

Simulation of the Pyrolysis Process of Cyclohexane-Containing Semi-Aromatic Polyamide Based on ReaxFF-MD

 ,
,
Abstract
1. Introduction
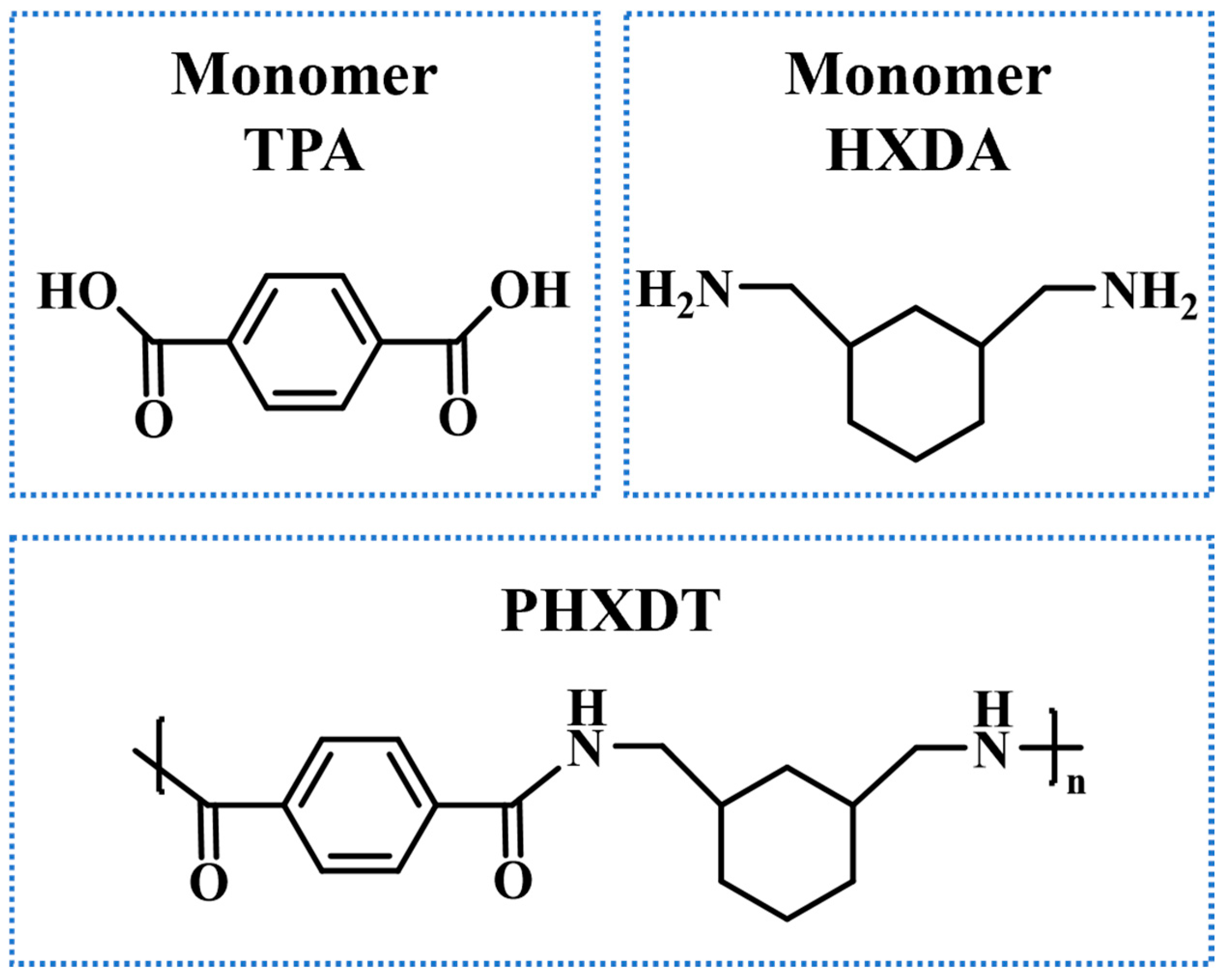
2. Computational Methods and Simulation Details
3. Results and Discussion
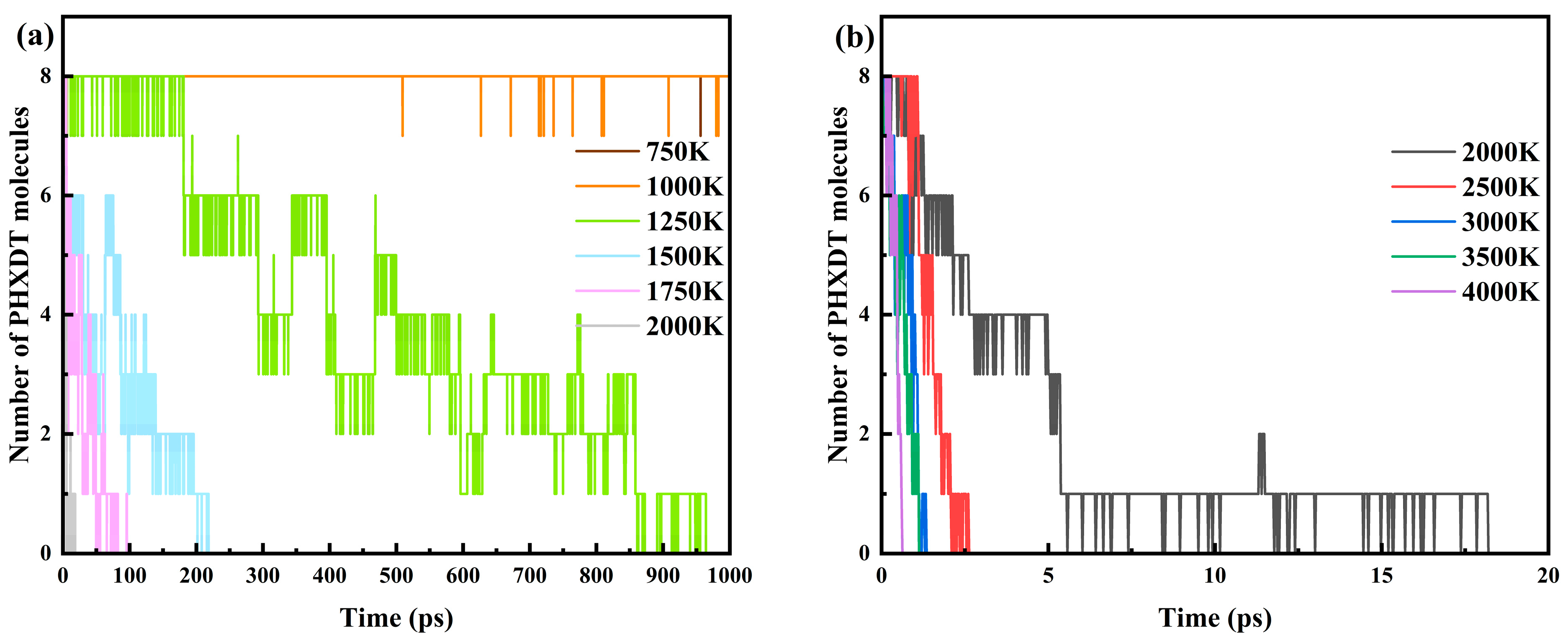
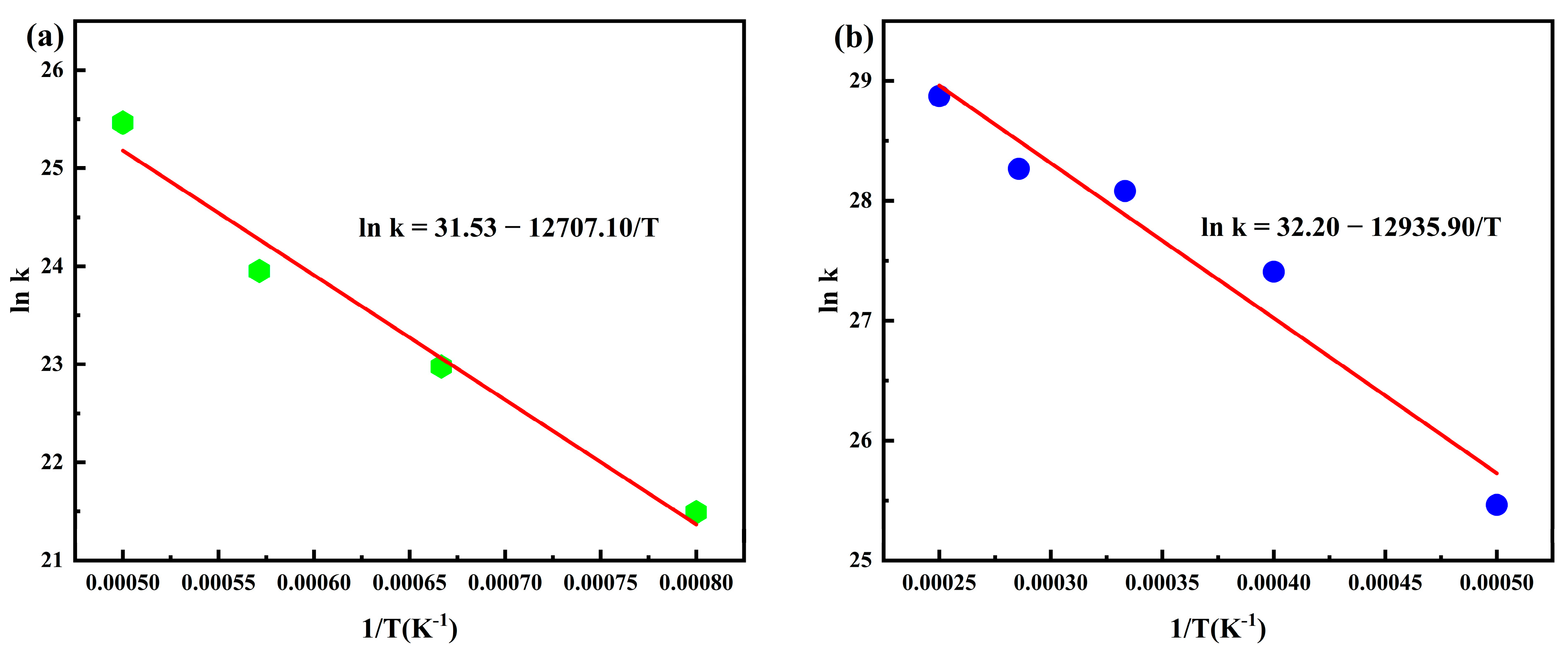
3.1. Calculation of PHXDT Pyrolysis Kinetics
3.2. Analysis of the PHXDT Pyrolysis Reaction Process
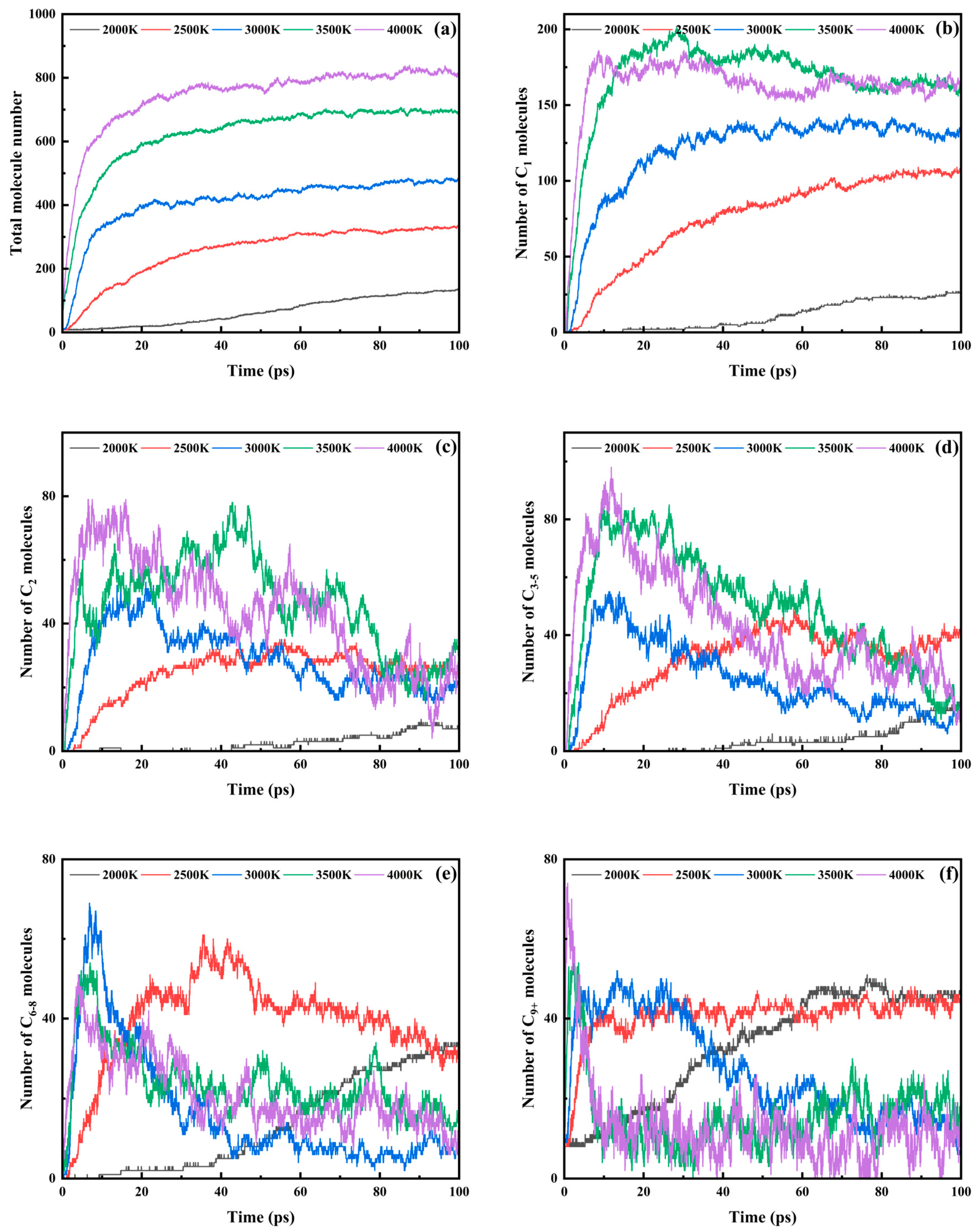
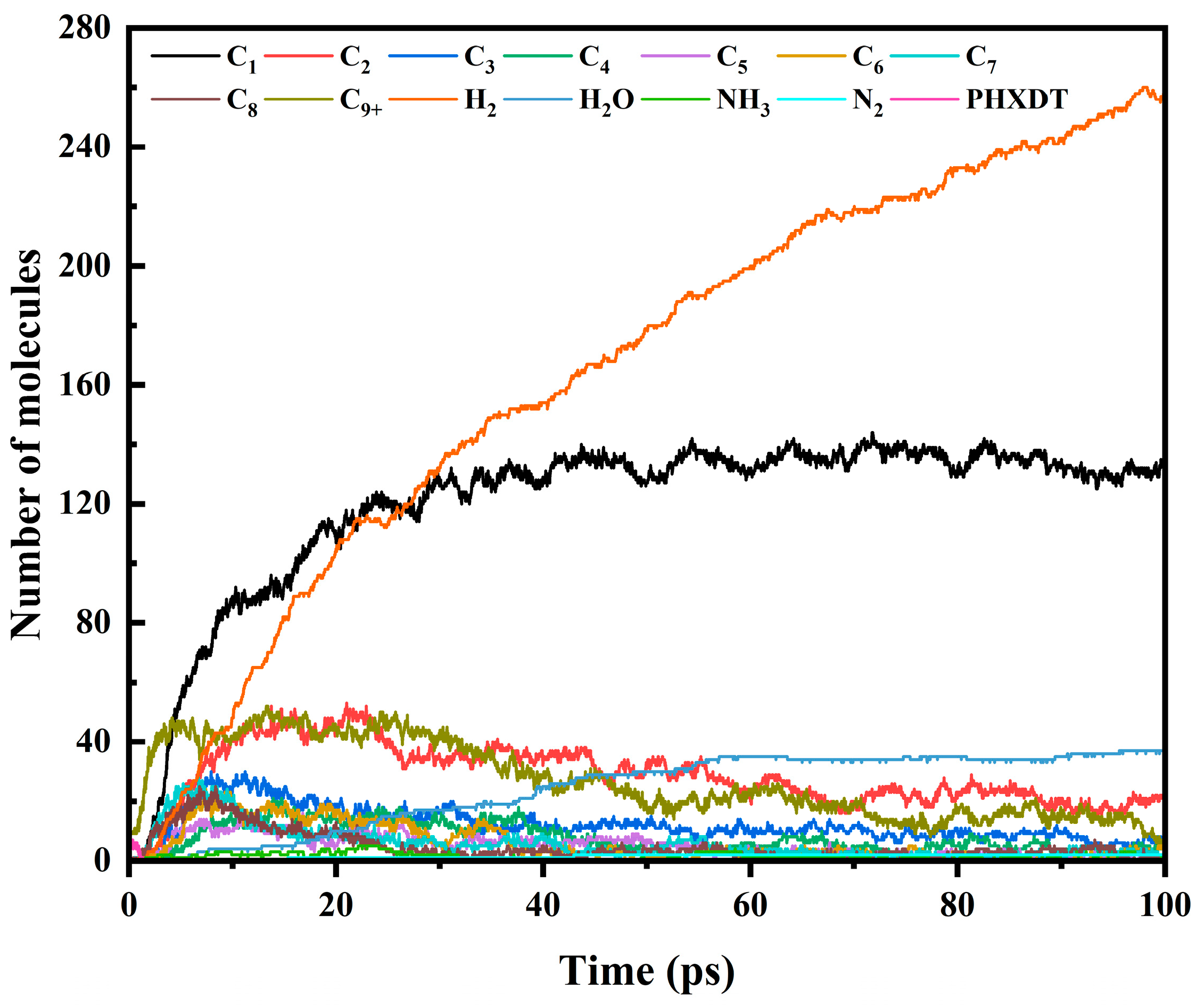
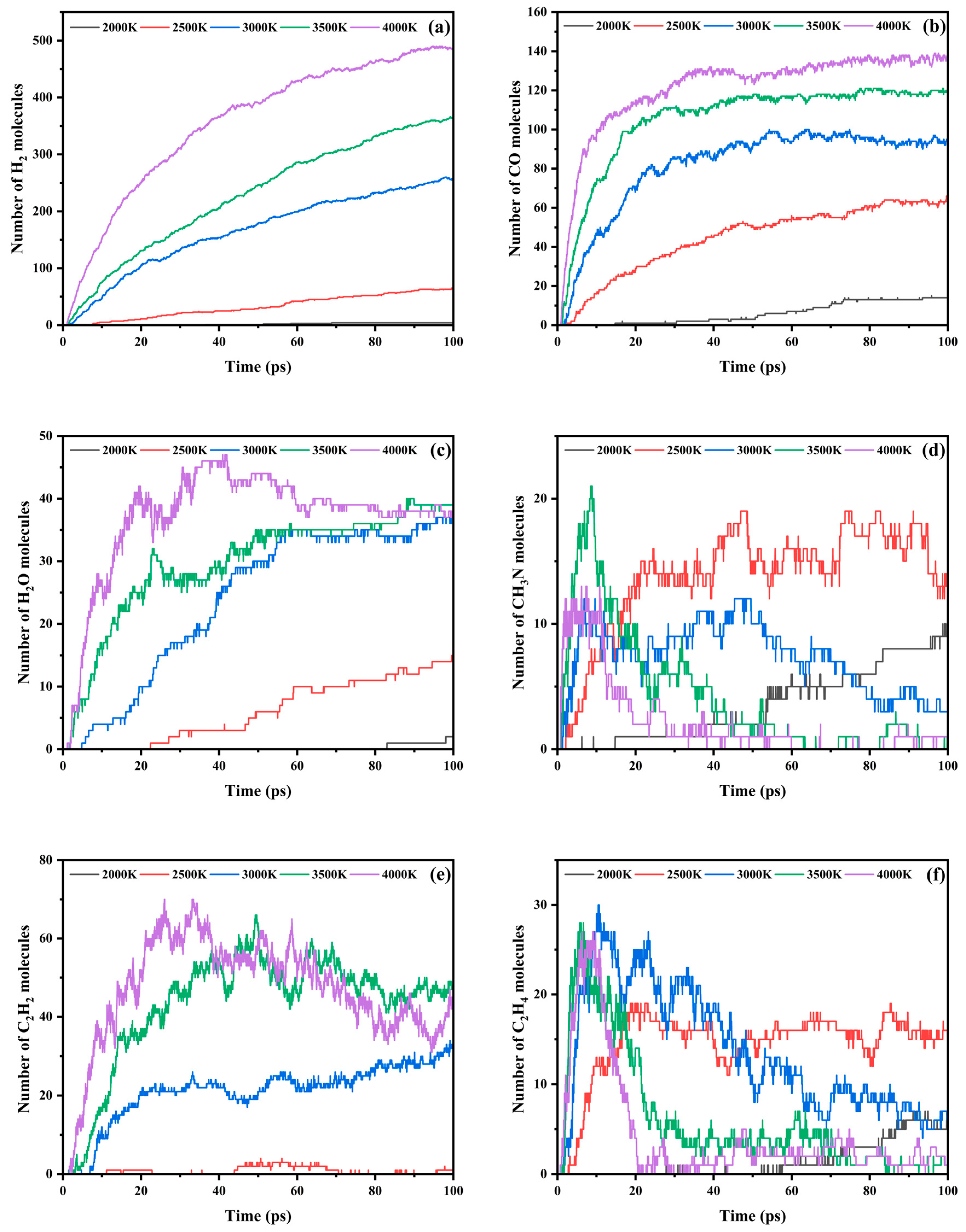
3.3. Trends in Major Pyrolysis Products
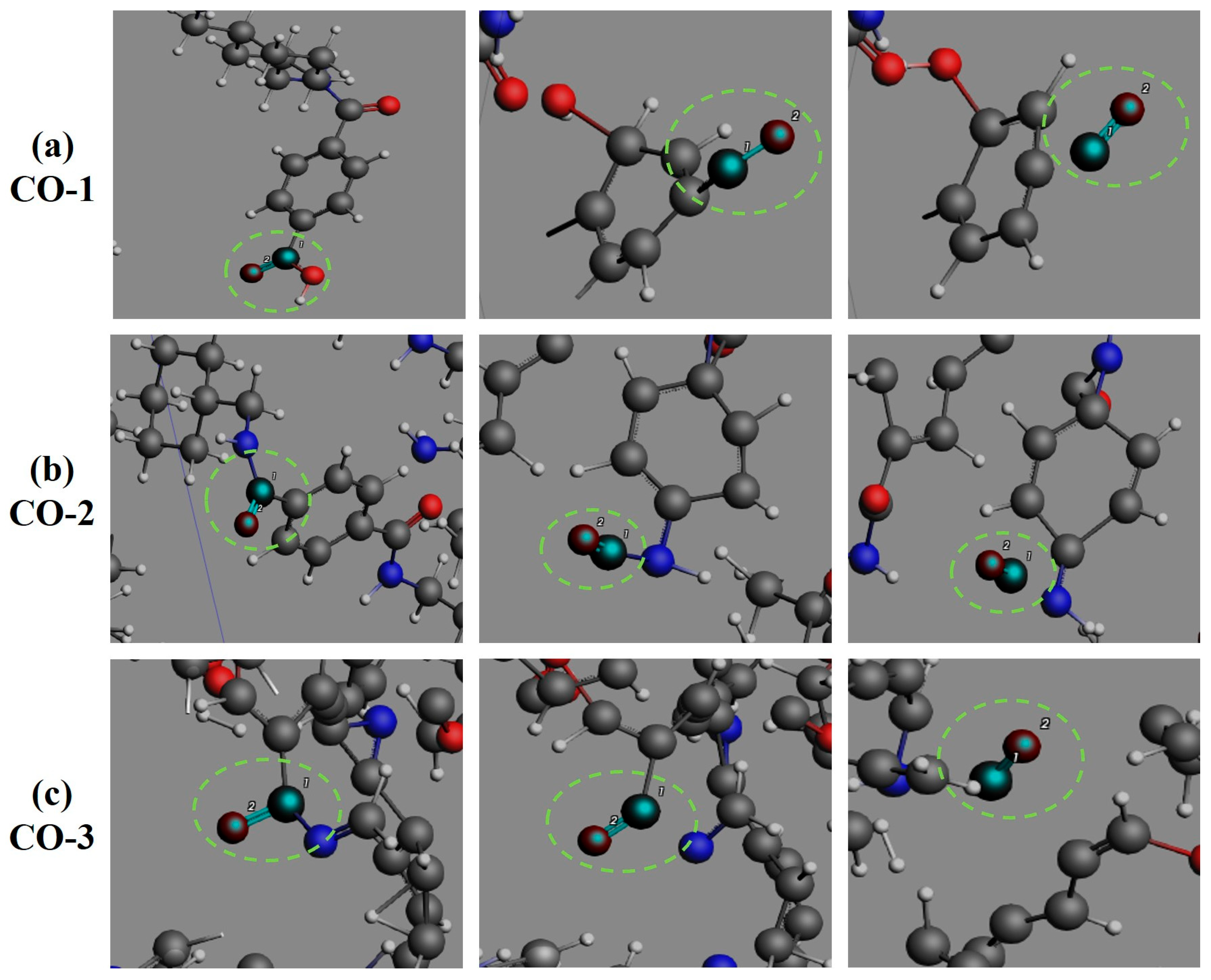
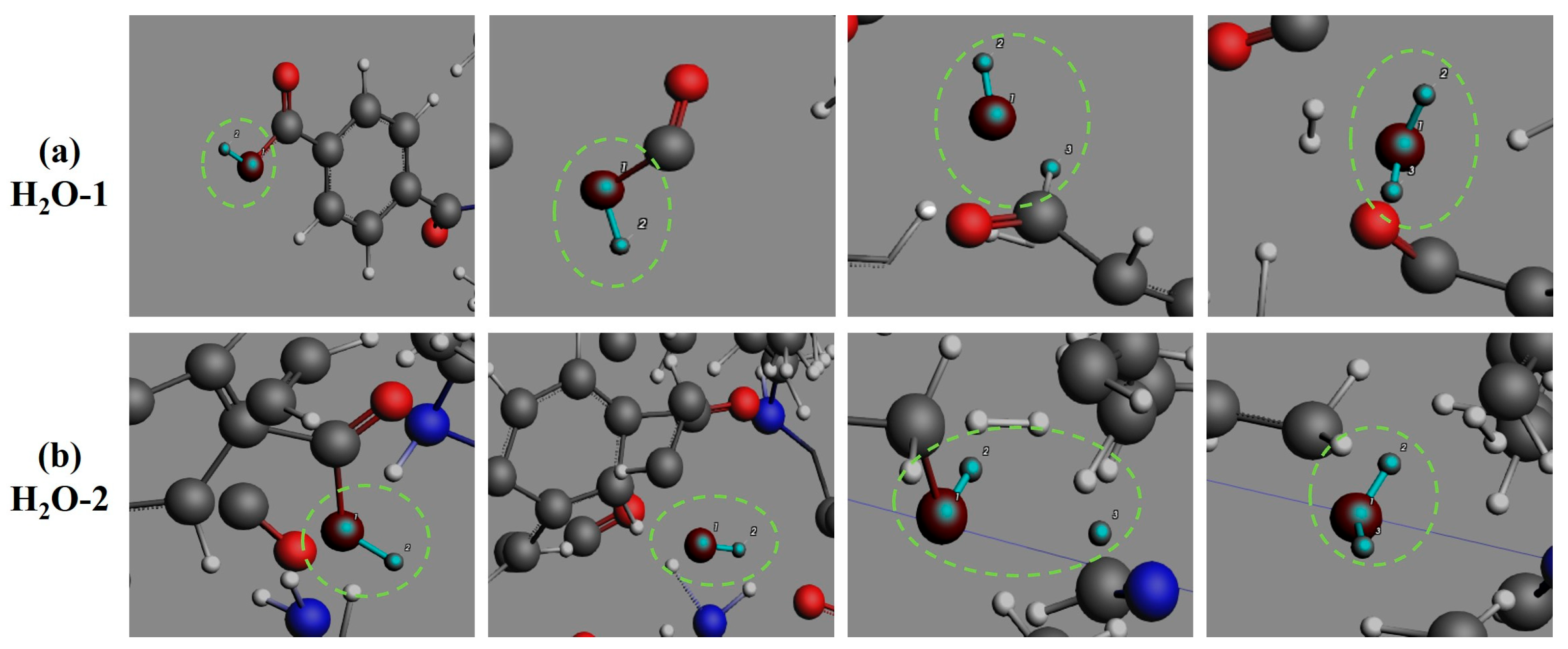
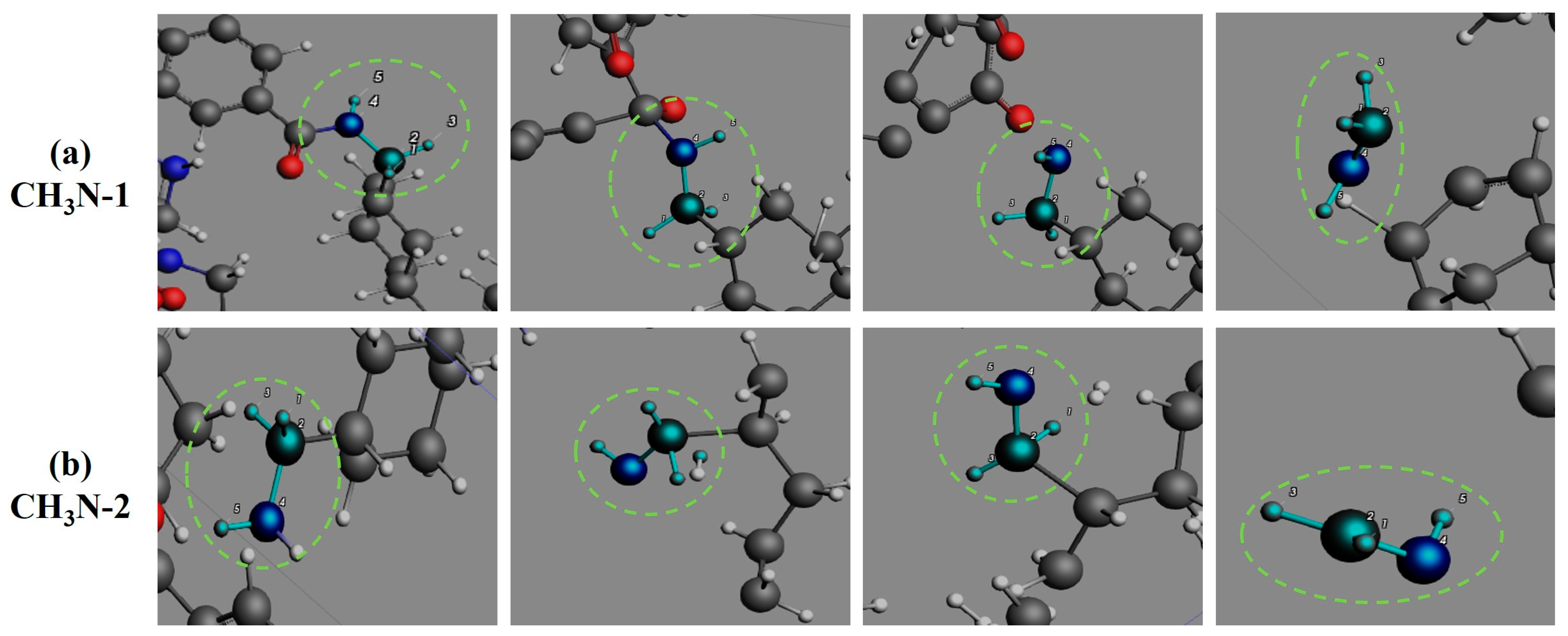
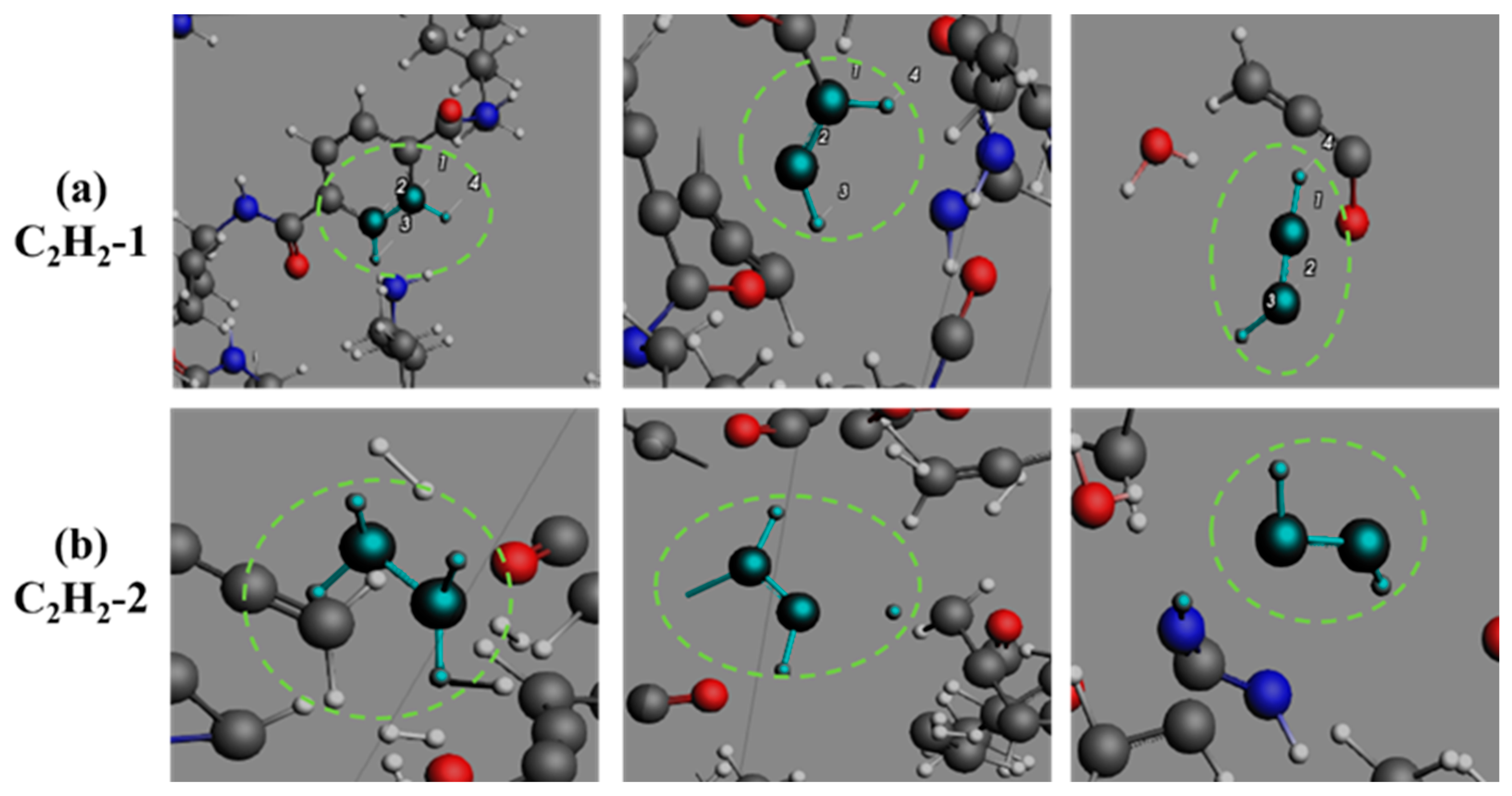
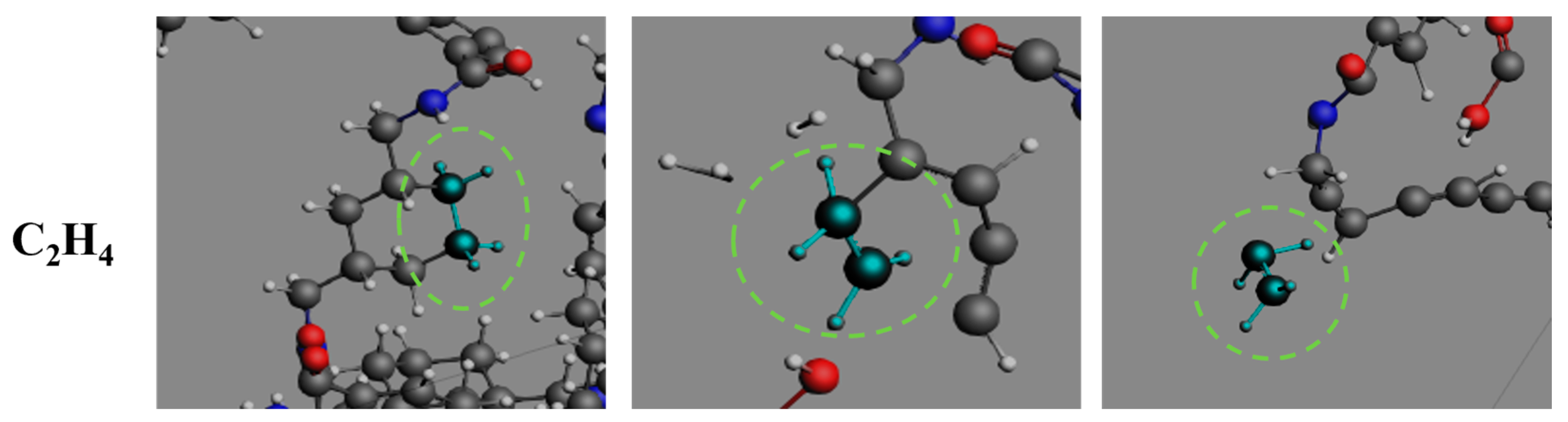
3.4. Formation Pathways of Major Pyrolysis Products
3.5. Role of Cyclohexane Structure in the Pyrolysis Process of PHXDT and Thermal Stability Design of c-SaPA Materials
4. Conclusions
- (1)
- ReaxFF-MD simulations revealed distinct pyrolysis kinetics for multi-chain PHXDT systems, with calculated activation energy (107.55 kJ/mol) and a pre-exponential factor (9.64 × 1013 s−1) within the 2000–4000 K range. The temperature-dependent kinetics followed Arrhenius behavior, demonstrating accelerated backbone scission and radical-driven fragmentation at elevated temperatures, where decomposition times decreased exponentially from 18.2 ps (2000 K) to 0.625 ps (4000 K).
- (2)
- Product distribution exhibited hierarchical fragmentation patterns: C1–2 products accumulated monotonically at low temperatures but stabilized rapidly under high thermal stress; C3–5 intermediates showed bimodal growth–decay behavior above 3000 K; C6–8 derivatives displayed accelerated peak emergence with reduced terminal quantities, indicating enhanced secondary cracking; C9+ macromolecular rapidly reached peak contents under elevated temperatures, followed by carbonization-driven stabilization at a diminished level. This progression confirmed a multi-stage mechanism: initial backbone rupture, intermediate macromolecular fragmentation, and terminal dehydrogenation and carbonization.
- (3)
- The pyrolysis product distribution exhibits strong thermal field dependence. H2 dominates at elevated temperatures, showing exponential yield increase with temperature. CO formation exhibits rapid initial growth followed by stabilization. This process derives oxygen from structural oxygen-containing groups, engaging in dynamic competition with H2O for oxygen allocation. Thermal energy preferentially drives oxygen transfer toward CO formation. The characteristic peak phenomena observed in small-molecule hydrocarbons such as CH3N, C2H2, and C2H4 unveil critical transitions in rate-limiting steps governing formation and decomposition pathways during pyrolysis.
- (4)
- The pyrolysis mechanism was governed by selective bond cleavage through radical chain reactions, generating dominant products (H2, CO, H2O, CH3N, C2H2, C2H4). Critical pathways included carboxyl group decarboxylation, amide bond dissociation, and transformations of N-C-O/C-C-O intermediates and OH·/H· radicals. These interconnected processes formed a reaction network where small-molecule release correlated with progressive carbon skeleton simplification, ultimately yielding thermally stable coke.
- (5)
- The cyclohexane structure, in particular, has been identified as a key active structure that plays a crucial role in the regulation of the rapid generation of small molecule gases (H2, C2H2, C2H4, CH3N, etc.) and the acceleration of the conversion of intermediates to char. This dynamic process exhibits significantly higher reactivity in comparison to the benzene-ring structure, thereby providing a structurally sensitive basis for the directional modulation of the pyrolysis products of polyamides. The initial pyrolysis energy barrier of cyclohexane structure can be increased by restricting their conformational transition and the chain reaction can be inhibited by combining an H· radical trapping agent.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Endo, T.; Higashihara, T. Direct Synthesis of Thermally Stable Semiaromatic Polyamides by Bulk Polymerization Using Aromatic Diamines and Aliphatic Dicarboxylic Acids. ACS Omega 2022, 7, 8753–8758. [Google Scholar] [CrossRef] [PubMed]
- García, J.M.; García, F.C.; Serna, F.; de la Peña, J.L. High-Performance Aromatic Polyamides. Prog. Polym. Sci. 2010, 35, 623–686. [Google Scholar] [CrossRef]
- Teng, C.; Li, H.; Liu, J.; Gu, H.; Kong, H.; Yu, M. Effect of High Molecular Weight PPTA on Liquid Crystalline Phase and Spinning Process of Aramid Fibers. Polymers 2020, 12, 1206. [Google Scholar] [CrossRef] [PubMed]
- Hernández, G.; Ferrero, S.; Reinecke, H.; Bartolomé, C.; Martinez-Ilarduya, J.M.; Álvarez, C.; Lozano, Á.E. New Insights in the Synthesis of High-Molecular-Weight Aromatic Polyamides-Improved Synthesis of Rod-like PPTA. Int. J. Mol. Sci. 2023, 24, 2734. [Google Scholar] [CrossRef]
- Liu, H.; Xu, M.; Li, X. Achievement of High-Reliability and High-Efficient Deposit of PA66 by Additive Friction Stir Deposition. Compos. Part B Eng. 2024, 284, 111682. [Google Scholar] [CrossRef]
- Mao, L.; Pan, L.; Ma, B.; He, Y. Synthesis and Characterization of Bio-Based Amorphous Polyamide From Dimethyl Furan-2,5-Dicarboxylate. J. Polym. Environ. 2022, 30, 1072–1079. [Google Scholar] [CrossRef]
- Yan, G.; Zhang, G.; Ren, H.; Li, Y.; Yang, J. Synthesis and Characterization of Semiaromatic Polyamides with Dicyclohexane Units. RSC Adv. 2016, 6, 76490–76497. [Google Scholar] [CrossRef]
- Zhang, C. Progress in Semicrystalline Heat-Resistant Polyamides. e-Polymers 2018, 18, 373–408. [Google Scholar] [CrossRef]
- Lin, X.B.; Du, S.L.; Long, J.W.; Chen, L.; Wang, Y.Z. A Novel Organophosphorus Hybrid with Excellent Thermal Stability: Core-Shell Structure, Hybridization Mechanism, and Application in Flame Retarding Semi-Aromatic Polyamide. ACS Appl. Mater. Interfaces 2016, 8, 881–890. [Google Scholar] [CrossRef]
- Zhang, G.; Yan, G.M.; Ren, H.H.; Li, Y.; Wang, X.J.; Yang, J. Effects of a Trans- or Cis-Cyclohexane Unit on the Thermal and Rheological Properties of Semi-Aromatic Polyamides. Polym. Chem. 2015, 7, 44–53. [Google Scholar] [CrossRef]
- Li, J.; Yi, Y.; Wang, C.; Lu, W.; Liao, M.; Jing, X.; Wang, W. An Intrinsically Transparent Polyamide Film with Superior Toughness and Great Optical Performance. Polymers 2024, 16, 599. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Dai, J.; Ji, X.; Chen, J.; Jiang, Y.; Li, S.; Mao, T.; Fan, X. Synthesis of Novel Poly(Arylene Ether Amide) Containing Aliphatic Ring for Optical Property. High Perform. Polym. 2022, 34, 264–272. [Google Scholar] [CrossRef]
- Zhong, H.; Deng, J. Preparation and Chiral Applications of Optically Active Polyamides. Macromol. Rapid Commun. 2021, 42, 2100341. [Google Scholar] [CrossRef] [PubMed]
- Li, P.H.; Wang, C.Y.; Li, G.; Jiang, J.M. Highly Organosoluble and Transparent Polyamides Containing Cyclohexane and Trifluoromethyl Moieties: Synthesis and Characterization. Express Polym. Lett. 2009, 3, 703–712. [Google Scholar] [CrossRef]
- Long, J.W.; Chen, L.; Liu, B.W.; Shi, X.H.; Lin, X.B.; Li, Y.M.; Wang, Y.Z. Tuning the Pendent Groups of Semiaromatic Polyamides toward High Performance. Macromolecules 2020, 53, 3504–3513. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.; Nikolaou, E.; Yamamoto, N. Novel Biodegradable Copolyamides Based on Adipic Acid, Bis(p-Aminocyclohexyl)Methane and Several α-Amino Acids: Synthesis, Characterization and Study of Their Degradability for Food Packaging Applications: 4. Polymer 1994, 35, 4678–4689. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, S.; Wu, Y.; Huang, T.; Yu, B.; Zhu, M.; Yu, H. One-Pot Synthesis of Copolyamide PA MXDT-MXD6 towards Oxygen-Barrier and High-Temperature Transparent Applications. Eur. Polym. J. 2024, 214, 113120. [Google Scholar] [CrossRef]
- Mutua, F.N.; Yang, T.; Gao, Y.; Zhu, B.; He, Y. Preparation, Analysis, and Isothermal Crystallization Behavior of Poly[1,3-Bis(Aminomethyl)Cyclohexamethylene Oxamide]. J. Appl. Polym. Sci. 2018, 135, 46345. [Google Scholar] [CrossRef]
- Dong, J.; Fan, Z. Nonisothermal Crystallization Behaviour of a Novel Cycloaliphatic Microcrystalline Poly(4,4′-Aminocyclohexyl Methylene Dodecanedicarboxylamide). Polym. Polym. Compos. 2014, 22, 401–408. [Google Scholar] [CrossRef]
- Guo, J.; Li, H.; Xue, B.; Zhu, D.; Zhang, X.; Zhao, H.; Qiao, X.; Cui, Z.; Fu, P.; Zhao, Q.; et al. A Synthesized Semi-Aromatic Copolyamaide through Synergy of Three Different Kinds of Monomers: Toward High Transparency, Excellent Heat Resistance and Melt Flowing Property. J. Appl. Polym. Sci. 2021, 138, 49678. [Google Scholar] [CrossRef]
- Li, P.H.; Wang, C.Y.; Li, G.; Jiang, J.M. Synthesis and Characterization of Novel Polyamides Derived from 1,4-Bis((4-Amino-2-(Trifluoromethyl)Phenoxy)Methyl)Cyclohexane and Aromatic Dicarboxylic Acids. Polym. Bull. 2010, 64, 127–140. [Google Scholar] [CrossRef]
- Murthy, N.S.; Bray, R.G. Structure and Properties of Polyamide 6 and 4-Aminomethylcyclohexane Carboxylic Acid Copolymers with an Unusually Short Helical Pitch for Nylons. Polymer 2003, 44, 5387–5396. [Google Scholar] [CrossRef]
- Bradler, P.R.; Fischer, J.; Wallner, G.M.; Lang, R.W. Characterization of Irradiation Crosslinked Polyamides for Solar Thermal Applications—Basic Thermo-Analytical and Mechanical Properties. Polymers 2018, 10, 969. [Google Scholar] [CrossRef] [PubMed]
- Bradler, P.R.; Fischer, J.; Wallner, G.M.; Lang, R.W. Characterization of Irradiation Crosslinked Polyamides for Solar Thermal Applications-Fatigue Properties. Compos. Sci. Technol. 2019, 175, 55–59. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, G.R.; Xiao, X.X.; Liu, Q.S.; Jiang, M.; Guo, D.M.; Zhao, H.B.; Li, W.D.; Chen, L.; Liu, B.W.; et al. Controllable Micro Cross-Linking towards Multifunctional Flame-Retardant Aliphatic Polyamide. Chem. Eng. J. 2023, 472, 144983. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Y.; Zheng, Z.; Lu, G.; Tang, Z.; Chen, X. Synthesis and Thermal Degradation Mechanism of a Semi-Aromatic Copolyamide from Renewable Sources. Polym. Degrad. Stab. 2022, 203, 110089. [Google Scholar] [CrossRef]
- Liu, M.; Li, K.; Yang, S.; Fu, P.; Wang, Y.; Zhao, Q. Synthesis and Thermal Decomposition of Poly(Dodecamethylene Terephthalamide). J. Appl. Polym. Sci. 2011, 122, 3369–3376. [Google Scholar] [CrossRef]
- Meng, C.; Liu, X. Study on Thermal Degradation Kinetics of Bio-Based Semi-Aromatic High-Temperature Polyamide PA5T/56 and the Effect of Benzene Ring. Iran. Polym. J. 2022, 31, 641–650. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, D.; Xu, X.; Yuan, Y. Pyrolysis Characteristics, Kinetics, Thermodynamics and Volatile Products of Waste Medical Surgical Mask Rope by Thermogravimetry and Online Thermogravimetry-Fourier Transform Infrared-Mass Spectrometry Analysis. Fuel 2021, 295, 120632. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, M.; Li, Y.; Xiong, Y.; Wu, C. Recycling and Degradation of Polyamides. Molecules 2024, 29, 1742. [Google Scholar] [CrossRef]
- Mao, Q.; Feng, M.; Jiang, X.Z.; Ren, Y.; Luo, K.H.; van Duin, A.C.T. Classical and Reactive Molecular Dynamics: Principles and Applications in Combustion and Energy Systems. Prog. Energy Combust. Sci. 2023, 97, 101084. [Google Scholar] [CrossRef]
- Zhang, F.; Yang, R.; Lu, D. Investigation of Polymer Aging Mechanisms Using Molecular Simulations: A Review. Polymers 2023, 15, 1928. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Sreedhar, I.; Patel, C.M. Molecular Dynamics Simulation of Polyamide-Based Materials-A Review. Comput. Mater. Sci. 2021, 200, 110853. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- AlAreeqi, S.; Bahamon, D.; Polychronopoulou, K.; Vega, L.F. Insights into the Thermal Stability and Conversion of Carbon-Based Materials by Using ReaxFF Reactive Force Field: Recent Advances and Future Directions. Carbon 2022, 196, 840–866. [Google Scholar] [CrossRef]
- Zheng, Y.B.; Zhang, Q.; Mei, S.N.; Wang, W.Q.; Shi, J.; Yu, Q.W.; Zhai, G.H.; Yang, J.M. Molecular Dynamic Simulation of LiH–H2O Reactions Using the ReaxFF Reactive Force Field. Int. J. Hydrogen Energy 2023, 48, 4329–4338. [Google Scholar] [CrossRef]
- Kowalik, M.; Ashraf, C.; Damirchi, B.; Akbarian, D.; Rajabpour, S.; van Duin, A.C.T. Atomistic Scale Analysis of the Carbonization Process for C/H/O/N-Based Polymers with the ReaxFF Reactive Force Field. J. Phys. Chem. B 2019, 123, 5357–5367. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF Reactive Force-Field: Development, Applications and Future Directions. Npj Comput. Mater. 2016, 2, 15011. [Google Scholar] [CrossRef]
- Yilmaz, D.E.; Woodward, W.H.; van Duin, A.C.T. Machine Learning-Assisted Hybrid ReaxFF Simulations. J. Chem. Theory Comput. 2021, 17, 6705–6712. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Shi, J.; Jia, Y.; Li, J.; Zhang, Q.; Wang, W.; Yu, Q. Molecular Dynamic Simulation of Ni–Al Alloy–H2O Reactions Using the ReaxFF Reactive Force Field. ACS Omega 2023, 8, 9807–9814. [Google Scholar] [CrossRef]
- Yin, F.; Tang, C.; Wang, Q.; Liu, X.; Tang, Y. Molecular Dynamics Simulations on the Thermal Decomposition of Meta-Aramid Fibers. Polymers 2018, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Fan, K.; Guo, Z.; Guo, W. Pyrolysis Behavior of Automotive Polypropylene Plastics: ReaxFF Molecular Dynamics Study on the Co-Pyrolysis of Polypropylene and EPDM/POE. Energy 2023, 280, 128202. [Google Scholar] [CrossRef]
- Li, X.; Han, Y.; Qu, J.; Chen, Q.; Wei, Y.; Hou, G.; Liu, J. ReaxFF Molecular Dynamics Simulation of the Thermal Decomposition Reaction of Bio-Based Polyester Materials. Phys. Chem. Chem. Phys. 2023, 25, 9445–9453. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhang, L.; Zou, L.; Ayubi, B.I.; Wang, Y. Mechanism Analysis and Potential Applications of Atomic Oxygen Erosion Protection for Kapton-Type Polyimide Based on Molecular Dynamics Simulations. Polymers 2024, 16, 1687. [Google Scholar] [CrossRef]
- Chen, T.B.Y.; Yuen, A.C.Y.; Lin, B.; Liu, L.; Lo, A.L.P.; Chan, Q.N.; Zhang, J.; Cheung, S.C.P.; Yeoh, G.H. Characterisation of Pyrolysis Kinetics and Detailed Gas Species Formations of Engineering Polymers via Reactive Molecular Dynamics (ReaxFF). J. Anal. Appl. Pyrolysis 2021, 153, 104931. [Google Scholar] [CrossRef]
- Zhao, T.; Li, T.; Xin, Z.; Zou, L.; Zhang, L. A ReaxFF-Based Molecular Dynamics Simulation of the Pyrolysis Mechanism for Polycarbonate. Energy Fuels 2018, 32, 2156–2162. [Google Scholar] [CrossRef]
- Deepa, P.; Sona, C.; Jayakannan, M. Synthesis and Investigation of the Effect of Nematic Phases on the Glass-Transition Behavior of Novel Cycloaliphatic Liquid-Crystalline Poly(Ester Amide)s. J. Polym. Sci. Part Polym. Chem. 2006, 44, 5557–5571. [Google Scholar] [CrossRef]
- Guo, W.; Fan, K.; Guo, G.; Wang, J. Atomic-Scale Insight into Thermal Decomposition Behavior of Polypropylene: A ReaxFF Method. Polym. Degrad. Stab. 2022, 202, 110038. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Kato, T.; Yamada, Y.; Nishikawa, Y.; Ishikawa, H.; Sato, S. Carbonization Mechanisms of Polyimide: Methodology to Analyze Carbon Materials with Nitrogen, Oxygen, Pentagons, and Heptagons. Carbon 2021, 178, 58–80. [Google Scholar] [CrossRef]
- Chen, J.; Meng, T.; Wang, Q.; Bai, Y.; Jiaqiang, E.; Leng, E.; Zhang, F.; Liao, G. Study on the Mechanisms of Epoxy Resin Gasification in Supercritical Water by Molecular Dynamics and Experimental Methods. Chem. Eng. J. 2022, 433, 133828. [Google Scholar] [CrossRef]
- Tan, S.; Do, D.D.; Nicholson, D. Consistency of NVT, NPT, µVT and Gibbs (NV2T and NPT) with Kinetic Monte Carlo Schemes. Chem. Eng. J. 2020, 401, 126056. [Google Scholar] [CrossRef]
- Bosko, J.T.; Todd, B.D.; Sadus, R.J. Molecular Simulation of Dendrimers and Their Mixtures under Shear: Comparison of Isothermal-Isobaric (NpT) and Isothermal-Isochoric (NVT) Ensemble Systems. J. Chem. Phys. 2005, 123, 034905. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Di, J.; Wu, Y.; Meng, X.; Ji, H.; Jiang, H.; Li, J.; Lu, Q. Insights into the Pyrolysis Mechanisms of Epoxy Resin Polymers Based on the Combination of Experiments and ReaxFF-MD Simulation. Chem. Eng. J. 2023, 473, 145404. [Google Scholar] [CrossRef]
- Zhang, F.; Cao, Y.; Liu, X.; Xu, H.; Lu, D.; Yang, R. How Small Molecules Affect the Thermo-Oxidative Aging Mechanism of Polypropylene: A Reactive Molecular Dynamics Study. Polymers 2021, 13, 1243. [Google Scholar] [CrossRef]
- Sai, T.; Ran, S.; Guo, Z.; Song, P.; Fang, Z. Recent Advances in Fire-Retardant Carbon-Based Polymeric Nanocomposites through Fighting Free Radicals. SusMat 2022, 2, 411–434. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Fragments | C1 | C2 | C3–5 | C6–8 | C9+ | |
|---|---|---|---|---|---|---|
| 2000 K | 135 | 26 | 8 | 15 | 34 | 47 |
| 2500 K | 327 | 103 | 24 | 35 | 32 | 46 |
| 3000 K | 484 | 136 | 21 | 14 | 7 | 6 |
| 3500 K | 690 | 158 | 31 | 15 | 15 | 13 |
| 4000 K | 816 | 162 | 22 | 19 | 9 | 16 |
| H2 | CO | H2O | CH3N | C2H2 | C2H4 | |
|---|---|---|---|---|---|---|
| 2000 K | 4 | 14 | 2 | 9 | 0 | 5 |
| 2500 K | 65 | 62 | 15 | 13 | 1 | 16 |
| 3000 K | 257 | 94 | 37 | 3 | 32 | 7 |
| 3500 K | 364 | 121 | 39 | 0 | 35 | 1 |
| 4000 K | 484 | 137 | 38 | 0 | 25 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zheng, Y.; Zhang, Q.; Wu, K.; Yu, Q.; Yang, J. Simulation of the Pyrolysis Process of Cyclohexane-Containing Semi-Aromatic Polyamide Based on ReaxFF-MD. Polymers 2025, 17, 1593. https://doi.org/10.3390/polym17121593
Zhang X, Zheng Y, Zhang Q, Wu K, Yu Q, Yang J. Simulation of the Pyrolysis Process of Cyclohexane-Containing Semi-Aromatic Polyamide Based on ReaxFF-MD. Polymers. 2025; 17(12):1593. https://doi.org/10.3390/polym17121593
Chicago/Turabian StyleZhang, Xiaotong, Yuanbo Zheng, Qian Zhang, Kai Wu, Qinwei Yu, and Jianming Yang. 2025. "Simulation of the Pyrolysis Process of Cyclohexane-Containing Semi-Aromatic Polyamide Based on ReaxFF-MD" Polymers 17, no. 12: 1593. https://doi.org/10.3390/polym17121593
APA StyleZhang, X., Zheng, Y., Zhang, Q., Wu, K., Yu, Q., & Yang, J. (2025). Simulation of the Pyrolysis Process of Cyclohexane-Containing Semi-Aromatic Polyamide Based on ReaxFF-MD. Polymers, 17(12), 1593. https://doi.org/10.3390/polym17121593