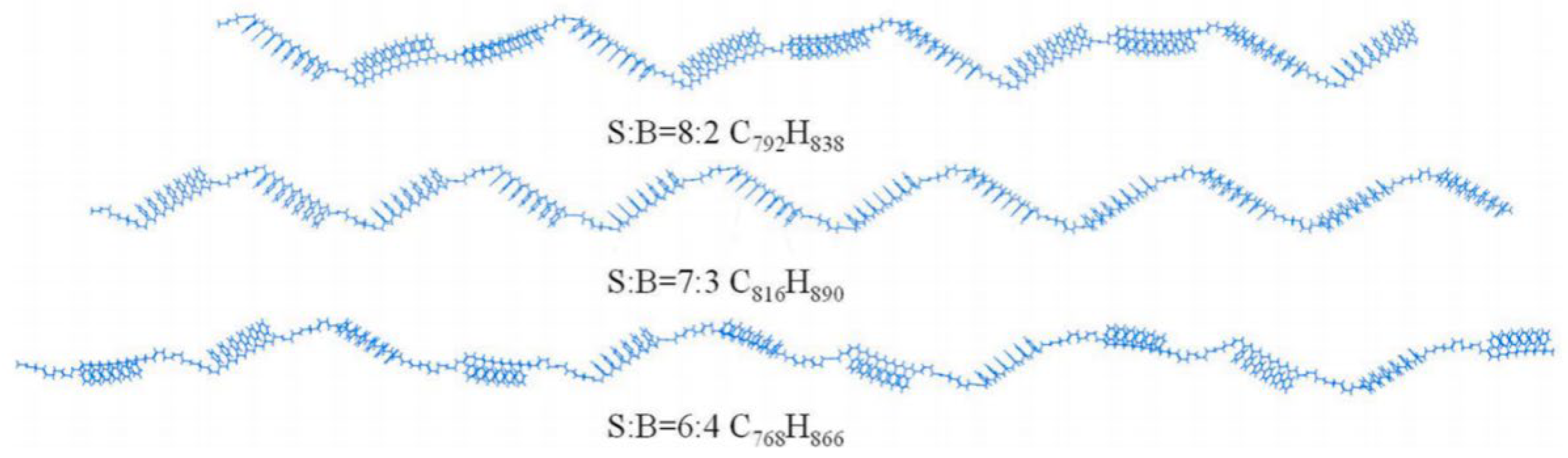
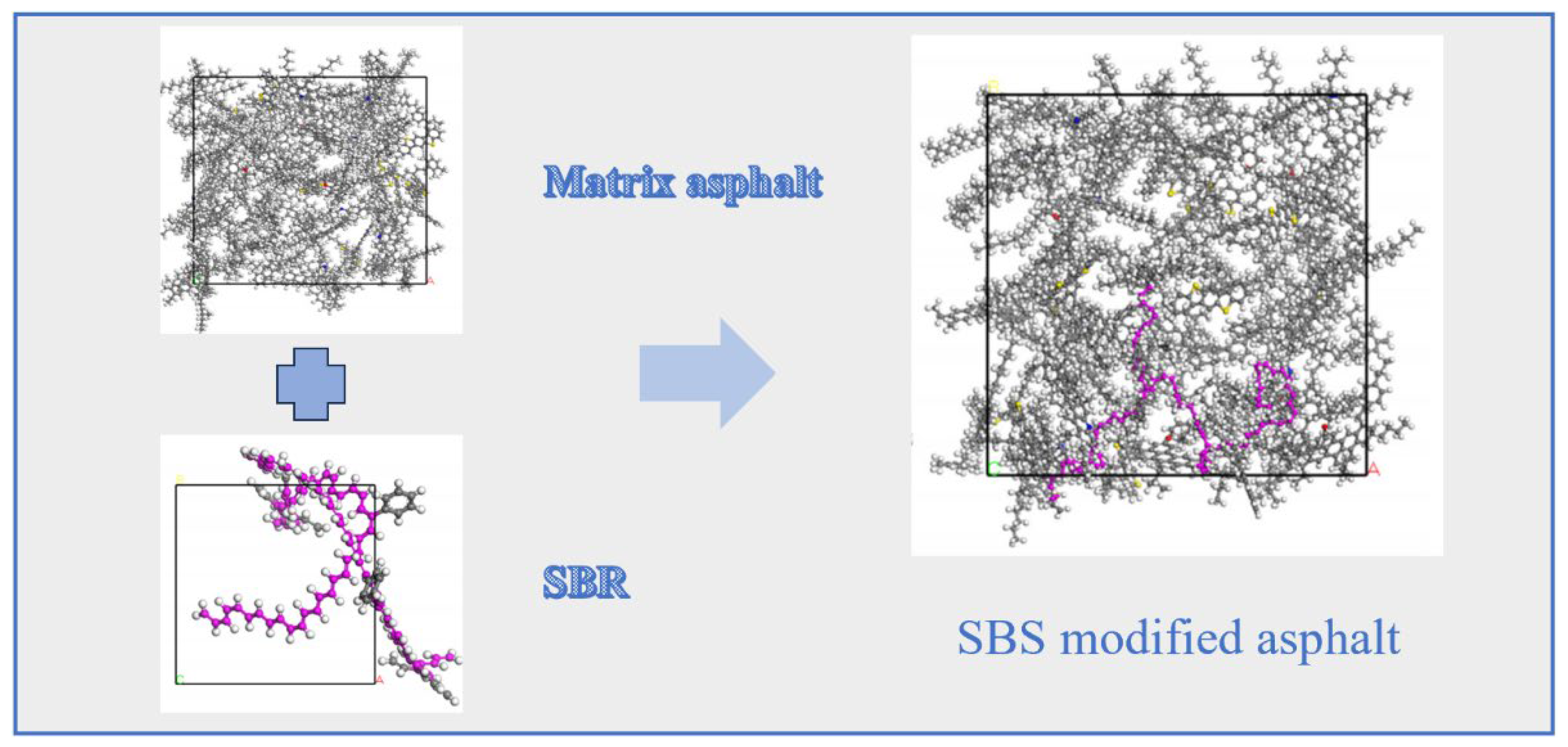
2.3.1. SBS Modifier Models
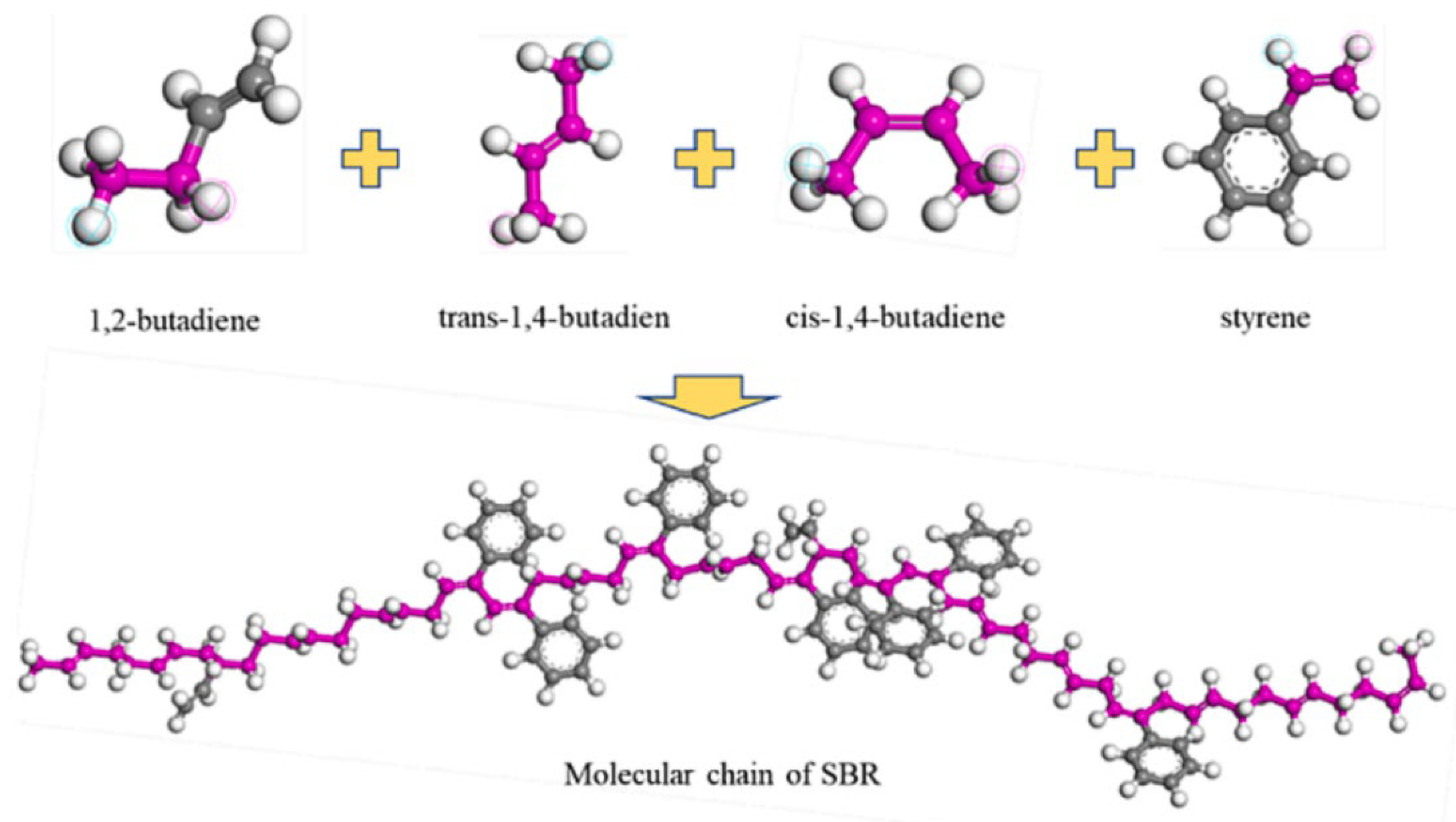
2.3.2. SBR Modifier Model
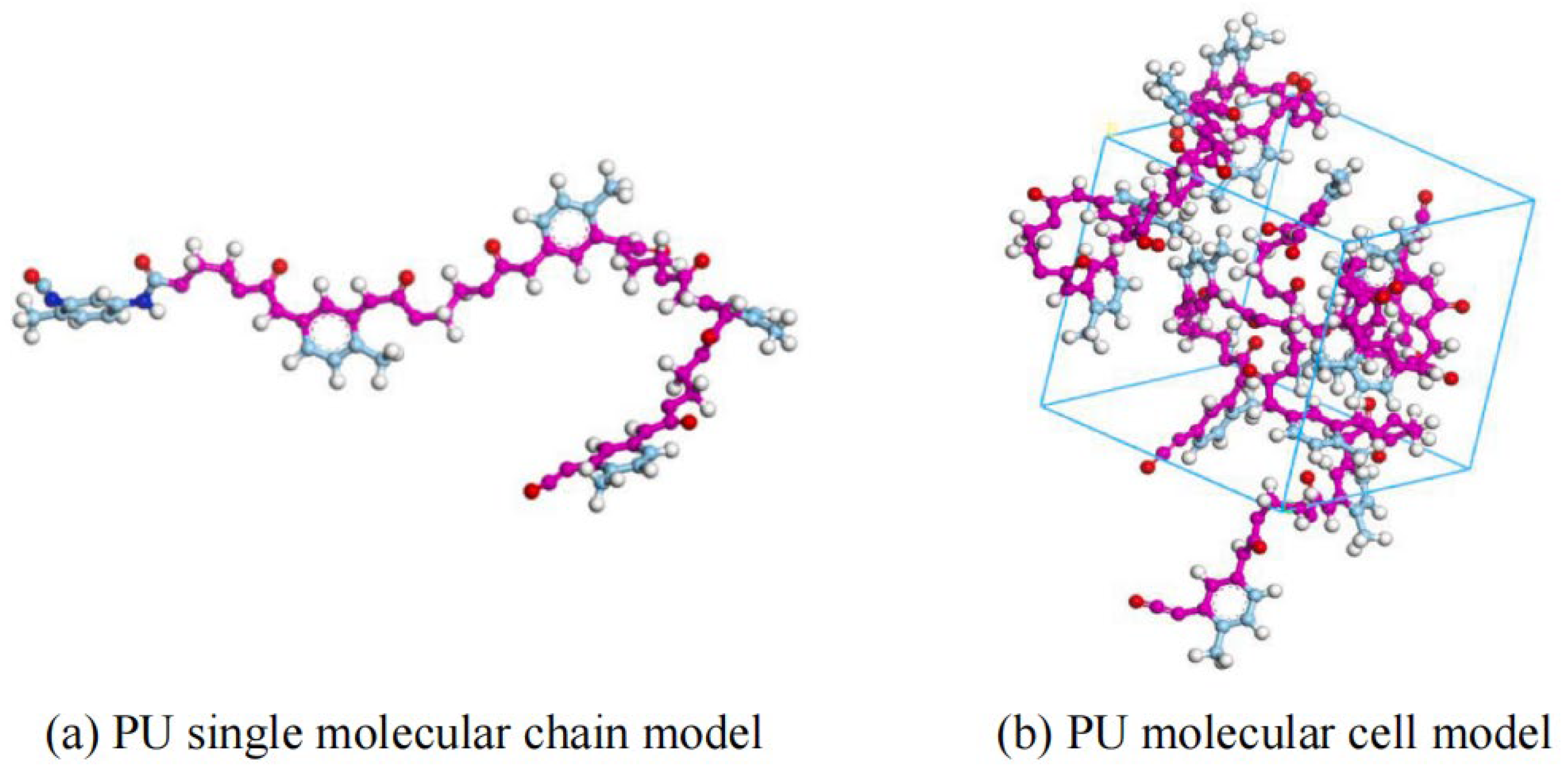
2.3.3. PU Modifier Model
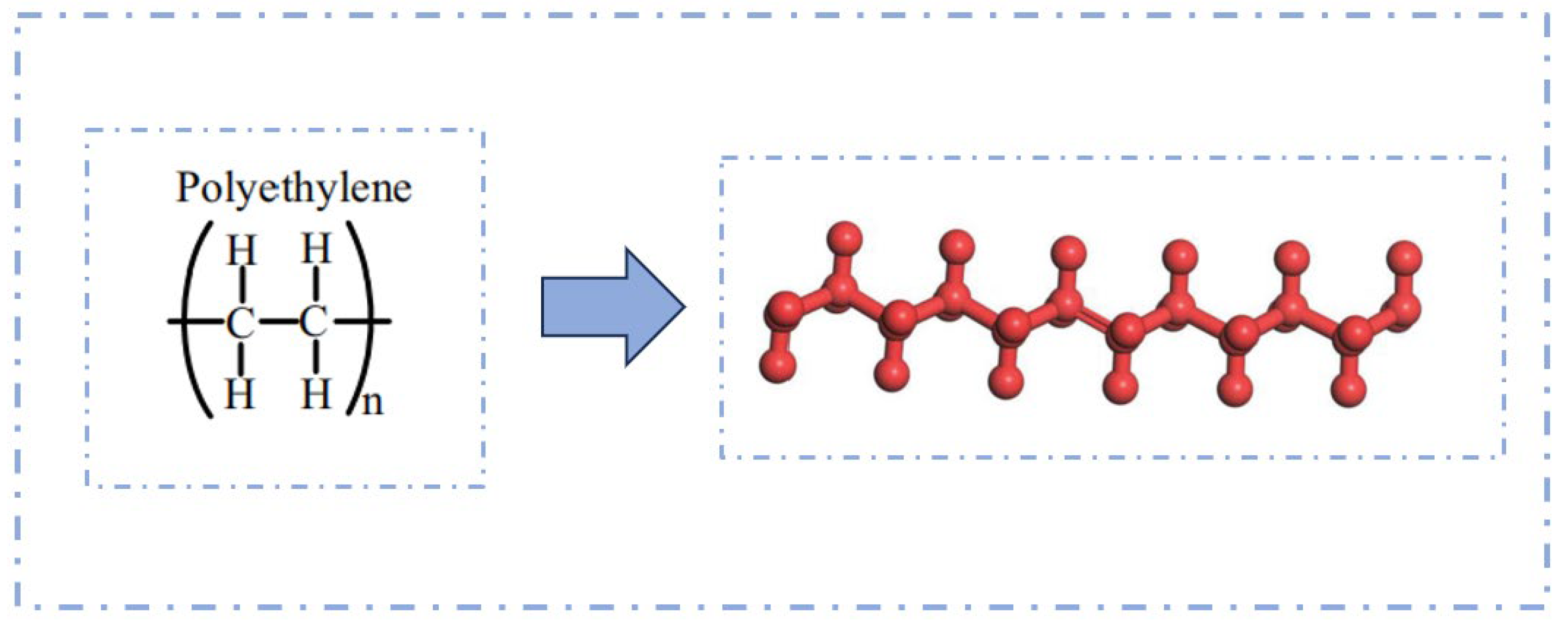
2.3.4. PE Modifier Model
2.4. Molecular Dynamics Simulation of Phase Separation and Compatibility in Polymer-Modified Asphalt
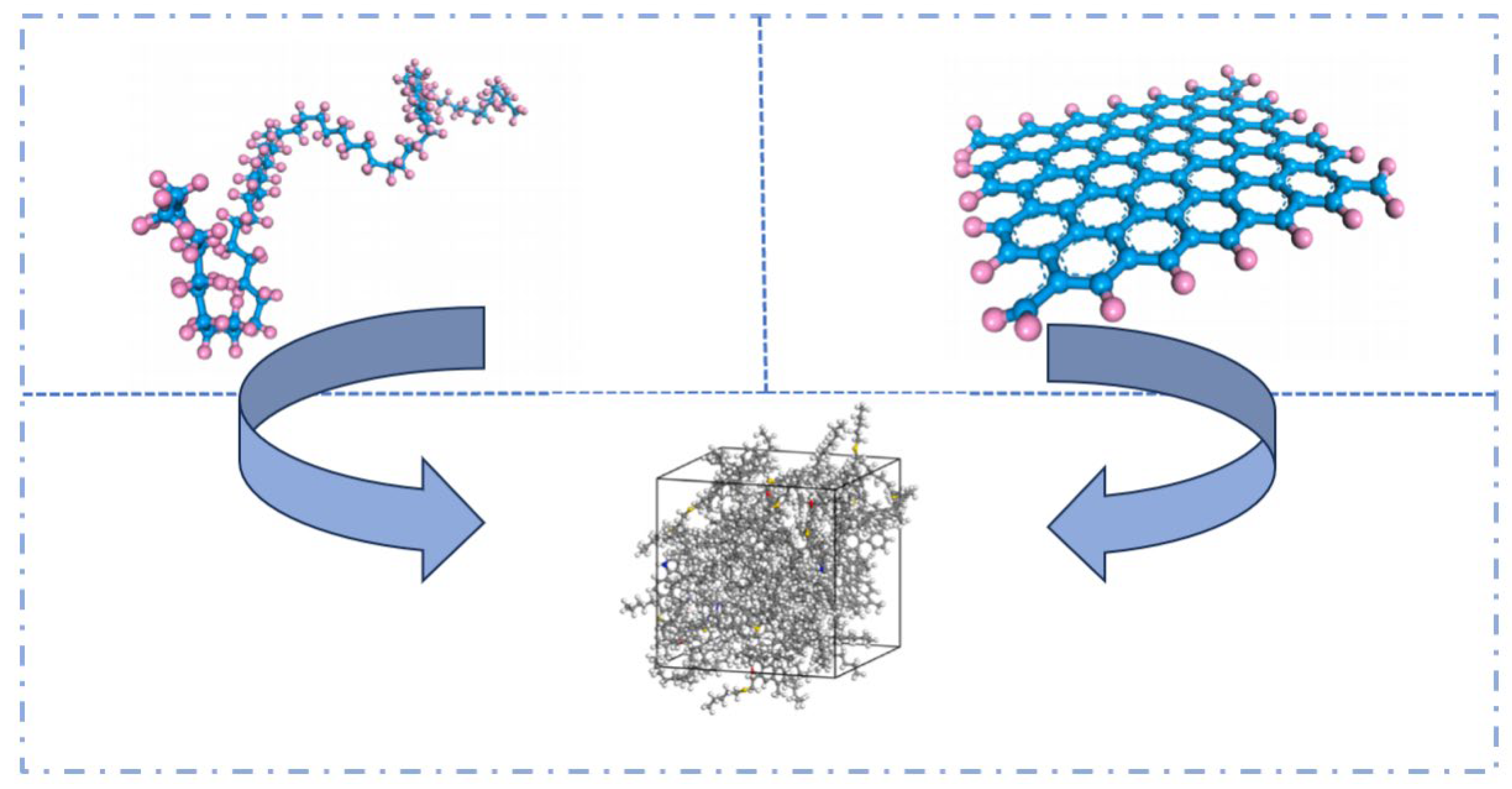
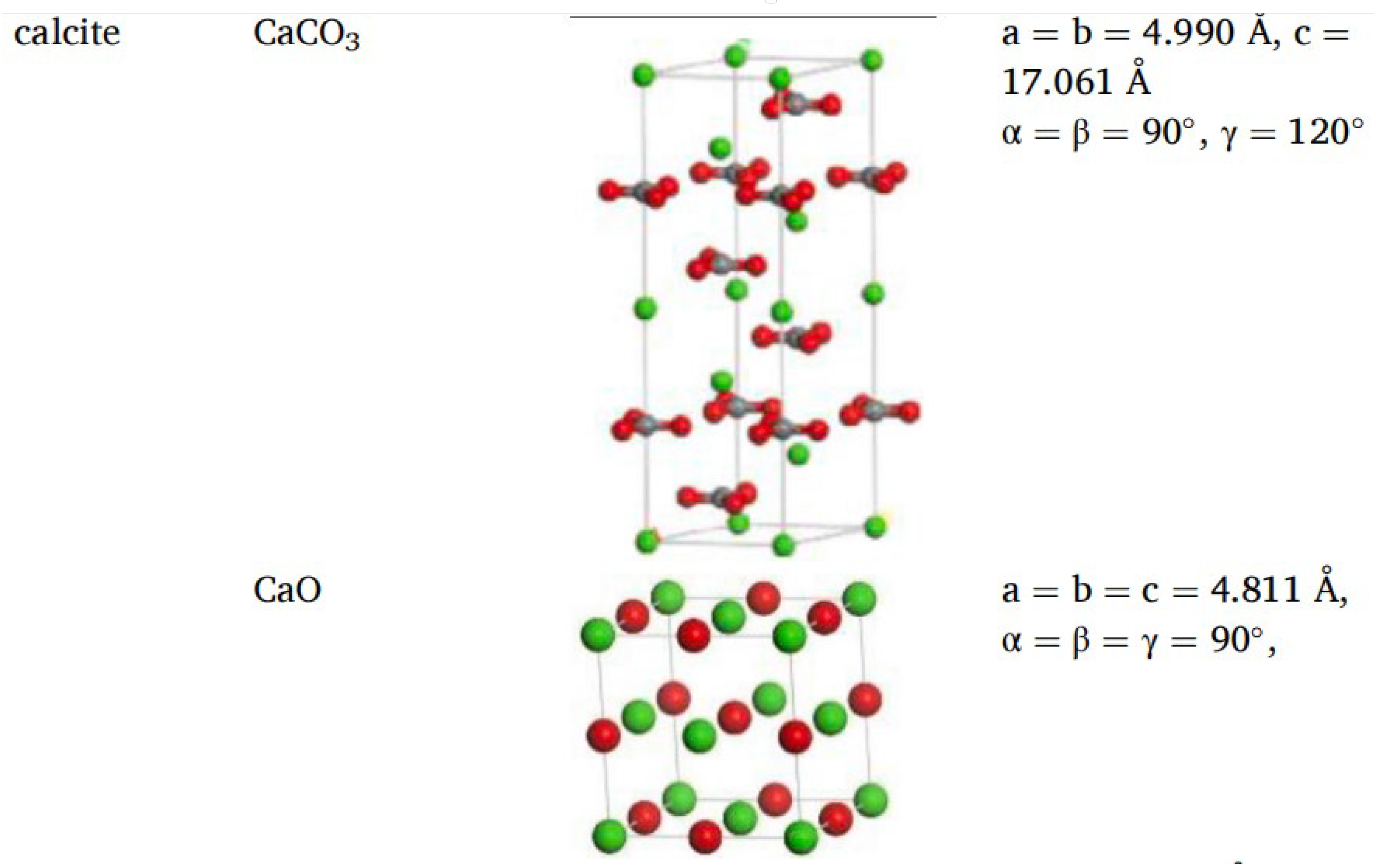
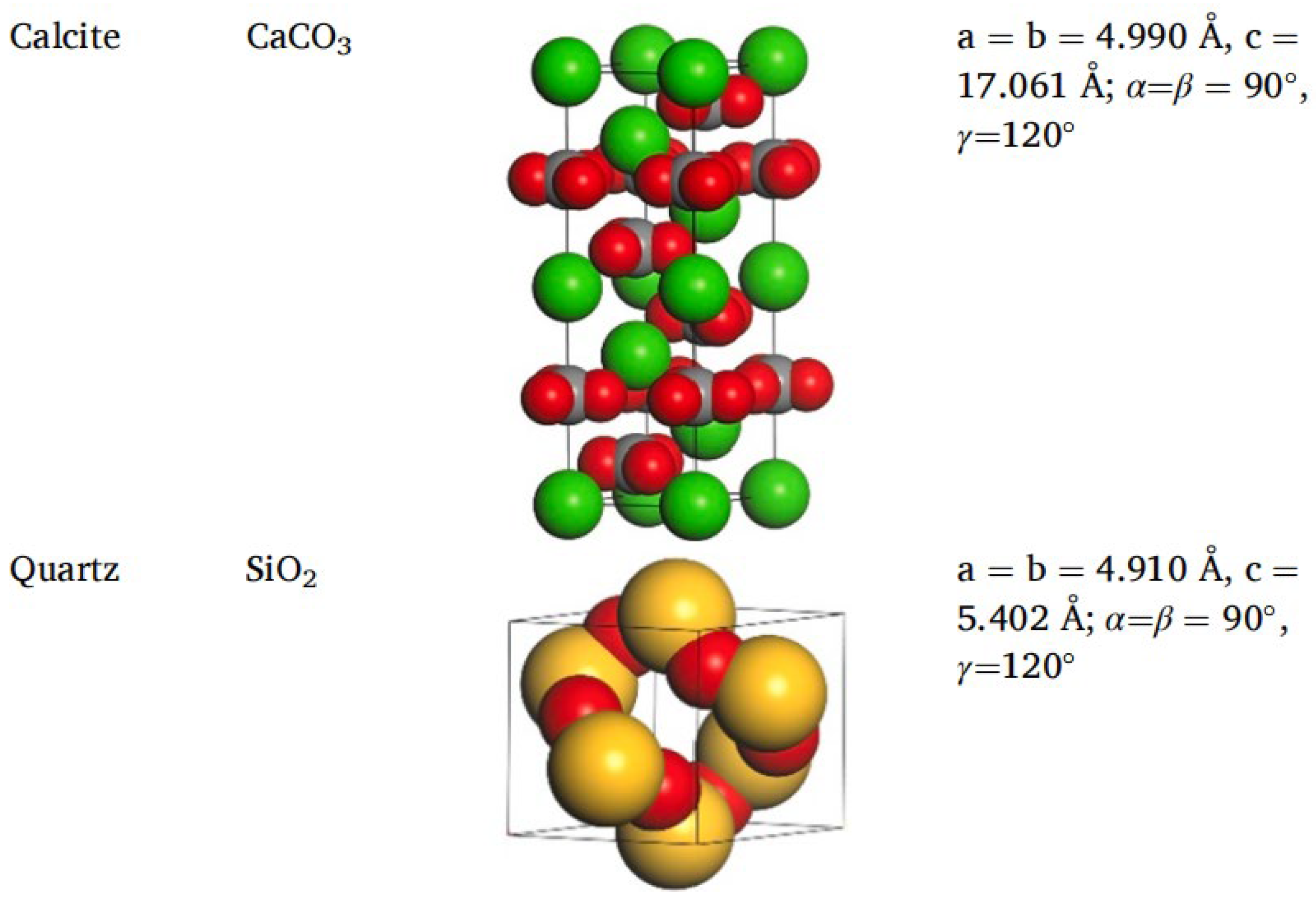
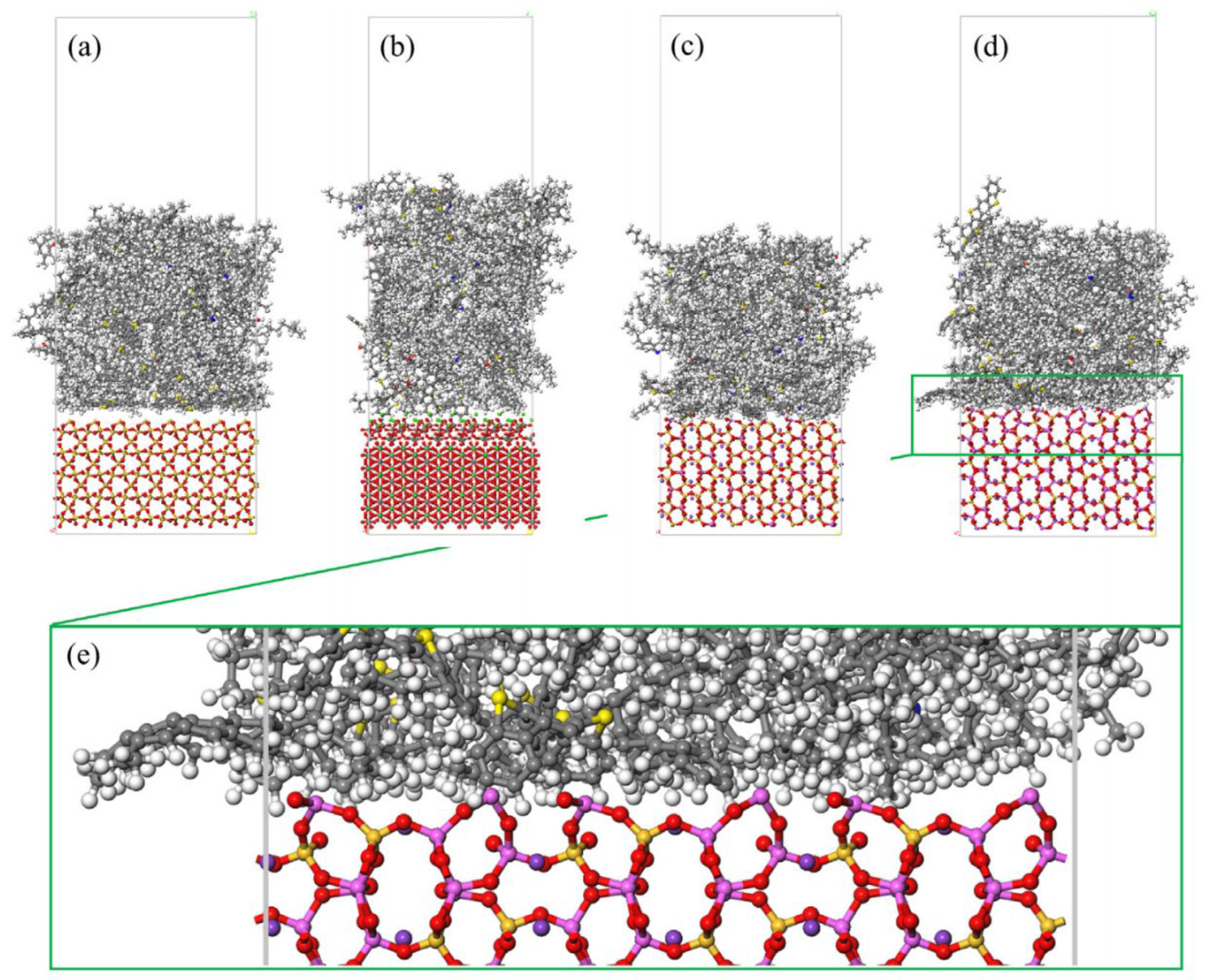
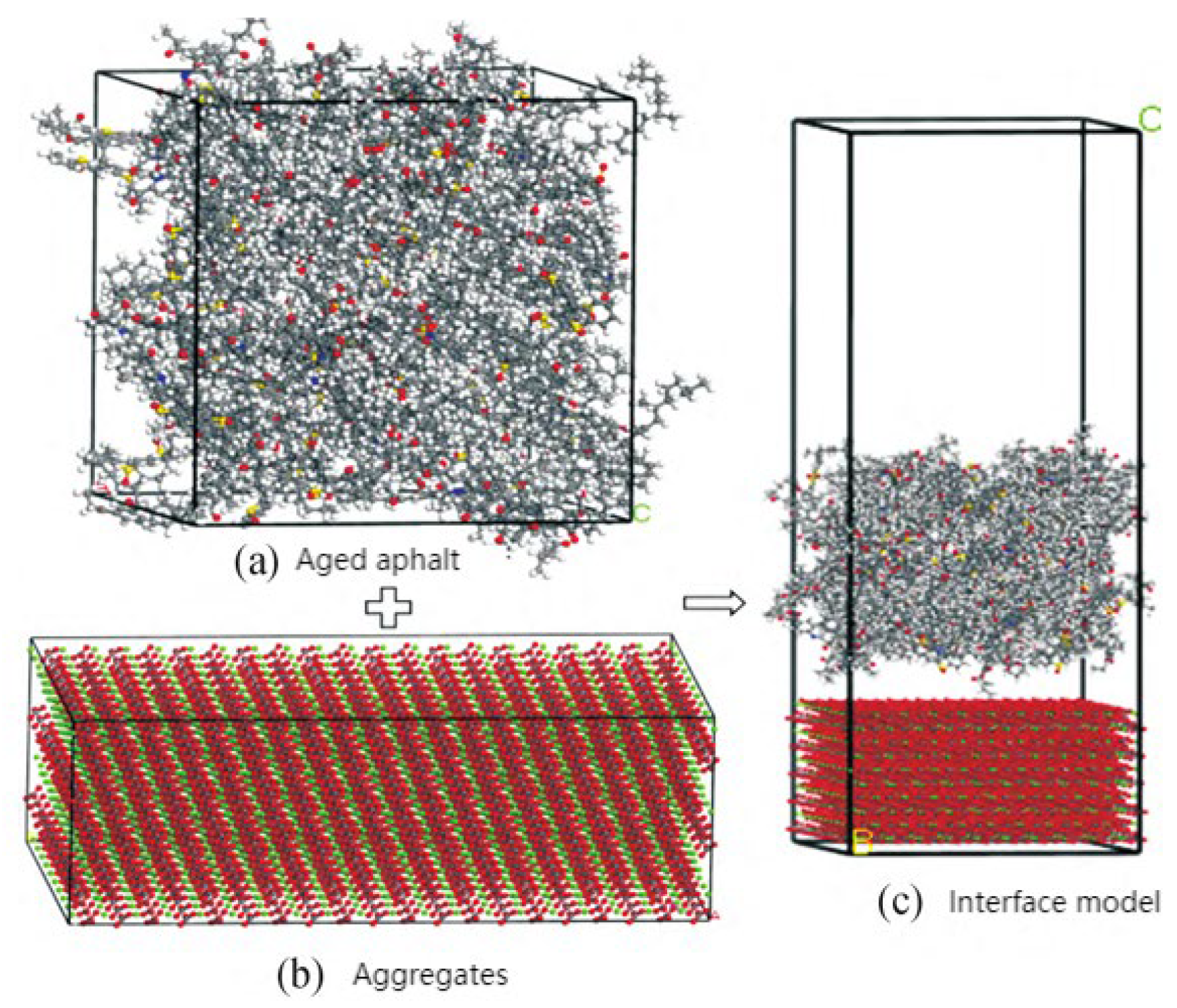
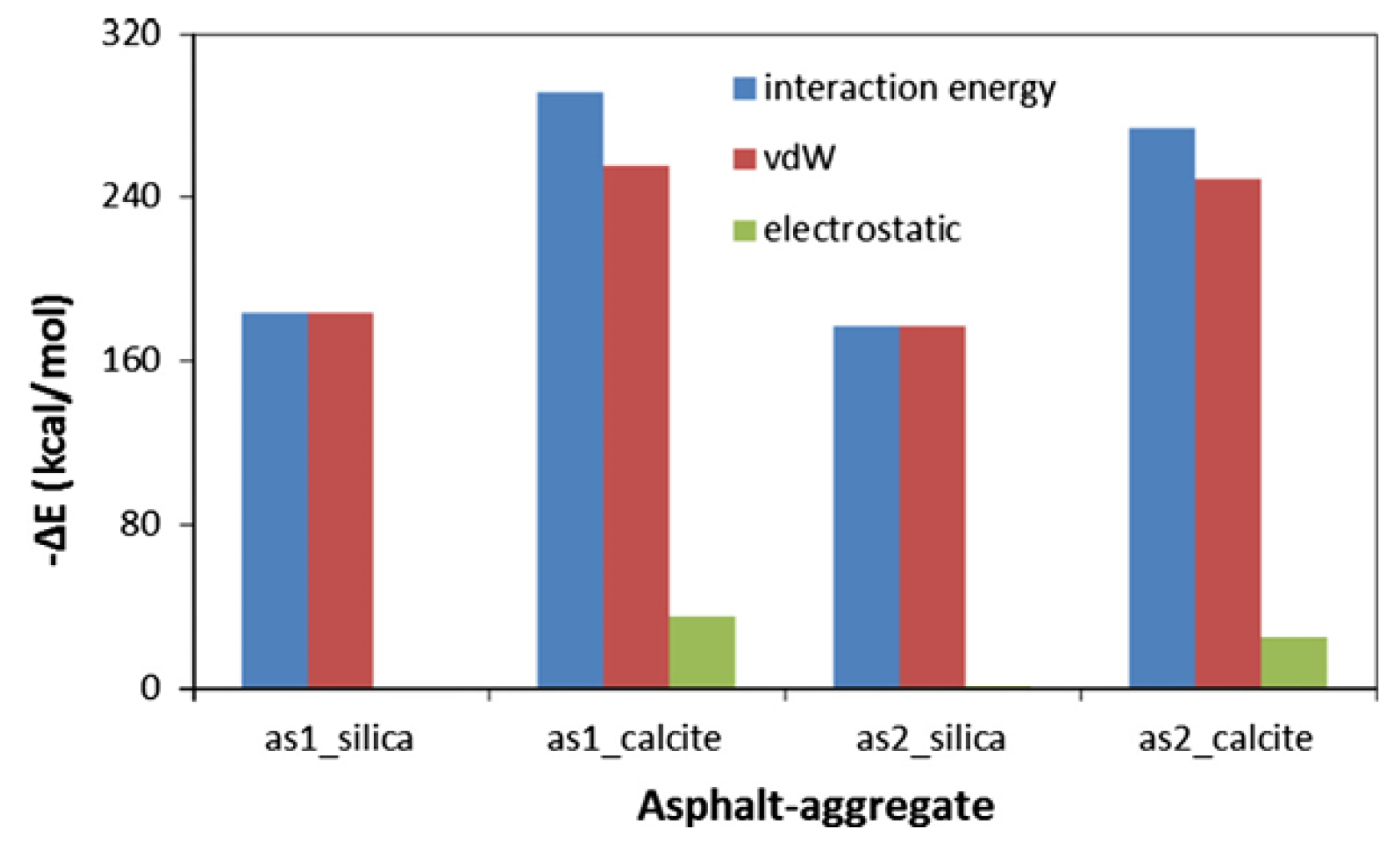
2.5. Molecular Models of Asphalt–Aggregate Interaction
3. Molecular Dynamics Simulation for the Study of Asphalt Material Properties
3.1. High-Temperature Performance of Asphalt
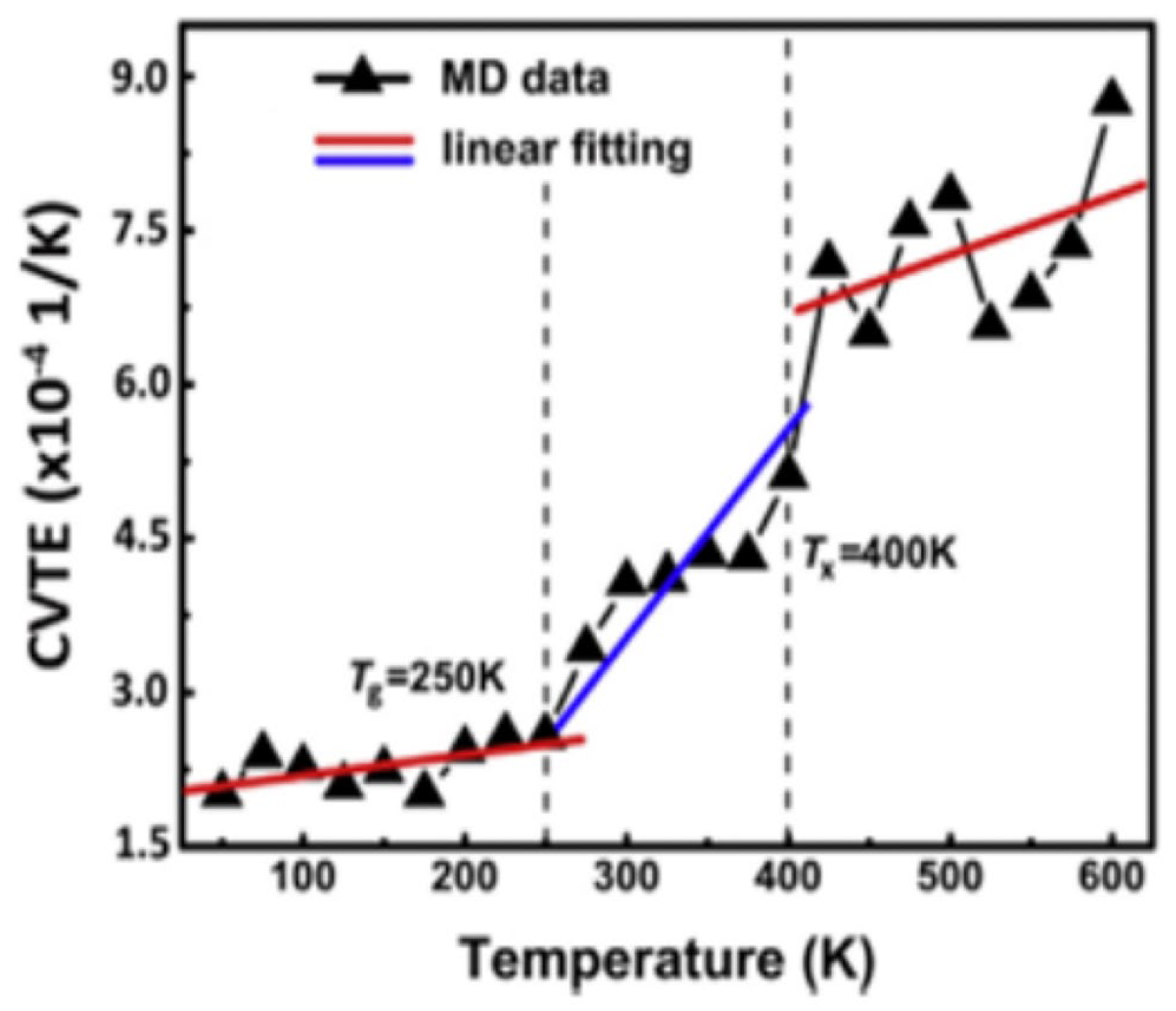
3.2. Low-Temperature Performance of Asphalt
3.3. Self-Healing Behavior of Asphalt Materials
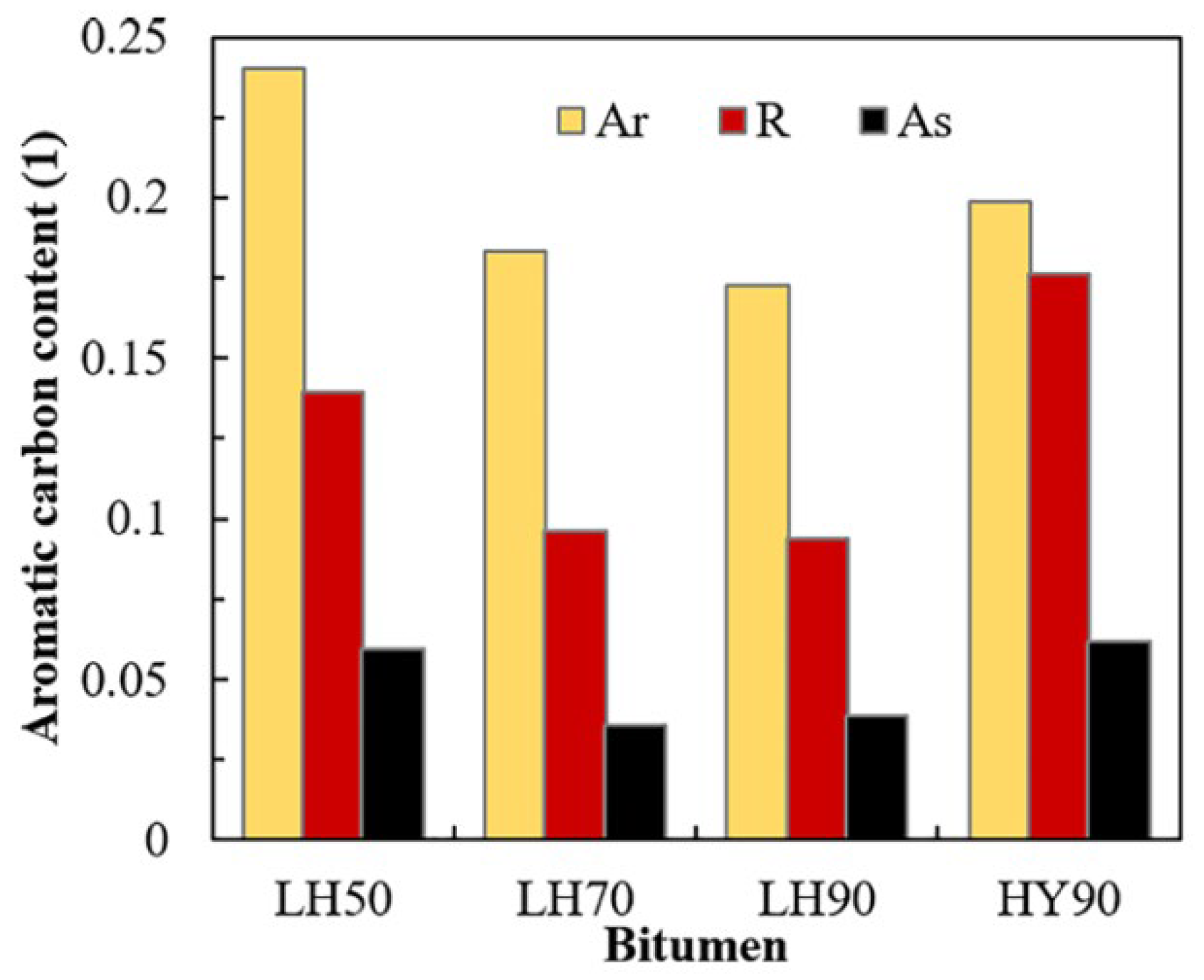
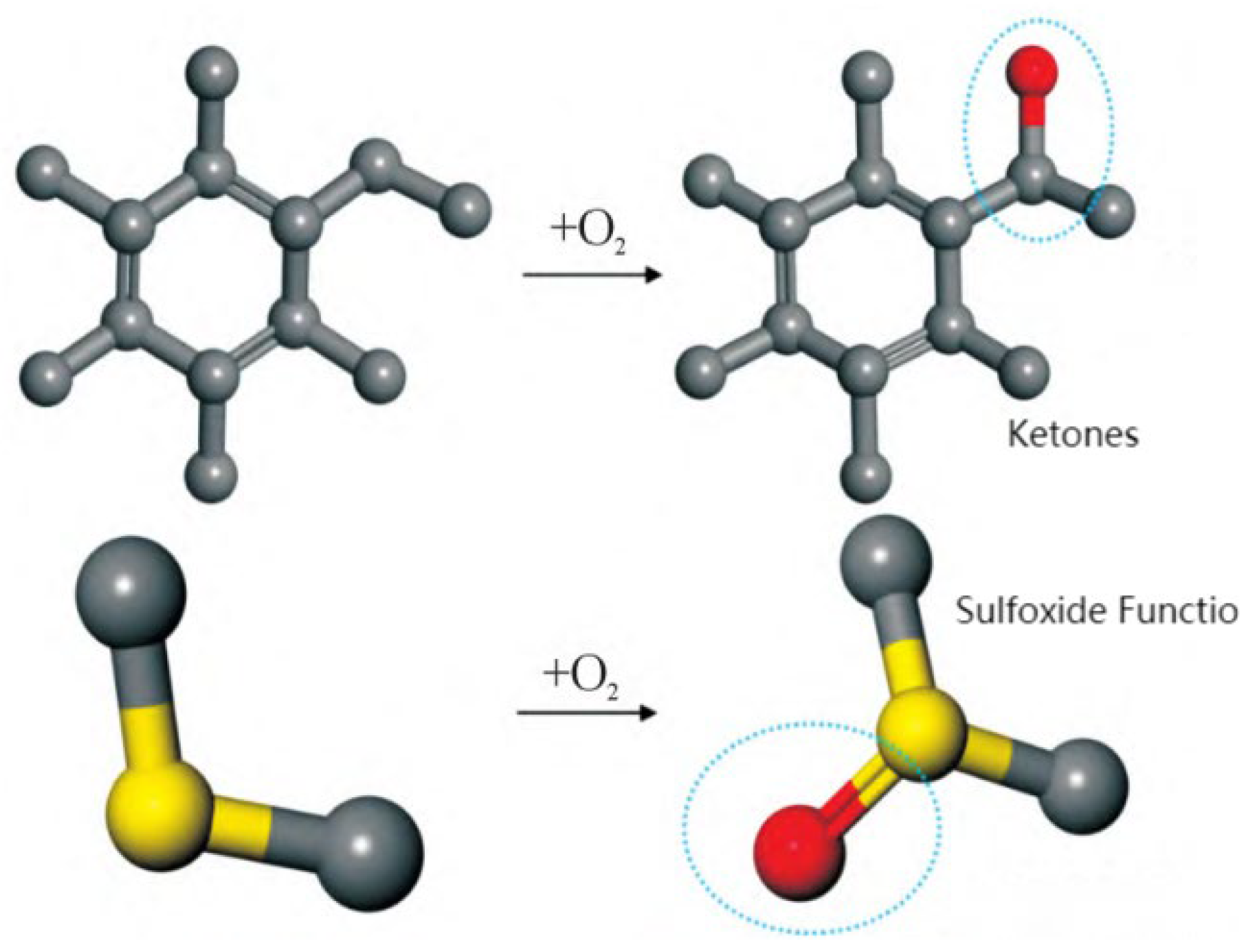
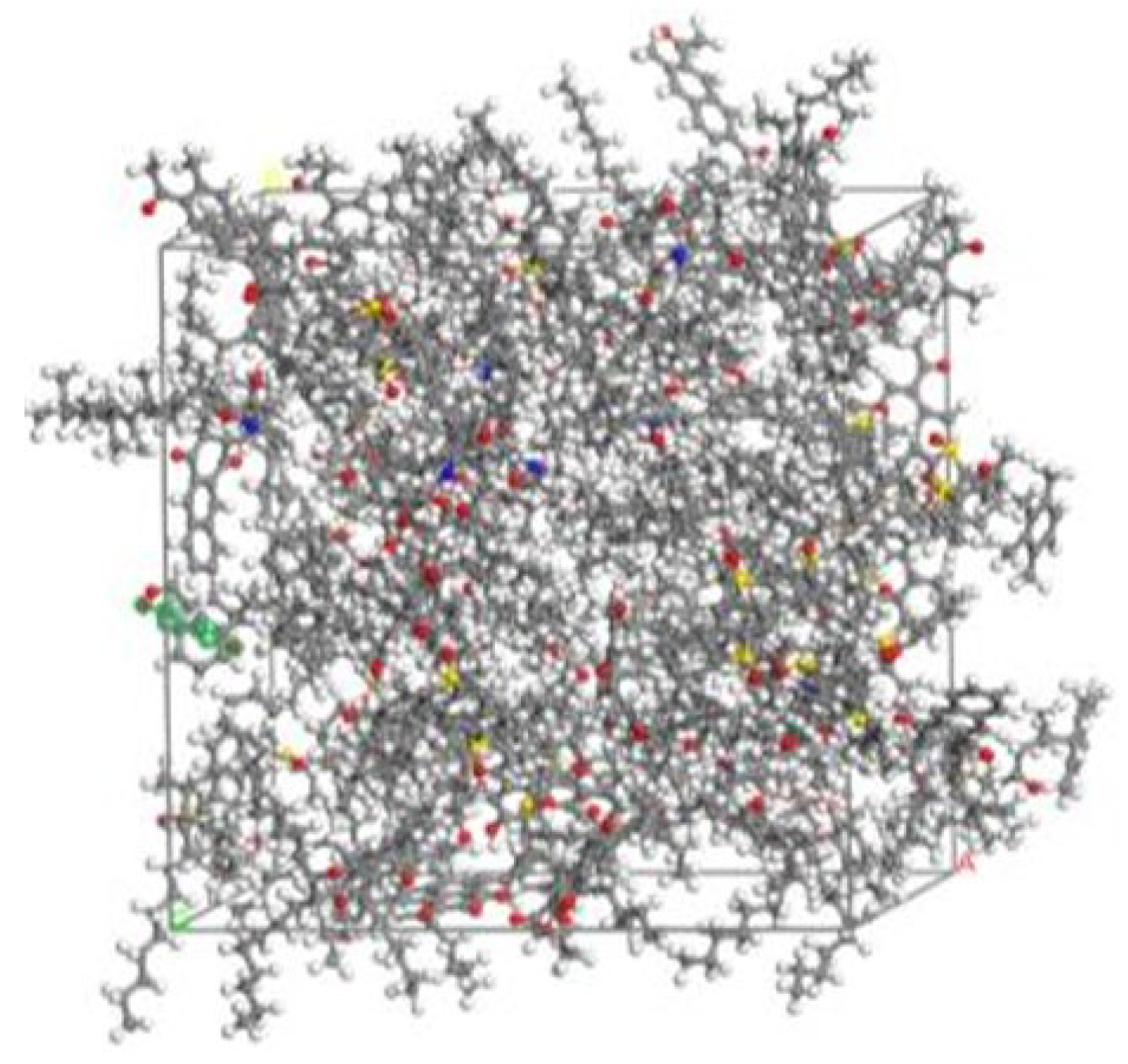
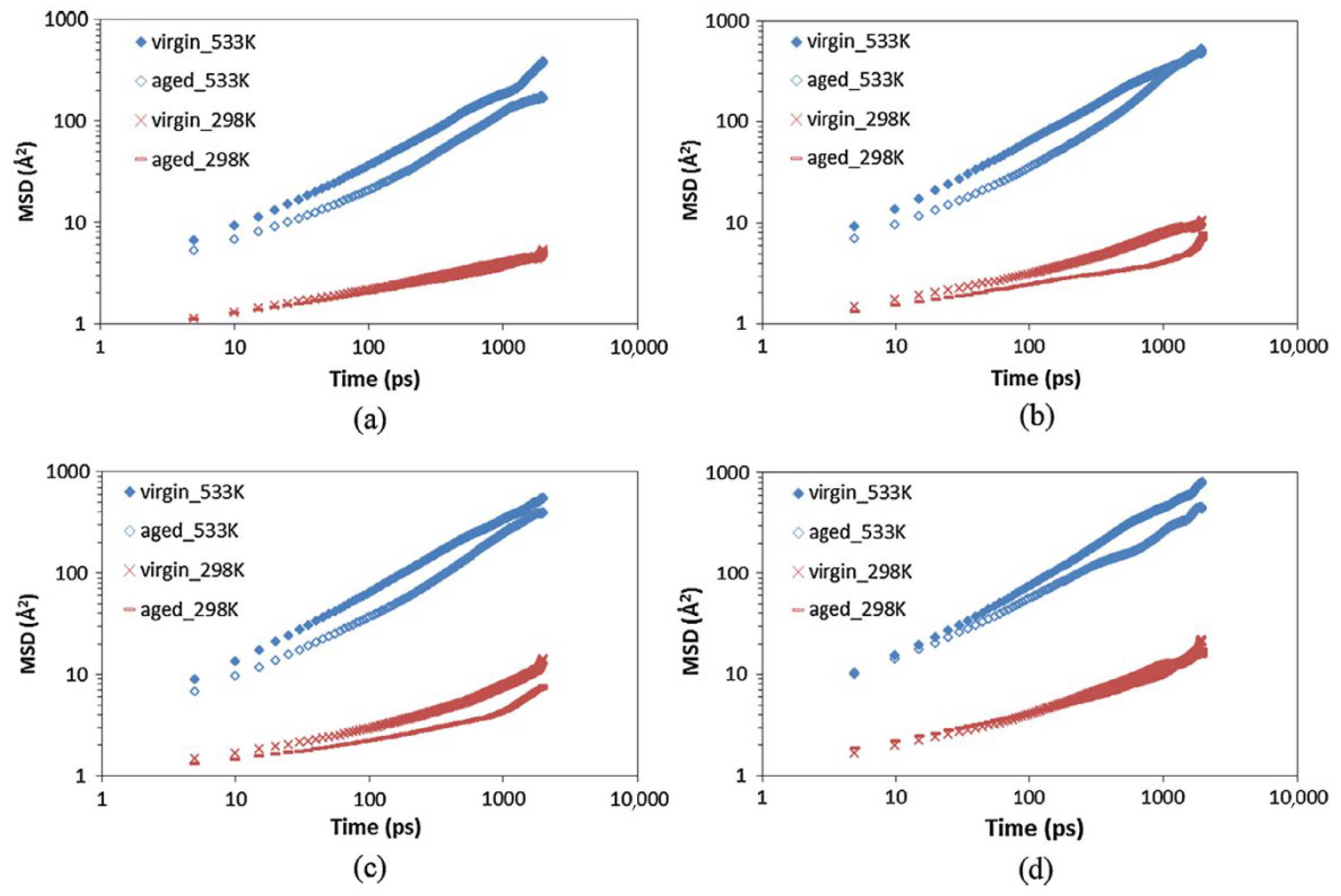
3.4. Aging Resistance of Asphalt
3.5. Adhesion Performance of the Asphalt–Aggregate Interface Model
4. Technical Challenges and Future Recommendations
- (1)
- Model Realism and Experimental Integration
- (2)
- Multiscale and Efficient Simulation Strategies
- (3)
- Chemical Reactivity, Modifier Behavior, and Interface Mechanics
5. Conclusions
- (1)
- Asphalt is a complex multi-component material whose molecular structure and chemical composition vary significantly depending on the asphalt grade, the type of modifier, and the modification method. This variability poses a major challenge for the accurate construction of molecular models. The current modeling approaches primarily include average molecular models and molecular assembly methods, yet both still fall short of fully capturing the true microscopic composition of asphalt.
- (2)
- Future research should focus on developing more refined, multiscale molecular models of asphalt and its modifiers to enhance the understanding of the roles of different chemical components at microscopic scales. Additionally, integrating experimental data for model optimization is essential to better reflect the actual composition and properties of asphalt materials.
- (3)
- MD simulations offer advantages in studying the physical properties of asphalt—such as diffusion, self-healing, aging resistance, and interfacial adhesion—yet face challenges including high computational resource demands, limited simulation timescales, and difficulties in linking microscopic behavior to macroscopic mechanical properties. The current simulations predominantly focus on microscopic scales, and establishing effective correlations between molecular-level phenomena and macroscopic performance remains a key challenge.
- (4)
- Future directions include advancing multiscale modeling techniques, such as combining coarse-grained MD (CGMD) with finite element methods (FEMs), to improve the simulation efficiency and achieve predictive capabilities spanning molecular to macroscopic levels. In asphalt–aggregate interface research, incorporating environmental factors (e.g., moisture, temperature, chemical corrosion) is necessary to enhance the model realism and applicability. Furthermore, coupling experimental data with high-resolution characterization techniques—such as X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), and scanning electron microscopy (SEM)—will improve the model reliability and engineering relevance.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, D.; Cao, J.; Gao, L.; Yi, J. Recent developments in asphalt-aggregate separation technology for reclaimed asphalt pavement. J. Road Eng. 2022, 2, 332–347. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, Y.; Zhang, J.; Ji, X.; Zhan, H. Molecular components and molecular modelling for asphalt: A review. Adv. Phys. Res. 2025, 4, 2400128. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Zhou, L.; Feng, Z.; Sun, J.; Hao, G.; Li, Z. Effect of rubber powder desulfurization on the compatibility between rubber and bitumen: Combination and insight of experiments and molecular dynamics simulation. Constr. Build. Mater. 2024, 414, 134966. [Google Scholar] [CrossRef]
- Song, Y.; Xu, H.; Wu, S.; Xie, J.; Chen, A.; Lv, Y.; Cheng, Y.; Li, Y. High-quality utilization of reclaimed asphalt pavement (RAP) in asphalt mixture with the enhancement of steel slag and epoxy asphalt. Constr. Build. Mater. 2024, 445, 137963. [Google Scholar] [CrossRef]
- Du, J.; Gui, Y.; Fu, C.; Li, G. Large-scale molecular dynamics simulation and aggregate behavior research on asphalt. Case Stud. Constr. Mater. 2024, 21, e03749. [Google Scholar] [CrossRef]
- Li, L.; Xin, C.; Guan, M.; Guo, M. Using Molecular Dynamics Simulation to Analyze the Feasibility of Using Waste Cooking Oil as an Alternative Rejuvenator for Aged Asphalt. Sustainability 2021, 13, 4373. [Google Scholar] [CrossRef]
- Xie, J.; He, W.; Zhao, X.-C.; Li, S.; Lu, Z.; Ding, Z. Research Progress on the Application of Molecular Dynamics Simulation in Asphalt Systems. Chem. Ind. Eng. Prog. 2024, 43, 4432–4449. (In Chinese) [Google Scholar]
- Yao, H.; Liu, J.; Xu, M.; Ji, J.; Dai, Q.; You, Z. Discussion on molecular dynamics (MD) simulations of the asphalt materials. Adv. Colloid Interface Sci. 2022, 299, 102565. [Google Scholar] [CrossRef]
- Su, M.; Si, C.; Zhang, Z.; Zhang, H. Molecular dynamics study on influence of Nano-ZnO/SBS on physical properties and molecular structure of asphalt binder. Fuel 2020, 263, 116777. [Google Scholar] [CrossRef]
- He, Y.; Mao, S.; Zhang, T.; Zeng, S.; Duan, H.; Zhuang, R.; Yu, J. Preparation and properties of self-healing SBS modified bitumen with dynamic acylhydrazone bonds. Eur. Polym. J. 2024, 210, 112932. [Google Scholar] [CrossRef]
- Wang, H.-N.; Ding, H.-Y.; Feng, P.-N.; Shao, L.-L.; Qu, X.; You, Z.-P. A Review of Molecular Simulation Techniques for Asphalt Mixtures. J. Traffic Transp. Eng. 2020, 20, 1–14. (In Chinese) [Google Scholar]
- Ma, D.; Shi, L.; Xia, Q.; Xu, T. Degradation behaviors of asphalt by microorganisms in asphalt pavement structure. Bioresour. Technol. 2025, 416, 131793. [Google Scholar] [CrossRef]
- Yao, H.; Zeng, J.; Liu, J.; Wang, Y.; Li, X.; Dai, Q.; You, Z. Perspectives on the Application of Waste Materials in Asphalt Based on Molecular Dynamics Simulations: A Review. Energy Fuels 2024, 38, 14839–14865. [Google Scholar] [CrossRef]
- Jennings, P.; Pribanic, J.; Desando, M.; Raub, M. Binder Characterization and Evaluation by Nuclear Magnetic Resonance Spectroscopy; Strategic Highway Research Program: Washington, DC, USA, 1993. [Google Scholar]
- An, D.; Qu, T.J. Seismic Behavior of Turbine-Generator Foundation under Strong Earthquake Action in Different Directions. Adv. Civ. Eng. 2018, 2018, 2506264. [Google Scholar] [CrossRef]
- Corbett, L.W. Composition of asphalt based on generic fractionation, using solvent deasphaltening, elution-adsorption chromatography, and densimetric characterization. Anal. Chem. 1969, 41, 576–579. [Google Scholar] [CrossRef]
- Koots, J.A.; Speight, J.G. Relation of petroleum resins to asphaltenes. Fuel 1975, 54, 179–184. [Google Scholar] [CrossRef]
- Murgich, J.; Rodríguez; Array, Y. Molecular recognition and molecular mechanics of micelles of some model asphaltenes and resins. Energy Fuels 1996, 10, 68–76. [Google Scholar] [CrossRef]
- Groenzin, H.; Mullins, O.C. Molecular size and structure of asphaltenes from various sources. Energy Fuels 2000, 14, 677–684. [Google Scholar] [CrossRef]
- Zhang, L.; Greenfield, M.L. Effects of polymer modification on properties and microstructure of model asphalt systems. Energy Fuels 2008, 22, 3363–3375. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, B.; Zhang, Y.; Wei, J.; Cao, X. Molecular dynamics simulation to investigate the influence of SBS on molecular agglomeration behavior of asphalt. J. Mater. Civ. Eng. 2015, 27, C4014004. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, J.; Pei, J.; Ma, W.; Hu, Z.; Guan, Y. Evaluation of the compatibility between rubber and asphalt based on molecular dynamics simulation. Front. Struct. Civ. Eng. 2020, 14, 435–445. [Google Scholar] [CrossRef]
- Rostler, F.S.; White, R.M. Influence of chemical composition of asphalts on performance, particularly durability. In Symposium on Road and Paving Materials—1959; ASTM International: West Conshohocken, PA, USA, 1960. [Google Scholar]
- Dong, X.-G.; Lei, Q.-F.; Yu, Q.-S. NMR determination of petroleum asphaltenes and their model molecules evaluation. J. Fuel Chem. Technol. 2004, 32, 668–672. [Google Scholar]
- Qi, B.F.; Cao, Z.B.; Chen, L.R.; Zhang, H.C.; Wang, L.J.; Que, G.H. Study on structure of resins and asphaltenes with UV absorption spectrum. J. Petrochem. Univ. 2001, 14, 14–17. [Google Scholar]
- Liu, Y.-R.; Tang, X.; Zeng, Q.; Lai, J.-P. Impacts of ultraviolet absorption by zinc oxide nanoparticle modifiers on asphalt aging. Sci. Rep. 2024, 14, 19918. [Google Scholar] [CrossRef]
- Zhang, L.; Greenfield, M.L. Analyzing properties of model asphalts using molecular simulation. Energy Fuels 2007, 21, 1712–1716. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Chemical compositions of improved model asphalt systems for molecular simulations. Fuel 2014, 115, 347–356. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. High internal energies of proposed asphaltene structures. Energy Fuels 2011, 25, 3698–3705. [Google Scholar] [CrossRef]
- Xu, M.; Yi, J.; Qi, P.; Wang, H.; Marasteanu, M.; Feng, D. Improved chemical system for molecular simulations of asphalt. Energy Fuels 2019, 33, 3187–3198. [Google Scholar] [CrossRef]
- Yu, H.; Ge, J.; Qian, G.; Zhang, C.; Dai, W.; Li, P. Evaluation on the rejuvenation and diffusion characteristics of waste cooking oil on aged SBS asphalt based on molecular dynamics method. J. Clean. Prod. 2023, 406, 136998. [Google Scholar] [CrossRef]
- Han, J.; Li, B.; Ji, H.; Guo, F.; Wei, D.; Cao, S.; Zhang, W.; Chen, X. Interfacial adhesion between recycled asphalt binder and aggregates considering aggregate surface anisotropy: A molecular dynamics simulation. Constr. Build. Mater. 2024, 438, 137176. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Huang, Z.; Ren, R.; Liu, K. Nano-micelle formation and aggregation in SBS-Modified asphalt induced by π-π interaction using molecular dynamics. Mater. Des. 2024, 237, 112571. [Google Scholar]
- Chen, M.; Geng, J.; Xia, C.; He, L.; Liu, Z. A review of phase structure of SBS modified asphalt: Affecting factors, analytical methods, phase models and improvements. Constr. Build. Mater. 2021, 294, 123610. [Google Scholar] [CrossRef]
- Schaur, A.; Unterberger, S.H.; Lackner, R. Impact of molecular structure of PP on thermo-rheological properties of polymer-modified bitumen. Constr. Build. Mater. 2021, 287, 122981. [Google Scholar] [CrossRef]
- Cai, J.; Song, C.; Zhou, B.; Tian, Y.; Li, R.; Zhang, J.; Pei, J. Investigation on high-viscosity asphalt binder for permeable asphalt concrete with waste materials. J. Clean. Prod. 2019, 228, 40–51. [Google Scholar] [CrossRef]
- Yan, C.; Huang, W.; Lin, P.; Zhang, Y.; Lv, Q. Chemical and rheological evaluation of aging properties of high content SBS polymer modified asphalt. Fuel 2019, 252, 417–426. [Google Scholar] [CrossRef]
- Fan, J.; Xu, G.; Ma, T.; Zhu, Y.; Huang, W. Multiscale Characterization of the Mechanism of High-Viscosity SBS-Modified Asphalt. J. Southeast Univ. (Nat. Sci. Ed.) 2023, 53, 377–385. (In Chinese) [Google Scholar]
- Li, C.; Fan, S.; Xu, T. Method for evaluating compatibility between SBS modifier and asphalt matrix using molecular dynamics models. J. Mater. Civ. Eng. 2021, 33, 04021207. [Google Scholar] [CrossRef]
- Li, G.; Gu, Z.; Tan, Y.; Xing, C.; Zhang, J.; Zhang, C. Research on the phase structure of Styrene-Butadiene-Styrene modified asphalt based on molecular dynamics. Constr. Build. Mater. 2022, 326, 126933. [Google Scholar] [CrossRef]
- Mouillet, V.; Lamontagne, J.; Durrieu, F.; Planche, J.-P.; Lapalu, L. Infrared microscopy investigation of oxidation and phase evolution in bitumen modified with polymers. Fuel 2008, 87, 1270–1280. [Google Scholar] [CrossRef]
- Lin, K.-S.; Mdlovu, N.V.; Wang, H.-P.; Hussain, A. Liquefaction of waste tire rubber chips used for the absorptive recycling of spilled oils. J. Environ. Chem. Eng. 2022, 10, 108680. [Google Scholar] [CrossRef]
- Sinkhonde, D. Modeling and experimental studies on energy consumption and properties of concrete containing waste brick powder and waste tire rubber for sustainable construction. Clean. Eng. Technol. 2023, 13, 100631. [Google Scholar] [CrossRef]
- Wiśniewska, P.; Wang, S.; Formela, K. Waste tire rubber devulcanization technologies: State-of-the-art, limitations and future perspectives. Waste Manag. 2022, 150, 174–184. [Google Scholar] [CrossRef]
- Liao, G.; Fang, X.; Wang, H.; Tang, J.; Szary, P.; Chen, J. Durability improvement of poroelastic road surface with treated rubber: Molecular dynamics simulation and experimental observations. J. Clean. Prod. 2022, 369, 133334. [Google Scholar] [CrossRef]
- Xie, T.; He, Z.; Yu, H.; Ma, Y.; Shi, C.; Zhang, C.; Ge, J.; Dai, W. Evaluation of styrene butadiene rubber asphalt modification mechanism and adhesion effect based on molecular simulation. Fuel 2024, 364, 131023. [Google Scholar] [CrossRef]
- Jiao, X.; Huang, D.; Zhao, S.; Ouyang, J. Study on the Compatibility of SBR and Asphalt Base Based on Molecular Simulation. Materials 2024, 17, 1175. [Google Scholar] [CrossRef]
- Li, T.; Gómez, N.H.C.; Lu, G.; Liang, D.; Wang, D.; Oeser, M. Use of polyurethane precursor–based modifier as an eco-friendly approach to improve performance of asphalt. J. Transp. Eng. Part B Pavements 2021, 147, 04021031. [Google Scholar] [CrossRef]
- Lu, P.; Ma, Y.; Ye, K.; Huang, S. Analysis of high-temperature performance of polymer-modified asphalts through molecular dynamics simulations and experiments. Constr. Build. Mater. 2022, 350, 128903. [Google Scholar] [CrossRef]
- Lu, P.; Huang, S.; Shen, Y.; Zhou, C.; Shao, L. Mechanical performance analysis of polyurethane-modified asphalt using molecular dynamics method. Polym. Eng. Sci. 2021, 61, 2323–2338. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, K.; Chen, Y.; Zhang, W.; Gillani, S.T.A.; Qiao, Z. Feasibility and environmental assessment of introducing waste polyurethane from wind turbine blades as a modifier for asphalt. Constr. Build. Mater. 2024, 446, 138052. [Google Scholar] [CrossRef]
- Li, M.; Chen, X.; Cong, P.; Luo, C.; Zhu, L.; Li, H.; Zhang, Y.; Chao, M.; Yan, L. Facile synthesis of polyethylene-modified asphalt by chain end-functionalization. Compos. Commun. 2022, 30, 101088. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, K.; Yu, C.; Yuan, D.; Ban, X. Study of the microscopic mechanism of natural rubber (Cis-1, 4-polyisoprene, NR)/polyethylene (PE) modified asphalt from the perspective of simulation. Polymers 2022, 14, 4087. [Google Scholar] [CrossRef]
- Li, E.D.; Xu, W.Y.; Zhang, Y. Microcosmic mechanism of PE modified asphalt based on molecular simulation. Aust. J. Civ. Eng. 2024, 1–10. [Google Scholar] [CrossRef]
- Hu, K.; Yu, C.; Yang, Q.; Li, Z.; Zhang, W.; Zhang, T.; Feng, Y. Mechanistic study of graphene reinforcement of rheological performance of recycled polyethylene modified asphalt: A new observation from molecular dynamics simulation. Constr. Build. Mater. 2022, 320, 126263. [Google Scholar] [CrossRef]
- Su, M.; Zhou, J.; Lu, J.; Chen, W.; Zhang, H. Using molecular dynamics and experiments to investigate the morphology and micro-structure of SBS modified asphalt binder. Mater. Today Commun. 2022, 30, 103082. [Google Scholar] [CrossRef]
- Li, N.; Liu, Z.; Yin, J.; Zhang, H.; Dou, H.; Li, B. Investigation of Compatibility Mechanisms and Diffusion Behavior of Polymer SBS-Modified Asphalt Compatibilizer Using Molecular Dynamics Simulation. Materials 2025, 18, 2238. [Google Scholar] [CrossRef]
- He, L.; Zhou, Z.; Ling, F.; Alexiadis, A.; Bergh, W.V.D.; Falchetto, A.C.; Balieu, R.; Zhu, J.; Valentin, J.; Kowalski, K.J.; et al. A coarse-grained molecular model for simulating self-healing of bitumen. Appl. Sci. 2022, 12, 10360. [Google Scholar] [CrossRef]
- Horgnies, M.; Darque-Ceretti, E.; Fezai, H.; Felder, E. Influence of the interfacial composition on the adhesion between aggregates and bitumen: Investigations by EDX, XPS and peel tests. Int. J. Adhes. Adhes. 2011, 31, 238–247. [Google Scholar] [CrossRef]
- Qiao, J.-G.; Zhang, S.-Z.; Li, H.-J. Study on Adhesive Healing Behavior of Aged Asphalt-Aggregate Interface Based on Molecular Dynamics Simulation. J. Chongqing Univ. Technol. (Nat. Sci. Ed.) 2024, 38, 176–183. (In Chinese) [Google Scholar]
- Zhang, H.; Huang, M.; Hong, J.; Lai, F.; Gao, Y. Molecular dynamics study on improvement effect of bis (2-hydroxyethyl) terephthalate on adhesive properties of asphalt-aggregate interface. Fuel 2021, 285, 119175. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Ma, B.; Mao, W.-J.; Si, W.; Wang, X. Review of interfacial adhesion between asphalt and aggregate based on molecular dynamics. Constr. Build. Mater. 2023, 362, 129642. [Google Scholar] [CrossRef]
- Chen, P.; Luo, X.; Gao, Y.; Zhang, Y. Modeling percentages of cohesive and adhesive debonding in bitumen-aggregate interfaces using molecular dynamics approaches. Appl. Surf. Sci. 2022, 571, 151318. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Gu, F.; Xu, T.; Wang, H. Impact of minerals and water on bitumen-mineral adhesion and debonding behaviours using molecular dynamics simulations. Constr. Build. Mater. 2018, 171, 214–222. [Google Scholar] [CrossRef]
- Ran, L.-F. Study on Aging Mechanism of SBS Modified Asphalt under Coupled Conditions of Heat, Light and Water and Development of High-Performance Rejuvenator. Ph.D. Thesis, Chongqing Jiaotong University, Chongqing, China, 2016. (In Chinese). [Google Scholar]
- Li, G.; Chen, Z.; Tan, Y.; Cong, X.; Dong, Y.; Xiao, S. Experimental and molecular dynamics simulation of hard asphalt microstructure. Constr. Build. Mater. 2023, 377, 131025. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Wang, D.; Guo, M.; Liu, P.; Yu, J. Characterization of bitumen micro-mechanical behaviors using AFM, phase dynamics theory and MD simulation. Materials 2017, 10, 208. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, D.; Xu, P. Molecular Dynamics Investigation of the Diffusion Mechanisms and Thermodynamic Behaviors in Warm Mix Recycled Asphalt Binders with and Without Rejuvenators. Materials 2025, 18, 703. [Google Scholar] [CrossRef]
- Song, M.; Gao, Y.; Li, G.; Lv, X.; Zhao, Y.; Zhang, X.; Luo, H. High-Temperature Rheological and Molecular Dynamics Analysis of Asphalt Modified with SiC Filler. Langmuir 2025, 41, 10818–10830. [Google Scholar] [CrossRef]
- Sultana, S.; Bhasin, A. Effect of chemical composition on rheology and mechanical properties of asphalt binder. Constr. Build. Mater. 2014, 72, 293–300. [Google Scholar] [CrossRef]
- Behnood, A.; Gharehveran, M.M. Morphology, rheology, and physical properties of polymer-modified asphalt binders. Eur. Polym. J. 2019, 112, 766–791. [Google Scholar] [CrossRef]
- Tan, T.; Ke, W.; Wang, Z. Study on asphalt damage characteristics in cold area with large temperature gradient. J. Phys. Conf. Ser. 2024, 2680, 012028. [Google Scholar] [CrossRef]
- Zhang, S.; Cui, Y.; Du, C.; Liu, L.; Chen, Q. Low-temperature performance and micro-structure of warm mix recycled composite aged asphalt. Constr. Build. Mater. 2024, 440, 137443. [Google Scholar] [CrossRef]
- Cao, L.-P.; Tan, Y.-Q.; Dong, Z.-J.; Sun, L.-J. Evaluation of Low-Temperature Performance of SBS Modified Asphalt Using Glass Transition Temperature. China J. Highw. Transp. 2006, 19, 1–6. (In Chinese) [Google Scholar]
- Kang, Y.; Zhou, D.; Wu, Q.; Liang, R.; Shangguan, S.; Liao, Z.; Wei, N. Molecular dynamics study on the glass forming process of asphalt. Constr. Build. Mater. 2019, 214, 430–440. [Google Scholar] [CrossRef]
- Liu, S.; Qi, X.; Shan, L. Effect of molecular structure on low-temperature properties of bitumen based on molecular dynamics. Constr. Build. Mater. 2022, 319, 126029. [Google Scholar] [CrossRef]
- Qu, X.; Wang, D.; Hou, Y.; Oeser, M.; Wang, L. Influence of paraffin on the microproperties of asphalt binder using MD Simulation. J. Mater. Civ. Eng. 2018, 30, 04018191. [Google Scholar] [CrossRef]
- Bhasin, A.; Bommavaram, R.; Greenfield, M.L.; Little, D.N. Use of molecular dynamics to investigate self-healing mechanisms in asphalt binders. J. Mater. Civ. Eng. 2011, 23, 485–492. [Google Scholar] [CrossRef]
- Bazin, P.; Saunier, J. Deformability, fatigue and healing properties of asphalt mixes. In Proceedings of the International Conference on the Structural Design of Asphalt Pavements, Ann Arbor, MI, USA, 7–11 August 1967. [Google Scholar]
- Luo, D.; Guo, M.; Tan, Y. Molecular simulation of minerals-asphalt interfacial interaction. Minerals 2018, 8, 176. [Google Scholar] [CrossRef]
- Traxler, R.N.; Coombs, C.E. The colloidal nature of asphalt as shown by its flow properties. J. Phys. Chem. 2002, 40, 1133–1147. [Google Scholar] [CrossRef]
- Smith, B.J.; Hesp, S.A. Crack pinning in asphalt mastic and concrete: Regular fatigue studies. Transp. Res. Rec. 2000, 1728, 75–81. [Google Scholar] [CrossRef]
- Meng, Y.-J.; Zhang, X.-N. Fatigue performances of modified asphalts based on cumulative dissipated energy ratio. J. South China Univ. Technol. 2012, 40, 99–103. [Google Scholar]
- Bhasin, A.; Little, D.N.; Bommavaram, R.; Vasconcelos, K. A framework to quantify the effect of healing in bituminous materials using material properties. Road Mater. Pavement Des. 2008, 9, 219–242. [Google Scholar] [CrossRef]
- Sun, D.; Lin, T.; Zhu, X.; Tian, Y.; Liu, F. Indices for self-healing performance assessments based on molecular dynamics simulation of asphalt binders. Comput. Mater. Sci. 2016, 114, 86–93. [Google Scholar] [CrossRef]
- Li, H.; Xing, C.; Zhu BZhang, X.; Gao, Y.; Tang, S.; Cheng, H. Comparative analysis of four styrene-butadiene-styrene (SBS) structure repair agents in the reju-venation of aged SBS-modified bitumen. Constr. Build. Mater. 2025, 476, 141232. [Google Scholar] [CrossRef]
- Xing, C.; Tang, S.; Chang, Z.; Han, Z.; Li, H.; Zhu, B. A comprehensive review on the plant-mixed cold recycling technology of emulsified asphalt: Raw materials and factors affecting performances. Constr. Build. Mater. 2024, 439, 137344. [Google Scholar] [CrossRef]
- Jones, D.R. SHRP Materials Reference Library: Asphalt Cements: A Concise Data Compilation; Strategic Highway Research Program; National Research Council: Washington, DC, USA, 1993. [Google Scholar]
- Yu, M.; Zhang, H.; Wang, J.; Sun, J.; Liu, D.; Sun, Y.; Chen, J. Multi-scale analysis of virgin/aged/recycled asphalt self-healing performance based on molecular simulation and micro-macro tests. Constr. Build. Mater. 2024, 431, 136535. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Yao, H.; Haq, Z.U.; Wang, S. Self-healing behavior of rubberized asphalt modulated by the degradation of crumb tire rubber. Constr. Build. Mater. 2024, 440, 137403. [Google Scholar] [CrossRef]
- Yuan, Y.; Lin, M.; Xu, S.; Wang, Y.; Chen, H.; Zheng, Z. Analysis of the self-healing mechanism of TB compound-modified asphalt based on molecular dynamics. Case Stud. Constr. Mater. 2024, 21, e03580. [Google Scholar] [CrossRef]
- Suo, Z.; Zhao, Z.; Yan, S.; Hu, J.; Tao, H.; Shijie, X.; Nie, L. Component Changes and Mechanism of Cold Regeneration of Aged Asphalt Using Waste Vegetable Oils. J. Mater. Civ. Eng. 2024, 36, 04024206. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Wong, S.Y.; Leong, L.T.; Farooqi, M.A.; Jaya, R.P.; Al-Saffar, Z.H.; Yaacob, H. Durability phenomena of Bitumen and bituminous pavement materials. Open Civ. Eng. J. 2021, 15, 279–289. [Google Scholar] [CrossRef]
- Farshidi, F.; Jones, D.; Harvey, J.T. Warm-Mix Asphalt Study: Evaluation of Hot and Warm Mix Asphalt with Respect to Binder Aging; University of California Pavement Research Center: Davis, CA, USA, 2013. [Google Scholar]
- Fernández-Gómez, W.D.; Rondón Quintana, H.; Reyes Lizcano, F. A review of asphalt and asphalt mixture aging: Una revisión. Ing. E Investig. 2013, 33, 5–12. [Google Scholar] [CrossRef]
- Qiu, Y.-J.; Su, T.; Zheng, P.-F.; Ding, H.-B. Study on the Physical Aging Mechanism of Asphalt Binder Based on Molecular Simulation. J. Build. Mater. 2020, 23, 1464–1470. (In Chinese) [Google Scholar]
- Sun, W.; Wang, H. Molecular dynamics simulation of diffusion coefficients between different types of rejuvenator and aged asphalt binder. Int. J. Pavement Eng. 2019, 21, 966–976. [Google Scholar] [CrossRef]
- Wang, Q. Study on the dynamic aging mechanism of asphalt based on molecular dynamics simulation. Taiyuan University of Technology: Taiyuan, China, 2023; (In Chinese). [Google Scholar] [CrossRef]
- Wang, P.Y.; Wen, Y.; Zhao, K.; Chong, D.; Wong, A.S. Evolution and locational variation of asphalt binder aging in long-life hot-mix asphalt pavements. Constr. Build. Mater. 2014, 68, 172–182. [Google Scholar]
- Xu, G.; Wang, H. Molecular dynamics study of oxidative aging effect on asphalt binder properties. Fuel 2017, 188, 1–10. [Google Scholar] [CrossRef]
- Li, N.; Zhao, X.; Sun, J.; Xiao, Q.Y. Study on aging mechanism of rubber-modified asphalt. J Highw Trans Res Dev 2015, 32, 18–27. [Google Scholar]
- Hu, C.; Sun, Z.; Xi, L.; Tian, W.; Zhang, H. Rheology and Phase Morphology Properties of Recycled Asphalt Modified with Waste Cooking Oil and Activated Crumb Rubber. Int. J. Pavement Res. Technol. 2024, 1–16. [Google Scholar] [CrossRef]
- Zhan, Y.; Wu, H.; Song, W.; Zhu, L. Molecular dynamics study of the diffusion between virgin and aged asphalt binder. Coatings 2022, 12, 403. [Google Scholar] [CrossRef]
- Cao, D.-G.; Zhang, W.; Zhang, W.-G.; Zhu, M.-Y. Self-Healing Performance of SBS Modified Asphalt under Aging Conditions Based on Molecular Simulation. J. Liaoning Tech. Univ. (Nat. Sci. Ed.) 2024, 43, 682–691. (In Chinese) [Google Scholar]
- Zaumanis, M.; Mallick, R.B. Review of very high-content reclaimed asphalt use in plant-produced pavements: State of the art. Int. J. Pavement Eng. 2015, 16, 39–55. [Google Scholar] [CrossRef]
- Cui, Y.; Li, X.; Zhang, S. Diffusion mechanism of regenerant aged asphalt based on molecular dynamics simulation. J Build Mater 2021, 24, 1105–1109. [Google Scholar]
- Yi, X.; Dong, R.; Tang, N. Development of a novel binder rejuvenator composed by waste cooking oil and crumb tire rubber. Constr. Build. Mater. 2020, 236, 117621. [Google Scholar] [CrossRef]
- Putri, L.D.; Soehardi, F.; Srihandayani, S.; Ratodi, F.; Hasibuan, A.; Bakhtiar, M.I.; Nanuru, R.F.; Iskandar, A.; Pudji, N.P.; Djanggih, H. The Composition Aggregate Uses: A Grain of Buton Asphalt. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2020; Volume 469, p. 012004. [Google Scholar]
- Liu, F.; Wang, Q.; Zhang, X.; Zhou, Z.; Guo, C. Effect of oxidation kinetic aging on the adhesion behaviour of asphalt-aggregate interfaces from molecular view. Int. J. Pavement Eng. 2024, 25, 2313499. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, J.; Chen, H. Study on Adhesion Characteristics of Asphalt–Aggregate System Based on Surface Energy Theory. J. Southeast Univ. (Nat. Sci. Ed.) 2014, 44, 183–187. (In Chinese) [Google Scholar]
- Zhao, H.; Li, G.; Ma, Y.; Yu, X.; Chen, Y.; Li, W. Long-term performance of chemically modified cotton straw fibers in micro-surfacing asphalt mixtures. Case Stud. Constr. Mater. 2024, 20, e03294. [Google Scholar] [CrossRef]
- Kanitpong, K.; Bahia, H. Relating adhesion and cohesion of asphalts to the effect of moisture on laboratory performance of asphalt mixtures. Transp. Res. Rec. 2005, 1901, 33–43. [Google Scholar] [CrossRef]
- Hossain, M.I.; Tarefder, R.A. Behavior of asphalt mastic films under laboratory controlled humidity conditions. Constr. Build. Mater. 2015, 78, 8–17. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Pinto, I.; Park, S.-W. Experimental evaluation of anti-stripping additives in bituminous mixtures through multiple scale laboratory test results. Constr. Build. Mater. 2012, 29, 386–393. [Google Scholar] [CrossRef]
- Xu, G.; Wang, H. Study of cohesion and adhesion properties of asphalt concrete with molecular dynamics simulation. Comput. Mater. Sci. 2016, 112, 161–169. [Google Scholar] [CrossRef]
- Cui, B.; Wang, H. Molecular interaction of Asphalt-Aggregate interface modified by silane coupling agents at dry and wet conditions. Appl. Surf. Sci. 2022, 572, 151365. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Su, J.-Z.; Sun, M.; Jia, C.-X.; Qiao, R.-Z. Research Progress on Moisture Stability of Asphalt Mixtures Based on AFM Technology. Mater. Rev. 2024, 38, 216–221. (In Chinese) [Google Scholar]
- Liu, N.; Liu, L.; Zhang, Z.; Li, M.; Sun, L. Study on cohesion and adhesion behaviors of the zeolite foamed asphalt-warm mix mixture based on molecular dynamics simulation. Case Stud. Constr. Mater. 2024, 21, e03424. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, K.; Liu, Z.; Liu, M.; Tao, X.; Chang, H. Effects of water and chloride salt erosion on adhesion behavior between recycle aggregate/asphalt interface: An experimental and molecular dynamics study. Colloids Surfaces A Physicochem. Eng. Asp. 2025, 711, 136374. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison Items | Three-Component Model | Four-Component 12-Molecule Model |
|---|---|---|
| Model Composition | Maltenes, Resins, Asphaltenes | Aromatics, Saturates, Resins, Asphaltenes |
| Typicality | Highly generalizable and structurally concise | More accurately reflects the complex composition of asphalt |
| Advantages | The model is simple, computationally efficient, and suitable for preliminary simulation analysis. | The chemical composition is more comprehensive, enabling more accurate prediction of properties such as density and viscosity. |
| Disadvantages | Ignoring resins and aromatic fractions makes it difficult to reflect the structural diversity. | The model is relatively complex, with high construction difficulty and significant computational resource requirements. |
| Applicability | Preliminary study on the microstructure and behavior of asphalt. | In-depth study of structure–property relationships, self-healing, aging, and related aspects. |
| Quantity of representative molecules | Usually three types (e.g., C22, 1,7-dimethylnaphthalene, and asphaltenes). | Commonly consists of 12 types (e.g., quinoline, thiopyrene, etc.), with some models developed with up to 14 or 20 types. |
| Density matching accuracy | Moderate, with some deviations from the experimental values. | The density error can be controlled within 0.06 g/cm3, showing high agreement with the measured values. |
| Future scalability | Can be coupled with some modifiers, but with limited compatibility. | More suitable for incorporating complex systems such as SBS, aging molecules, and nanoparticles. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, C.; Xiong, Z.; Lu, T.; Li, H.; Zhou, W.; Li, C. Performance of Asphalt Materials Based on Molecular Dynamics Simulation: A Review. Polymers 2025, 17, 2051. https://doi.org/10.3390/polym17152051
Xing C, Xiong Z, Lu T, Li H, Zhou W, Li C. Performance of Asphalt Materials Based on Molecular Dynamics Simulation: A Review. Polymers. 2025; 17(15):2051. https://doi.org/10.3390/polym17152051
Chicago/Turabian StyleXing, Chengwei, Zhihang Xiong, Tong Lu, Haozongyang Li, Weichao Zhou, and Chen Li. 2025. "Performance of Asphalt Materials Based on Molecular Dynamics Simulation: A Review" Polymers 17, no. 15: 2051. https://doi.org/10.3390/polym17152051
APA StyleXing, C., Xiong, Z., Lu, T., Li, H., Zhou, W., & Li, C. (2025). Performance of Asphalt Materials Based on Molecular Dynamics Simulation: A Review. Polymers, 17(15), 2051. https://doi.org/10.3390/polym17152051