
Structure–Property Relationship in Isotactic Polypropylene Under Contrasting Processing Conditions

 ,
,  ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterization Techniques
3. Results and Discussion
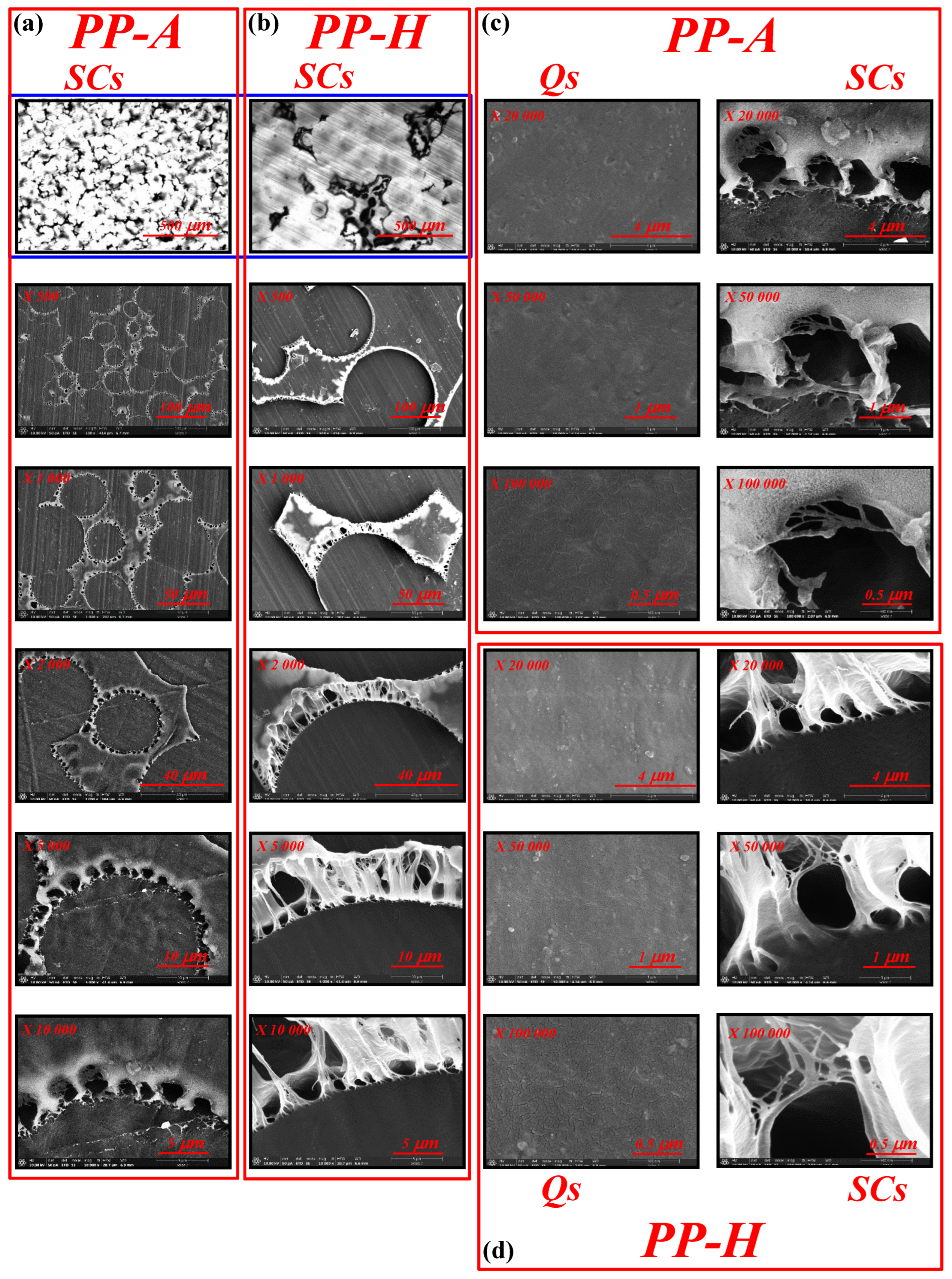
3.1. Microstructure Investigation
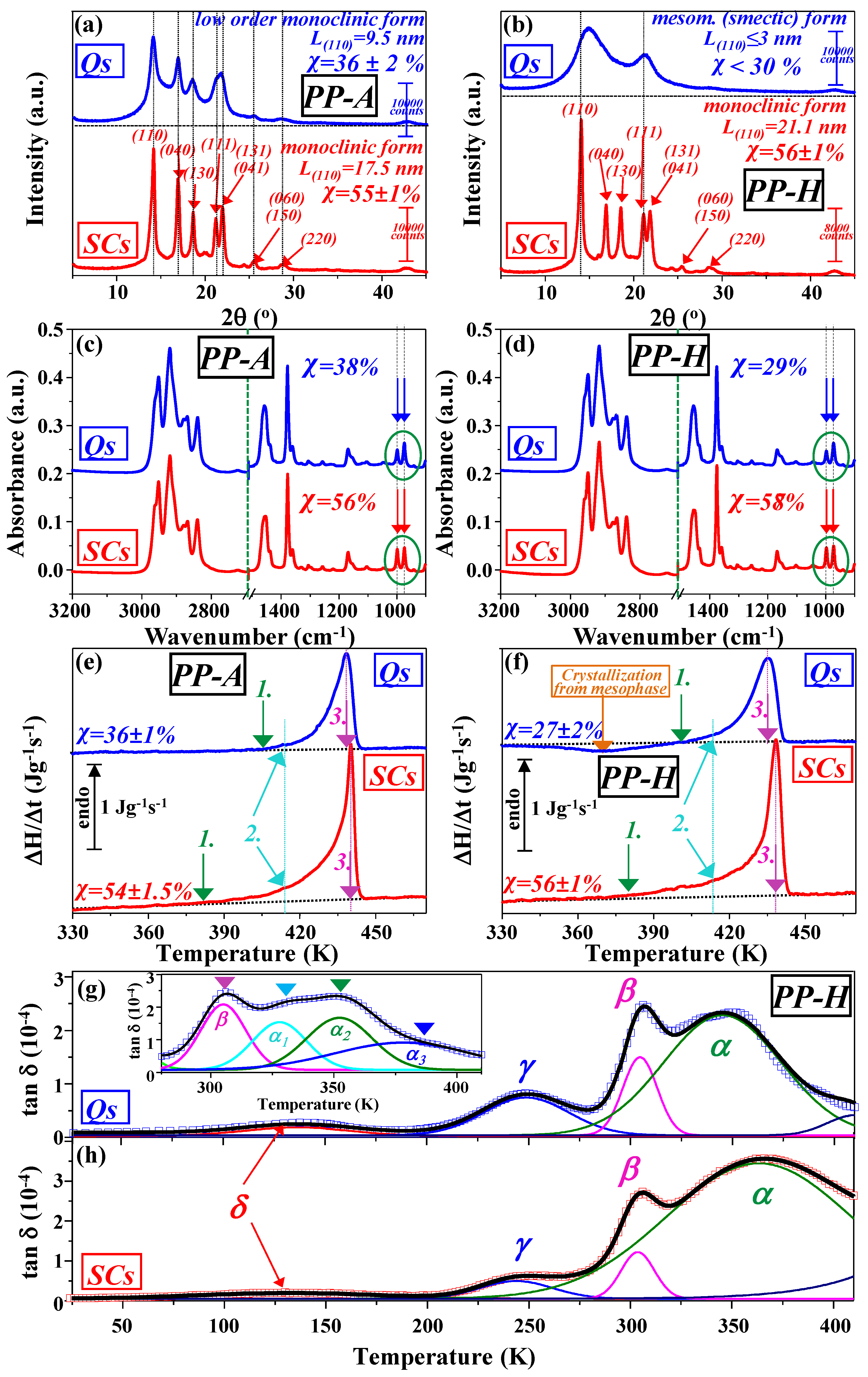
3.2. WAXD Study
3.3. ATR-FTIR Spectroscopy
3.4. Calorimetric Study
3.5. DDS Study
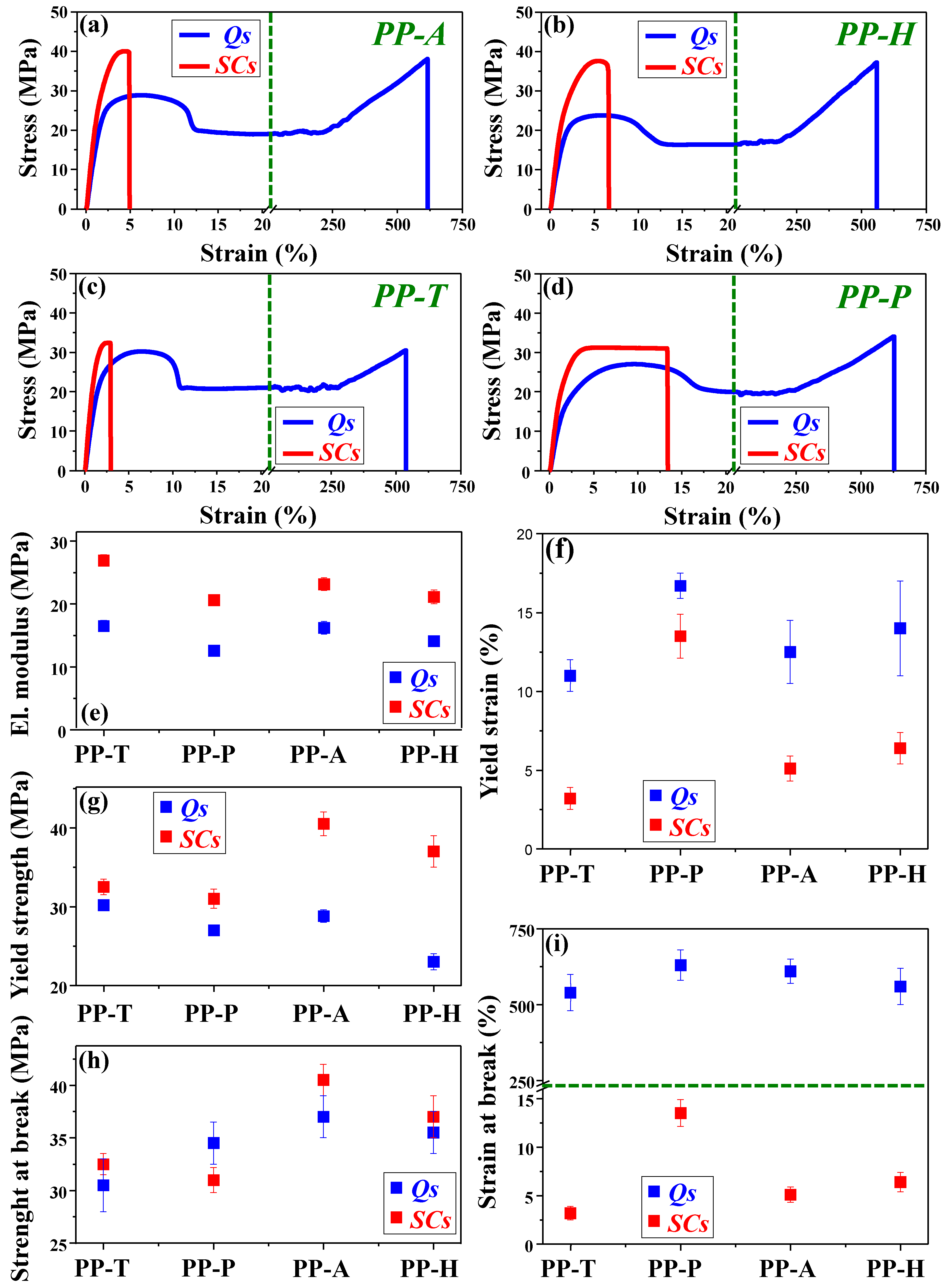
3.6. Mechanical Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mileva, D.; Tranchida, D.; Gahleitner, M. Designing polymer crystallinity: An industrial perspective. Polym. Cryst. 2018, 1, e10009. [Google Scholar] [CrossRef]
- Amer, I.; van Reenen, A.; Mokrani, T. Molecular weight and tacticity effect on morphological and mechanical properties of Ziegler–Natta catalyzed isotactic polypropylenes. Polímeros 2015, 25, 556–563. [Google Scholar] [CrossRef]
- Moore, E.P. Polypropylene Handbook: Polymerization, Characterization, Properties, Processing, Applications; Hanser Publishers: Cincinnati, OH, USA, 1996. [Google Scholar]
- Ozzetti, R.A.; De Oliveira Filho, A.P.; Schuchardt, U.; Mandelli, D. Determination of tacticity in polypropylene by FTIR with multivariate calibration. J. Appl. Polym. Sci. 2002, 85, 734–745. [Google Scholar] [CrossRef]
- Arranz-Andrés, J.; Peña, B.; Benavente, R.; Pérez, E.; Cerrada, M.L. Influence of isotacticity and molecular weight on the properties of metallocenic isotactic polypropylene. Eur. Polym. J. 2007, 43, 2357–2370. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Janimak, J.J.; Zhang, A.; Hsieh, E.T. Isotacticity effect on crystallization and melting in polypropylene fractions: 1. Crystalline structures and thermodynamic property changes. Polymer 1991, 32, 648–655. [Google Scholar] [CrossRef]
- Paukkeri, R.; Lehtinen, A. Thermal behaviour of polypropylene fractions: 2. The multiple melting peaks. Polymer 1993, 34, 4083–4088. [Google Scholar] [CrossRef]
- Paukkeri, R.; Lehtinen, A. Thermal behaviour of polypropylene fractions: 1. Influence of tacticity and molecular weight on crystallization and melting behaviour. Polymer 1993, 34, 4075–4082. [Google Scholar] [CrossRef]
- Fukuda, Y.; Kida, T.; Yamaguchi, M. Mechanical properties of isotactic polypropylene with nodular or spherulite morphologies. Polym. Eng. Sci. 2023, 63, 4043–4050. [Google Scholar] [CrossRef]
- Polypropylene Handbook; Pasquini, N., Ed.; Carl Hanser Verlag: Munich, Germany, 2005. [Google Scholar]
- Yamada, K.; Matsumoto, S.; Tagashira, K.; Hikosaka, M. Isotacticity dependence of spherulitic morphology of isotactic polypropylene. Polymer 1998, 39, 5327–5333. [Google Scholar] [CrossRef]
- Tripathi, D. Practical Guide to Polypropylene; Rapra Publishing: Shrewsbury, UK, 2001. [Google Scholar]
- Addeo, A. Polypropylene Handbook; Hanser Gardner: Cincinnati, OH, USA, 2005. [Google Scholar]
- Ariff, Z.; Ariffin, A.; Jikan, S.; Abdul Rahim, N. Rheological Behaviour of Polypropylene Through Extrusion and Capillary Rheometry. In Polypropylene; Dogan, F., Ed.; InTech: Houston, TX, USA, 2012; pp. 29–48. [Google Scholar]
- Maddah, H. Polypropylene as a Promising Plastic: A Review. Am. J. Polym. Sci. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Suljovrujic, E.; Milicevic, D.; Stolic, A.; Dudic, D.; Vasalic, D.; Dzunuzovic, E.; Stamboliev, G. Thermal, mechanical, and dielectric properties of radiation sterilized mesomorphic PP: Comparison between gamma and electron beam irradiation modalities. Polym. Degrad. Stab. 2024, 229, 110940. [Google Scholar] [CrossRef]
- Natta, G. Une nouvelle classe de polymeres d’α-olefines ayant une régularité de structure exceptionnelle. J. Polym. Sci. 1955, 16, 143–154. [Google Scholar] [CrossRef]
- Bogoeva-Gaceva, G. Advances in polypropylene based materials. Contrib. Sect. Nat. Math. Biotech. Sci. 2014, 35, 121–138. [Google Scholar] [CrossRef]
- Brückner, S.; Meille, S.V.; Petraccone, V.; Pirozzi, B. Polymorphism in isotactic polypropylene. Prog. Polym. Sci. 1991, 16, 361–404. [Google Scholar] [CrossRef]
- Seguela, R.; Staniek, E.; Escaig, B.; Fillon, B. Plastic deformation of polypropylene in relation to crystalline structure. J. Appl. Polym. Sci. 1999, 71, 1873–1885. [Google Scholar] [CrossRef]
- Shang, Y.; Zhao, J.; Li, J.; Wu, Z.; Jiang, S. Investigations in annealing effects on structure and properties of β-isotactic polypropylene with X-ray synchrotron experiments. Colloid Polym. Sci. 2014, 292, 3205–3221. [Google Scholar] [CrossRef]
- Arvidson, S.A.; Khan, S.A.; Gorga, R.E. Mesomorphic−α-Monoclinic Phase Transition in Isotactic Polypropylene: A Study of Processing Effects on Structure and Mechanical Properties. Macromolecules 2010, 43, 2916–2924. [Google Scholar] [CrossRef]
- Sharaf, M.A.; Kloczkowski, A. Evolution of the Deformation- and Flow-Induced Crystallization and Characterization of the Microstructure of a Single Spherulite, Lamella, and Chain of Isotactic Polypropylene. Macromol. Chem. Phys. 2024, 225, 2300203. [Google Scholar] [CrossRef]
- Suljovrujic, E. Radiation Modification of the Physical Properties of Polyolefins; University of Belgrade: Belgrade, Serbia, 2000. [Google Scholar]
- van der Meer, D.W. Structure-Property Relationships in Isotactic Polypropylene. Ph.D. Thesis, Twente University, Enschede, The Netherlands, 2003. [Google Scholar]
- Scoti, M.; De Stefano, F.; Di Girolamo, R.; Malafronte, A.; Talarico, G.; De Rosa, C. Crystallization Behavior and Properties of Propylene/4-Methyl-1-pentene Copolymers from a Metallocene Catalyst. Macromolecules 2023, 56, 1446–1460. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Circelli, T.; Waymouth, R.M. Crystallization of the α and γ Forms of Isotactic Polypropylene as a Tool To Test the Degree of Segregation of Defects in the Polymer Chains. Macromolecules 2002, 35, 3622–3629. [Google Scholar] [CrossRef]
- Suljovrujic, E.; Trifunovic, S.; Milicevic, D. The influence of gamma radiation on the dielectric relaxation behaviour of isotactic polypropylene. The α relaxation. Polym. Degrad. Stab. 2010, 95, 164–171. [Google Scholar] [CrossRef]
- Vittoria, V.; Perullo, A. Effect of quenching temperature on the structure of isotactic polypropylene films. J. Macromol. Sci. Part B 1986, 25, 267–281. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C.; Corradini, P. Solid Mesophases in Semicrystalline Polymers: Structural Analysis by DiffractionTechniques. In Interphases and Mesophases in Polymer Crystallization II; Allegra, G., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–74. [Google Scholar]
- Mileva, D.; Androsch, R.; Radusch, H.-J. Effect of structure on light transmission in isotactic polypropylene and random propylene-1-butene copolymers. Polym. Bull. 2009, 62, 561–571. [Google Scholar] [CrossRef]
- Stupp, S.I.; Supan, T.J.; Belton, D.J. Ice-water quenching technique for polypropylene. Orthot. Prosthet. 1979, 33, 16–21. [Google Scholar]
- Kim, Y.C.; Ahn, W.; Kim, C.Y. A study on multiple melting of isotactic polypropylene. Polym. Eng. Sci. 1997, 37, 1003–1011. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Bárány, T. Polypropylene Handbook Morphology, Blends and Composites: Morphology, Blends and Composites; Springer Nature: Cham, Switzerland, 2019. [Google Scholar]
- Rungswang, W.; Jarumaneeroj, C.; Patthamasang, S.; Phiriyawirut, P.; Jirasukho, P.; Soontaranon, S.; Rugmai, S.; Hsiao, B.S. Influences of tacticity and molecular weight on crystallization kinetic and crystal morphology under isothermal crystallization: Evidence of tapering in lamellar width. Polymer 2019, 172, 41–51. [Google Scholar] [CrossRef]
- Bassett, D.C.; Olley, R.H. On the lamellar morphology of isotactic polypropylene spherulites. Polymer 1984, 25, 935–943. [Google Scholar] [CrossRef]
- Ryan, A.J.; Stanford, J.L.; Bras, W.; Nye, T.M.W. A synchrotron X-ray study of melting and recrystallization in isotactic polypropylene. Polymer 1997, 38, 759–768. [Google Scholar] [CrossRef]
- Chan, C.-M.; Li, L. Direct Observation of the Growth of Lamellae and Spherulites by AFM. In Intrinsic Molecular Mobility and Toughness of Polymers II; Kausch, H.-H., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–41. [Google Scholar]
- Cong, Y.; Hong, Z.; Zhou, W.; Chen, W.; Su, F.; Li, H.; Li, X.; Yang, K.; Yu, X.; Qi, Z.; et al. Conformational Ordering on the Growth Front of Isotactic Polypropylene Spherulite. Macromolecules 2012, 45, 8674–8680. [Google Scholar] [CrossRef]
- Jiang, C.; Miao, C.; Zhou, J.; Yuan, M. Insights into damage mechanisms and advances in numerical simulation of spherulitic polymers. Polymer 2025, 318, 128001. [Google Scholar] [CrossRef]
- Michaeli, W.; Gutberlet, D.; Glißmann, M. Characterisation of the spherulite structure of polypropylene using light-microscope methods. Polym. Test. 2001, 20, 459–467. [Google Scholar] [CrossRef]
- Park, J.; Eom, K.; Kwon, O.; Woo, S. Chemical Etching Technique for the Investigation of Melt-crystallized Isotactic Polypropylene Spherulite and Lamellar Morphology by Scanning Electron Microscopy. Microsc. Microanal. 2001, 7, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-G.; Zhang, L.-Q.; Chen, C.; Cui, J.; Zeng, X.-b.; Liu, L.; Liu, F.; Ungar, G. 3D Morphology of Different Crystal Forms in β-Nucleated and Fiber-Sheared Polypropylene: α-Teardrops, α-Teeth, and β-Fans. Macromolecules 2023, 56, 5502–5511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-J.; Liu, J.-G.; Yan, S.-K.; Dong, J.-Y.; Li, L.; Chan, C.-M.; Schultz, J.M. Atomic force microscopy study of the lamellar growth of isotactic polypropylene. Polymer 2005, 46, 4077–4087. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, L.; Zhen, W.; Sun, X.; Ren, Z.; Li, H.; Yan, S. An abnormal melting behavior of isotactic polypropylene spherulites grown at low temperatures. Polymer 2017, 111, 183–191. [Google Scholar] [CrossRef]
- Raimo, M.; Silvestre, C. Topographic Analysis of Isotactic Polypropylene Spherulites by Atomic Force Microscopy. J. Scanning Probe Microsc. 2009, 4, 45–47. [Google Scholar] [CrossRef]
- Schönherr, H.; Snétivy, D.; Vansco, G.J. A nanoscopic view at the spherulitic morphology of isotactic polypropylene by atomic force microscopy. Polym. Bull. 1993, 30, 567–574. [Google Scholar] [CrossRef]
- Bassett, D.C. Principles of Polymer Morphology; Cambridge: New York, NY, USA, 1981. [Google Scholar]
- Bassett, D.C.; Keller, A.; Mitsuhashi, S. New features in polymer crystal growth from concentrated solutions. J. Polym. Sci. Part A Gen. Pap. 1963, 1, 763–788. [Google Scholar] [CrossRef]
- Bassett, D.C.; Vaughan, A.S. On the lamellar morphology of melt-crystallized isotactic polystyrene. Polymer 1985, 26, 717–725. [Google Scholar] [CrossRef]
- Imai, M.; Kaji, K. Polymer crystallization from the metastable melt: The formation mechanism of spherulites. Polymer 2006, 47, 5544–5554. [Google Scholar] [CrossRef]
- Norton, D.R.; Keller, A. The spherulitic and lamellar morphology of melt-crystallized isotactic polypropylene. Polymer 1985, 26, 704–716. [Google Scholar] [CrossRef]
- Padden, F.J., Jr.; Keith, H.D. Spherulitic Crystallization in Polypropylene. J. Appl. Phys. 1959, 30, 1479–1484. [Google Scholar] [CrossRef]
- Stachurski, Z.H.; Macnicol, J. The geometry of spherulite boundaries. Polymer 1998, 39, 5717–5724. [Google Scholar] [CrossRef]
- Androsch, R.; Di Lorenzo, M.L.; Schick, C.; Wunderlich, B. Mesophases in polyethylene, polypropylene, and poly(1-butene). Polymer 2010, 51, 4639–4662. [Google Scholar] [CrossRef]
- Kida, T.; Yamaguchi, M. Role of Rigid–Amorphous chains on mechanical properties of polypropylene solid using DSC, WAXD, SAXS, and Raman spectroscopy. Polymer 2022, 249, 124834. [Google Scholar] [CrossRef]
- Schawe, J.E.K. Mobile amorphous, rigid amorphous and crystalline fractions in isotactic polypropylene during fast cooling. J. Therm. Anal. Calorim. 2017, 127, 931–937. [Google Scholar] [CrossRef]
- Zia, Q.; Mileva, D.; Androsch, R. Rigid Amorphous Fraction in Isotactic Polypropylene. Macromolecules 2008, 41, 8095–8102. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. Crystallization-induced formation of rigid amorphous fraction. Polym. Cryst. 2018, 1, e10023. [Google Scholar] [CrossRef]
- Zuo, F.; Keum, J.K.; Chen, X.; Hsiao, B.S.; Chen, H.; Lai, S.-Y.; Wevers, R.; Li, J. The role of interlamellar chain entanglement in deformation-induced structure changes during uniaxial stretching of isotactic polypropylene. Polymer 2007, 48, 6867–6880. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Q.; Zhao, J.; Li, L. Molecular and thermodynamics descriptions of flow-induced crystallization in semi-crystalline polymers. J. Appl. Phys. 2020, 127, 241101. [Google Scholar] [CrossRef]
- Sigalas, N.I.; Van Kraaij, S.A.T.; Lyulin, A.V. Effect of Temperature on Flow-Induced Crystallization of Isotactic Polypropylene: A Molecular-Dynamics Study. Macromolecules 2023, 56, 8417–8427. [Google Scholar] [CrossRef]
- Hine, P.; Broome, V.; Ward, I. The incorporation of carbon nanofibres to enhance the properties of self reinforced, single polymer composites. Polymer 2005, 46, 10936–10944. [Google Scholar] [CrossRef]
- Laura, D.M.; Keskkula, H.; Barlow, J.W.; Paul, D.R. Effect of rubber particle size and rubber type on the mechanical properties of glass fiber reinforced, rubber-toughened nylon 6. Polymer 2003, 44, 3347–3361. [Google Scholar] [CrossRef]
- Haggenmueller, R.; Guthy, C.; Lukes, J.R.; Fischer, J.E.; Winey, K.I. Single Wall Carbon Nanotube/Polyethylene Nanocomposites: Thermal and Electrical Conductivity. Macromolecules 2007, 40, 2417–2421. [Google Scholar] [CrossRef]
- Tangirala, R.; Baer, E.; Hiltner, A.; Weder, C. Photopatternable reflective films produced by nanolayer extrusion. Adv. Funct. Mater. 2004, 14, 595–604. [Google Scholar] [CrossRef]
- Dudić, D.; Kostoski, D.; Djoković, V.; Dramićanin, M.D. Formation and behaviour of low-temperature melting peak of quenched and annealed isotactic polypropylene. Polym. Int. 2002, 51, 111–116. [Google Scholar] [CrossRef]
- Brucato, V.; Piccarolo, S.; La Carrubba, V. An experimental methodology to study polymer crystallization under processing conditions. The influence of high cooling rates. Chem. Eng. Sci. 2002, 57, 4129–4143. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Tarallo, O.; Malafronte, A.; Di Girolamo, R.; Esposito, S.; Piemontesi, F.; Liguori, D.; Morini, G. The “Nodular” α Form of Isotactic Polypropylene: Stiff and Strong Polypropylene with High Deformability. Macromolecules 2017, 50, 5434–5446. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhao, Y.; Zhang, C.; Yang, J.; Xu, Y.; Wang, D. In-situ investigation on the structural evolution of mesomorphic isotactic polypropylene in a continuous heating process. Polymer 2016, 105, 133–143. [Google Scholar] [CrossRef]
- Mollova, A.; Androsch, R.; Mileva, D.; Gahleitner, M.; Funari, S.S. Crystallization of isotactic polypropylene containing beta-phase nucleating agent at rapid cooling. Eur. Polym. J. 2013, 49, 1057–1065. [Google Scholar] [CrossRef]
- Fu, X.; Jia, W.; Li, X.; Wang, Y.; Wang, Z.; Liu, C.; Shen, C.; Shao, C. Phase transitions of the rapid-compression-induced mesomorphic isotactic polypropylene under high-pressure annealing. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 651–661. [Google Scholar] [CrossRef]
- Hendra, P.J.; Vile, J.; Willis, H.A.; Zichy, V.; Cudby, M.E.A. The effect of cooling rate upon the morphology of quenched melts of isotactic polypropylenes. Polymer 1984, 25, 785–790. [Google Scholar] [CrossRef]
- Konishi, T.; Nishida, K.; Kanaya, T.; Kaji, K. Effect of Isotacticity on Formation of Mesomorphic Phase of Isotactic Polypropylene. Macromolecules 2005, 38, 8749–8754. [Google Scholar] [CrossRef]
- Ferrero, A.; Ferracini, E.; Mazzavillani, A.; Malta, V. A New X-Ray Study of the Quenched Isotactic Polypropylene Transition by Annealing. J. Macromol. Sci. Part B 2000, 39, 109–129. [Google Scholar] [CrossRef]
- Di Sacco, F.; Saidi, S.; Hermida-Merino, D.; Portale, G. Revisiting the Mechanism of the Meso-to-α Transition of Isotactic Polypropylene and Ethylene–Propylene Random Copolymers. Macromolecules 2021, 54, 9681–9691. [Google Scholar] [CrossRef]
- Lei, C.; Huang, W.; Xu, R.; Xu, Y. The correlation between the lower temperature melting plateau endotherm and the stretching-induced pore formation in annealed polypropylene films. J. Plast. Film. Sheeting 2012, 28, 151–164. [Google Scholar] [CrossRef]
- Nitta, K.-h.; Odaka, K. Influence of structural organization on tensile properties in mesomorphic isotactic polypropylene. Polymer 2009, 50, 4080–4088. [Google Scholar] [CrossRef]
- Zia, Q.; Radusch, H.-J.; Androsch, R. Deformation behavior of isotactic polypropylene crystallized via a mesophase. Polym. Bull. 2009, 63, 755–771. [Google Scholar] [CrossRef]
- Androsch, R. In Situ Atomic Force Microscopy of the Mesomorphic−Monoclinic Phase Transition in Isotactic Polypropylene. Macromolecules 2008, 41, 533–535. [Google Scholar] [CrossRef]
- Miller, R.L. On the existence of near-range order in isotactic polypropylenes. Polymer 1960, 1, 135–143. [Google Scholar] [CrossRef]
- Martorana, A.; Piccarolo, S.; Sapoundjieva, D. SAXS/WAXS study of the annealing process in quenched samples of isotactic poly(propylene). Macromol. Chem. Phys. 1999, 200, 531–540. [Google Scholar] [CrossRef]
- Hanna, L.A.; Hendra, P.J.; Maddams, W.; Willis, H.A.; Zichy, V.; Cudby, M.E.A. Vibrational spectroscopic study of structural changes in isotactic polypropylene below the melting point. Polymer 1988, 29, 1843–1847. [Google Scholar] [CrossRef]
- Konishi, T.; Nishida, K.; Kanaya, T. Crystallization of Isotactic Polypropylene from Prequenched Mesomorphic Phase. Macromolecules 2006, 39, 8035–8040. [Google Scholar] [CrossRef]
- Zia, Q.; Androsch, R.; Radusch, H.-J.; Piccarolo, S. Morphology, reorganization and stability of mesomorphic nanocrystals in isotactic polypropylene. Polymer 2006, 47, 8163–8172. [Google Scholar] [CrossRef]
- Gomez, M.A.; Tanaka, H.; Tonelli, A.E. High-resolution solid-state 13C nuclear magnetic resonance study of isotactic polypropylene polymorphs. Polymer 1987, 28, 2227–2232. [Google Scholar] [CrossRef]
- Suljovrujic, E.; Stojanovic, Z.; Dudic, D.; Milicevic, D. Radiation, thermo-oxidative and storage induced changes in microstructure, crystallinity and dielectric properties of (un)oriented isotactic polypropylene. Polym. Degrad. Stab. 2021, 188, 109564. [Google Scholar] [CrossRef]
- Lanyi, F.J.; Wenzke, N.; Kaschta, J.; Schubert, D.W. On the Determination of the Enthalpy of Fusion of α-Crystalline Isotactic Polypropylene Using Differential Scanning Calorimetry, X-Ray Diffraction, and Fourier-Transform Infrared Spectroscopy: An Old Story Revisited. Adv. Eng. Mater. 2020, 22, 1900796. [Google Scholar] [CrossRef]
- Lanyi, F.J.; Wenzke, N.; Kaschta, J.; Schubert, D.W. A method to reveal bulk and surface crystallinity of Polypropylene by FTIR spectroscopy—Suitable for fibers and nonwovens. Polym. Test. 2018, 71, 49–55. [Google Scholar] [CrossRef]
- Tarani, E.; Arvanitidis, I.; Christofilos, D.; Bikiaris, D.N.; Chrissafis, K.; Vourlias, G. Calculation of the degree of crystallinity of HDPE/GNPs nanocomposites by using various experimental techniques: A comparative study. J. Mater. Sci. 2023, 58, 1621–1639. [Google Scholar] [CrossRef]
- Reddy, K.R.; Tashiro, K.; Sakurai, T.; Yamaguchi, N.; Sasaki, S.; Masunaga, H.; Takata, M. Isothermal Crystallization Behavior of Isotactic Polypropylene H/D Blends as Viewed from Time-Resolved FTIR and Synchrotron SAXS/WAXD Measurements. Macromolecules 2009, 42, 4191–4199. [Google Scholar] [CrossRef]
- Stojanović, Z.; Kačarević-Popović, Z.; Galović, S.; Miličević, D.; Suljovrujić, E. Crystallinity changes and melting behavior of the uniaxially oriented iPP exposed to high doses of gamma radiation. Polym. Degrad. Stab. 2005, 87, 279–286. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, Q.; Ren, Z.; Sun, X.; Li, H.; Li, H.; Yan, S. The αβ-iPP growth transformation of commercial-grade iPP during non-isothermal crystallization. CrystEngComm 2015, 17, 9221–9227. [Google Scholar] [CrossRef]
- Caldas, V.; Brown, G.R.; Nohr, R.S.; MacDonald, J.G.; Raboin, L.E. The structure of the mesomorphic phase of quenched isotactic polypropylene. Polymer 1994, 35, 899–907. [Google Scholar] [CrossRef]
- Cohen, Y.; Saraf, R. A direct correlation function for mesomorphic polymers and its application to the ‘smectic’ phase of isotactic polpropylene. Polymer 2001, 42, 5865–5870. [Google Scholar] [CrossRef]
- An, H.; Li, X.; Geng, Y.; Wang, Y.; Wang, X.; Li, L.; Li, Z.; Yang, C. Shear-Induced Conformational Ordering, Relaxation, and Crystallization of Isotactic Polypropylene. J. Phys. Chem. B 2008, 112, 12256–12262. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wang, G.; Cong, Y.; Bai, L.; Li, L.; Yang, C. Shear-Induced Nucleation and Growth of Long Helices in Supercooled Isotactic Polypropylene. Macromolecules 2009, 42, 4751–4757. [Google Scholar] [CrossRef]
- Qian, C.; Zhao, Y.; Wang, Z.; Liu, L.; Wang, D. Probing the difference of crystalline modifications and structural disorder of isotactic polypropylene via high-resolution FTIR spectroscopy. Polymer 2021, 224, 123722. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, D.; Fang, Y. In Situ FTIR Spectroscopic Study of the Conformational Change of Isotactic Polypropylene during the Crystallization Process. J. Phys. Chem. B 2001, 105, 12461–12463. [Google Scholar] [CrossRef]
- Luongo, J.P. Infrared study of polypropylene. J. Appl. Polym. Sci. 1960, 3, 302–309. [Google Scholar] [CrossRef]
- Kissin, Y.V. Isospecific Polymerization of Olefins with Heterogeneous Ziegler-Natta Catalysts; Springer: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Cong, Y.; Hong, Z.; Qi, Z.; Zhou, W.; Li, H.; Liu, H.; Chen, W.; Wang, X.; Li, L. Conformational Ordering in Growing Spherulites of Isotactic Polypropylene. Macromolecules 2010, 43, 9859–9864. [Google Scholar] [CrossRef]
- Burfield, D.R.; Loi, P.S.T. The use of infrared spectroscopy for determination of polypropylene stereoregularity. J. Appl. Polym. Sci. 1988, 36, 279–293. [Google Scholar] [CrossRef]
- Huy, T.A.; Adhikari, R.; Lüpke, T.; Henning, S.; Michler, G.H. Molecular deformation mechanisms of isotactic polypropylene in α- and β-crystal forms by FTIR spectroscopy. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4478–4488. [Google Scholar] [CrossRef]
- Kilic, A.; Jones, K.; Shim, E.; Pourdeyhimi, B. Surface crystallinity of meltspun isotactic polypropylene filaments. Macromol. Res. 2016, 24, 25–30. [Google Scholar] [CrossRef]
- Heinen, W. Infrared determination of the crystallinity of polypropylene. J. Polym. Sci. 1959, 38, 545–547. [Google Scholar] [CrossRef]
- Li, L.; Liu, T.; Zhao, L.; Yuan, W.-k. Effect of compressed CO2 on the melting behavior and βα-recrystallization of β-form in isotactic polypropylene. J. Supercrit. Fluids 2011, 60, 137–143. [Google Scholar] [CrossRef]
- Quynn, R.G.; Riley, J.L.; Young, D.A.; Noether, H.D. Density, crystallinity, and heptane insolubility in isotactic polypropylene. J. Appl. Polym. Sci. 1959, 2, 166–173. [Google Scholar] [CrossRef]
- Lamberti, G.; Brucato, V. Real-time orientation and crystallinity measurements during the isotactic polypropylene film-casting process. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 998–1008. [Google Scholar] [CrossRef]
- Tadokoro, H.; Kobayashi, M.; Ukita, M.; Yasufuku, K.; Murahashi, S.; Torii, T. Normal Vibrations of the Polymer Molecules of Helical Conformation. V. Isotactic Polypropylene and Its Deuteroderivatives. J. Chem. Phys. 1965, 42, 1432–1449. [Google Scholar] [CrossRef]
- Wu, H.Y.; Li, X.X.; Xiang, F.M.; Huang, T.; Shi, Y.Y.; Wang, Y. Microstructure evolution of isotactic polypropylene during annealing: Effect of poly(ethylene oxide). Chin. J. Polym. Sci. 2012, 30, 199–208. [Google Scholar] [CrossRef]
- Milicevic, D.; Micic, M.; Stamboliev, G.; Leskovac, A.; Mitric, M.; Suljovrujic, E. Microstructure and crystallinity of polyolefins oriented via solid-state stretching at an elevated temperature. Fibers Polym. 2012, 13, 466–470. [Google Scholar] [CrossRef]
- Milicevic, D.; Trifunovic, S.; Galovic, S.; Suljovrujic, E. Thermal and crystallization behaviour of gamma irradiated PLLA. Radiat. Phys. Chem. 2007, 76, 1376–1380. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) Polymer Handbook; Wiley-Interscience: New York, NY, USA, 1999. [Google Scholar]
- Wunderlich, B. Thermal Analysis; Academic Press, Inc.: Cambridge, MA, USA, 1990. [Google Scholar]
- Chang, B.; Schneider, K.; Vogel, R.; Heinrich, G. Influence of Annealing on Mechanical αc-Relaxation of Isotactic Polypropylene: A Study from the Intermediate Phase Perspective. Macromol. Mater. Eng. 2017, 302, 1700291. [Google Scholar] [CrossRef]
- Rodriguez-Arnold, J.; Zhang, A.; Cheng, S.Z.D.; Lovinger, A.J.; Hsieh, E.T.; Chu, P.; Johnson, T.W.; Honnell, K.G.; Geerts, R.G.; Palackal, S.J.; et al. Crystallization, melting and morphology of syndiotactic polypropylene fractions: 1. Thermodynamic properties, overall crystallization and melting. Polymer 1994, 35, 1884–1895. [Google Scholar] [CrossRef]
- Lee, M.; Kim, C.-H.; Koo, C.-S.; Kim, B.-R.; Lee, Y. The variation of structure and physical properties of XLPE during thermal aging process. Polymer 2003, 27, 249–254. [Google Scholar]
- Haftka, S.; Könnecke, K. Physical properties of syndiotactic polypropylene. J. Macromol. Sci. Part B 1991, 30, 319–334. [Google Scholar] [CrossRef]
- Farrow, G. Crystallinity, ‘crystallite size’ and melting point of polypropylene. Polymer 1963, 4, 191–197. [Google Scholar] [CrossRef]
- Teodorescu, G.M.; Vuluga, Z.; Ion, R.M.; Fistoș, T.; Ioniță, A.; Slămnoiu-Teodorescu, S.; Paceagiu, J.; Nicolae, C.A.; Gabor, A.R.; Ghiurea, M. The Effect of Thermoplastic Elastomer and Fly Ash on the Properties of Polypropylene Composites with Long Glass Fibers. Polymers 2024, 16, 1238. [Google Scholar] [CrossRef]
- Weeks, J.J. Melting Temperature and Change of Lamellar Thickness with Time for Bulk Polyethylene. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1963, 67a, 441–451. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Z.; Fu, L.; Lu, Y.; Men, Y. Lamellar Thickness and Stretching Temperature Dependency of Cavitation in Semicrystalline Polymers. PLoS ONE 2014, 9, e97234. [Google Scholar] [CrossRef]
- Huang, S.; Li, H.; Jiang, S. Crystal structure and unique lamellar thickening for poly(l-lactide) induced by high pressure. Polymer 2019, 175, 81–86. [Google Scholar] [CrossRef]
- Ronkay, F.; Molnár, B.; Nagy, D.; Szarka, G.; Iván, B.; Kristály, F.; Mertinger, V.; Bocz, K. Melting temperature versus crystallinity: New way for identification and analysis of multiple endotherms of poly(ethylene terephthalate). J. Polym. Res. 2020, 27, 372. [Google Scholar] [CrossRef]
- Tencé-Girault, S.; Lebreton, S.; Bunau, O.; Dang, P.; Bargain, F. Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition. Crystals 2019, 9, 271. [Google Scholar] [CrossRef]
- Guleria, D.; Ge, S.; Cardon, L.; Vervoort, S.; den Doelder, J. Impact of resin density and short-chain branching distribution on structural evolution and enhancement of tensile modulus of MDO-PE films. Polym. Test. 2024, 139, 108560. [Google Scholar] [CrossRef]
- Banford, H.M.; Fouracre, R.A.; Faucitano, A.; Buttafava, A.; Martinotti, F. The influence of chemical structure on the dielectric behavior of polypropylene. IEEE Trans. Dielectr. Electr. Insul. 1996, 3, 594–598. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H. Polymer Handbook; Wiley: New York, NY, USA, 1975. [Google Scholar]
- Castejón, M.L.; Tiemblo, P.; Gómez-Elvira, J.M. Photo-oxidation of thick isotactic polypropylene films. II. Evolution of the low temperature relaxations and of the melting endotherm along the kinetic stages. Polym. Degrad. Stab. 2001, 71, 99–111. [Google Scholar] [CrossRef]
- Hara, T. Dielectric Property of Some Polymers in Low Temperature Region. Jpn. J. Appl. Phys. 1967, 6, 147–150. [Google Scholar] [CrossRef]
- Hoyos, M.; Tiemblo, P.; Gómez-Elvira, J.M. The role of microstructure, molar mass and morphology on local relaxations in isotactic polypropylene. The α relaxation. Polymer 2007, 48, 183–194. [Google Scholar] [CrossRef]
- Fouracre, R.A.; MacGregor, S.J.; Judd, M.; Banford, H.M. Condition monitoring of irradiated polymeric cables. Radiat. Phys. Chem. 1999, 54, 209–211. [Google Scholar] [CrossRef]
- Gitsas, A.; Floudas, G. Pressure Dependence of the Glass Transition in Atactic and Isotactic Polypropylene. Macromolecules 2008, 41, 9423–9429. [Google Scholar] [CrossRef]
- Jourdan, C.; Cavaille, J.Y.; Perez, J. Mechanical relaxations in polypropylene: A new experimental and theoretical approach. J. Polym. Sci. Part B Polym. Phys. 1989, 27, 2361–2384. [Google Scholar] [CrossRef]
- McCrum, N.G. Density-independent relaxations in polypropylene. J. Polym. Sci. Part B Polym. Lett. 1964, 2, 495–498. [Google Scholar] [CrossRef]
- Olivares, N.; Tiemblo, P.; Gomez-Elvira, J.M. Physicochemical processes along the early stages of the thermal degradation of isotactic polypropylene I. Evolution of the γ relaxation under oxidative conditions. Polym. Degrad. Stab. 1999, 65, 297–302. [Google Scholar] [CrossRef]
- Perepechko, I.I. Svoistva Polimerov Pri Nizkih Temperaturah; Khimiya: Moscow, Russia, 1977. [Google Scholar]
- Pluta, M.; Kryszewski, M. Studies of alpha-relaxation process in spherulitic and non-spherulitic samples of isotactic polypropylene with different molecular ordering. Acta Polym. 1987, 38, 42–52. [Google Scholar] [CrossRef]
- Read, B.E. Mechanical relaxation in isotactic polypropylene. Polymer 1990, 30, 1439–1445. [Google Scholar] [CrossRef]
- Quijada-Garrido, I.; Barrales-Rienda, J.M.; Pereña, J.M.; Frutos, G. Dynamic mechanical and dielectric behavior of erucamide (13-Cis-Docosenamide), isotactic poly(propylene), and their blends. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 1473–1482. [Google Scholar] [CrossRef]
- Sakai, A.; Tanaka, K.; Fujii, Y.; Nagamura, T.; Kajiyama, T. Structure and thermal molecular motion at surface of semi-crystalline isotactic polypropylene films. Polymer 2005, 46, 429–437. [Google Scholar] [CrossRef]
- Starkweather, H.W.; Avakian, P.; Matheson, R.R.; Fontanella, J.J.; Wintersgill, M.C. Ultralow temperature dielectric relaxations in polyolefins. Macromolecules 1992, 25, 6871–6875. [Google Scholar] [CrossRef]
- Suljovrujic, E. Gel production, oxidative degradation and dielectric properties of isotactic polypropylene irradiated under various atmospheres. Polym. Degrad. Stab. 2009, 94, 521–526. [Google Scholar] [CrossRef]
- Tiemblo, P.; Gomez-Elvira, J.M.; García Beltrán, S.; Matisova-Rychla, L.; Rychly, J. Melting and α relaxation effects on the kinetics of polypropylene thermooxidation in the range 80–170 °C. Macromolecules 2002, 35, 5922–5926. [Google Scholar] [CrossRef]
- Umemura, T.; Suzuki, T.; Kashiwazaki, T. Impurity Effect of the Dielectric Properties of Isotactic Polypropylene. IEEE Trans. Electr. Insul. 1982, EI-17, 300–305. [Google Scholar]
- Dintilhac, N.; Lewandowski, S.; Planes, M.; Lectez, A.S.; Dantras, E. Tuning dielectric response of polyethylene by low gamma dose: Molecular mobility study improvement by dipolar probes implementation. J. Non·Cryst. Solids 2023, 621, 122606. [Google Scholar] [CrossRef]
- Suljovrujic, E. Some aspects of structural electrophysics of irradiated polyethylenes. Polymer 2005, 46, 6353–6359. [Google Scholar] [CrossRef]
- Suljovrujic, E. Complete relaxation map of polypropylene: Radiation-induced modification as dielectric probe. Polym. Bull. 2012, 68, 2033–2047. [Google Scholar] [CrossRef]
- Suljovrujic, E. Dielectric studies of molecular β-relaxation in low density polyethylene: The influence of drawing and ionizing radiation. Polymer 2002, 43, 5969–5978. [Google Scholar] [CrossRef]
- Wang, Y.; Bao, Z.; Ding, S.; Jia, J.; Dai, Z.; Li, Y.; Shen, S.; Chu, S.; Yin, Y.; Li, X. γ-Ray Irradiation Significantly Enhances Capacitive Energy Storage Performance of Polymer Dielectric Films. Adv. Mater. 2024, 36, e2308597. [Google Scholar] [CrossRef]
- Hedvig, P. Dielectric Spectroscopy of Polymers; Academia Kiado: Budapest, Hungary, 1977. [Google Scholar]
- Suljovrujic, E.; Kostoski, D.; Kacarevic-Popovic, Z.; Dojcilovic, J. Effect of gamma irradiation on the dielectric relaxation of uniaxially oriented low density polyethylene. Polym. Int. 1999, 48, 1193–1196. [Google Scholar] [CrossRef]
- Zhuravlev, S.P.; Zhuravleva, N.M.; Polonskij, Y.A. Deformation characteristics of polypropylene film and thermal stability of capacitor insulation made on the base of polypropylene film. Elektrotekhnika 2002, 11, 36–40. [Google Scholar]
- Fournie, R. All film power capacitors. Endurance tests and degradation mechanisms. Bulletin de la Direction des etudes et recherches. Bull. Dir. Etudes Rech. Ser. B 1990, 1, 1–31. [Google Scholar]
- Montanari, G.C.; Fabiani, D.; Palmieri, F.; Kaempfer, D.; Thomann, R.; Mulhaupt, R. Modification of electrical properties and performance of EVA and PP insulation through nanostructure by organophilic silicates. IEEE Trans. Dielectr. Electr. Insul. 2004, 11, 754–762. [Google Scholar] [CrossRef]
- Jia, C.; Das, P.; Kim, I.; Yoon, Y.-J.; Tay, C.Y.; Lee, J.-M. Applications, treatments, and reuse of plastics from electrical and electronic equipment. J. Ind. Eng. Chem. 2022, 110, 84–99. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Y.; Wang, X.; Liu, H.; Cheng, L.; Liu, W.; Li, S.; Guo, J.; Xu, Y. Failure mechanism of metallized film capacitors under DC field superimposed AC harmonic: From equipment to material. High Volt. 2024, 9, 1081–1089. [Google Scholar] [CrossRef]
- Zhang, Y.-T.; Hou, S.; Li, D.-L.; Cao, Y.-J.; Zhan, Y.-P.; Jia, L.; Fu, M.-l.; Huang, H.-D. Hierarchical Structural Evolution, Electrical and Mechanical Performance of Polypropylene Containing Intrinsic Elastomers under Stretching and Annealing for Cable Insulation Applications. Ind. Eng. Chem. Res. 2024, 63, 11982–11991. [Google Scholar] [CrossRef]
- Zhang, C.; Dai, X.-Y.; Xing, Z.-L.; Guo, S.-W.; Li, F.; Chen, X.; Zhou, J.-J.; Li, L. Investigation on the Structure and Performance of Polypropylene Sheets and Bi-axially Oriented Polypropylene Films for Capacitors. Chin. J. Polym. Sci. 2022, 40, 1688–1696. [Google Scholar] [CrossRef]
- Suljovrujic, E.; Kostoski, D.; Dojcilovic, J. Charge trapping in gamma irradiated low-density polyethylene. Polym. Degrad. Stab. 2001, 74, 167–170. [Google Scholar] [CrossRef]
- Suljovrujic, E.; Micic, M.; Milicevic, D. Structural Changes and Dielectric Relaxation Behavior of Uniaxially Oriented High Density Polyethylene. J. Eng. Fibers Fabr. 2013, 8, 155892501300800316. [Google Scholar] [CrossRef]
- Kostoski, D.; Galovic, S.; Suljovrujic, E. Charge trapping and dielectric relaxations of gamma irradiated radiolytically oxidized highly oriented LDPE. Radiat. Phys. Chem. 2004, 69, 245–248. [Google Scholar] [CrossRef]
- Bohning, M.; Goering, H.; Fritz, A.; Brzezinka, K.-W.; Turky, G.; Schönhals, A.; Schartel, B. Dielectric study of molecular mobility in poly(propylene-graft-maleic anhydride)/clay nanocomposites. Macromolecules 2005, 38, 2764–2774. [Google Scholar] [CrossRef]
- Beuguel, Q.; Mija, A.; Vergnes, B.; Peuvrel-Disdier, E. Structural, thermal, rheological and mechanical properties of polypropylene/graphene nanoplatelets composites: Effect of particle size and melt mixing conditions. Polym. Eng. Sci. 2018, 58, 1937–1944. [Google Scholar] [CrossRef]
- Banford, H.M.; Fouracre, R.; Faucitano, A.; Buttafava, A.; Martinotti, F. The influence of γ-irradiation and chemical structure on the dielectric properties of PP. Radiat. Phys. Chem. 1996, 48, 129–130. [Google Scholar] [CrossRef]
- Milicevic, D.; Micic, M.; Suljovrujic, E. Radiation-induced modification of dielectric relaxation spectra of polyolefins: Polyethylenes vs. polypropylene. Polym. Bull. 2014, 71, 2317–2334. [Google Scholar] [CrossRef]
- Qian, S.; Igarashi, T.; Nitta, K.-h. Thermal degradation behavior of polypropylene in the melt state: Molecular weight distribution changes and chain scission mechanism. Polym. Bull. 2011, 67, 1661–1670. [Google Scholar] [CrossRef]
- Nitta, K.h.; Yamaguchi, N. Influence of Morphological Factors on Tensile Properties in the Pre-yield Region of Isotactic Polypropylenes. Polym. J. 2006, 38, 122–131. [Google Scholar] [CrossRef]
- Nitta, K.-H. Tensile Properties in β-Modified Isotactic Polypropylene. In Polypropylene—Polymerization and Characterization of Mechanical and Thermal Properties; Wang, W., Zeng, Y., Eds.; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar]
- Kida, T.; Fukuda, Y.; Yamaguchi, M.; Otsuki, Y.; Kimura, T.; Mizukawa, T.; Murakami, T.; Hato, K.; Okawa, T. Morphological transformation of extruded isotactic polypropylene film from the Mesophase to α-form crystals. React. Funct. Polym. 2023, 191, 105682. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Z.; Li, T.; Peng, X.; Jiang, S.; Turng, L.-S. Quantification of the Young’s modulus for polypropylene: Influence of initial crystallinity and service temperature. J. Appl. Polym. Sci. 2020, 137, 48581. [Google Scholar] [CrossRef]
- Makarewicz, C.; Safandowska, M.; Idczak, R.; Rozanski, A. Plastic Deformation of Polypropylene Studied by Positron Annihilation Lifetime Spectroscopy. Macromolecules 2022, 55, 10062–10076. [Google Scholar] [CrossRef]
- Peterson, J.M. Thermal Initiation of Screw Dislocations in Polymer Crystal Platelets. J. Appl. Phys. 1966, 37, 4047–4050. [Google Scholar] [CrossRef]
- Peterson, J.M. Peierls Stress for Screw Dislocations in Polyethylene. J. Appl. Phys. 1968, 39, 4920–4928. [Google Scholar] [CrossRef]
- Peterlin, A. Molecular model of drawing polyethylene and polypropylene. J. Mater. Sci. 1971, 6, 490–508. [Google Scholar] [CrossRef]
- Morosoff, N.; Peterlin, A. Plastic deformation of polypropylene. IV. Wide-angle x-ray scattering in the neck region. J. Polym. Sci. Part A-2: Polym. Phys. 1972, 10, 1237–1254. [Google Scholar] [CrossRef]
- Suljovrujic, E. The influence of molecular orientation on the crosslinking/oxidative behaviour of iPP exposed to gamma radiation. Eur. Polym. J. 2009, 45, 2068–2078. [Google Scholar] [CrossRef]
- Milicevic, D.; Trifunovic, S.; Popovic, M.; Vukasinovic-Milic, T.; Suljovrujic, E. The influence of orientation on the radiation-induced crosslinking/oxidative behavior of different PEs. Nucl. Instrum. Methods Phys. Res. Sect. B 2007, 260, 603–612. [Google Scholar] [CrossRef]
- Butler, M.F.; Donald, A.M.; Bras, W.; Mant, G.R.; Derbyshire, G.E.; Ryan, A.J. A Real-Time Simultaneous Small- and Wide-Angle X-ray Scattering Study of In-Situ Deformation of Isotropic Polyethylene. Macromolecules 1995, 28, 6383–6393. [Google Scholar] [CrossRef]
- Furuta, M.; Kojima, K. Morphological study of deformation process for linear polyethylene. J. Macromol. Sci. Part B 1986, 25, 349–364. [Google Scholar] [CrossRef]
- Liu, T.-M.; Juska, T.D.; Harrison, I.R. Plastic deformation of polypropylene. Polymer 1986, 27, 247–249. [Google Scholar] [CrossRef]
- Aboulfaraj, M.; G’Sell, C.; Ulrich, B.; Dahoun, A. In situ observation of the plastic deformation of polypropylene spherulites under uniaxial tension and simple shear in the scanning electron microscope. Polymer 1995, 36, 731–742. [Google Scholar] [CrossRef]
- G’sell, C.; Favier, V.; Hiver, J.M.; Dahoun, A.; Philippe, M.J.; Canova, G.R. Microstructure transformation and stress-strain behavior of isotactic polypropylene under large plastic deformation. Polym. Eng. Sci. 1997, 37, 1702–1711. [Google Scholar] [CrossRef]
- Ariyama, T.; Mori, Y.; Kaneko, K. Tensile properties and stress relaxation of polypropylene at elevated temperatures. Polym. Eng. Sci. 1997, 37, 81–90. [Google Scholar] [CrossRef]
- Chodák, I. High modulus polyethylene fibres: Preparation, properties and modification by crosslinking. Prog. Polym. Sci. 1998, 23, 1409–1442. [Google Scholar] [CrossRef]
- Séguéla, R. Dislocation approach to the plastic deformation of semicrystalline polymers: Kinetic aspects for polyethylene and polypropylene. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- Na, B.; Lv, R. Effect of cavitation on the plastic deformation and failure of isotactic polypropylene. J. Appl. Polym. Sci. 2007, 105, 3274–3279. [Google Scholar] [CrossRef]
- Pawlak, A.; Rozanski, A.; Galeski, A. Thermovision studies of plastic deformation and cavitation in polypropylene. Mech. Mater. 2013, 67, 104–118. [Google Scholar] [CrossRef]
- Chen, W.; Li, X.-y.; Liu, Y.-p.; Li, J.; Zhou, W.-m.; Chen, L.; Li, L.-b. The spatial correlation between crystalline and amorphous orientations of isotactic polypropylene during plastic deformation: An in situ observation with FTIR imaging. Chin. J. Polym. Sci. 2015, 33, 613–620. [Google Scholar] [CrossRef]
- Kim, M.; Park, T.Y.; Hong, S. Experimental determination of the plastic deformation and fracture behavior of polypropylene composites under various strain rates. Polym. Test. 2021, 93, 107010. [Google Scholar] [CrossRef]
- Liparoti, S.; Sorrentino, A.; Speranza, V. Morphology-Mechanical Performance Relationship at the Micrometrical Level within Molded Polypropylene Obtained with Non-Symmetric Mold Temperature Conditioning. Polymers 2021, 13, 462. [Google Scholar] [CrossRef] [PubMed]
- Shirinbayan, M.; Nouira, S.; Imaddahen, M.-A.; Fitoussi, J. Microstructure-sensitive investigation on the plastic deformation and damage initiation of fiber-reinforced polypropylene composite. Compos. Part B Eng. 2024, 286, 111790. [Google Scholar] [CrossRef]
- An, Y.; Wang, S.; Li, R.; Shi, D.; Gao, Y.; Song, L. Effect of different nucleating agent on crystallization kinetics and morphology of polypropylene. e-Polymers 2019, 19, 32–39. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suljovrujic, E.; Milicevic, D.; Djordjevic, K.; Rogic Miladinovic, Z.; Stamboliev, G.; Galovic, S. Structure–Property Relationship in Isotactic Polypropylene Under Contrasting Processing Conditions. Polymers 2025, 17, 1889. https://doi.org/10.3390/polym17141889
Suljovrujic E, Milicevic D, Djordjevic K, Rogic Miladinovic Z, Stamboliev G, Galovic S. Structure–Property Relationship in Isotactic Polypropylene Under Contrasting Processing Conditions. Polymers. 2025; 17(14):1889. https://doi.org/10.3390/polym17141889
Chicago/Turabian StyleSuljovrujic, Edin, Dejan Milicevic, Katarina Djordjevic, Zorana Rogic Miladinovic, Georgi Stamboliev, and Slobodanka Galovic. 2025. "Structure–Property Relationship in Isotactic Polypropylene Under Contrasting Processing Conditions" Polymers 17, no. 14: 1889. https://doi.org/10.3390/polym17141889
APA StyleSuljovrujic, E., Milicevic, D., Djordjevic, K., Rogic Miladinovic, Z., Stamboliev, G., & Galovic, S. (2025). Structure–Property Relationship in Isotactic Polypropylene Under Contrasting Processing Conditions. Polymers, 17(14), 1889. https://doi.org/10.3390/polym17141889