
Impact of Acid Hydrolysis on Morphology, Rheology, Mechanical Properties, and Processing of Thermoplastic Starch

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Hydrolyzed Starch
2.3. Preparation of Thermoplastic Starch
2.3.1. TPS Prepared by Single-Step Solution Casting
2.3.2. TPS Prepared by Two-Step Protocol: Solution Casting Followed by Melt Mixing
2.3.3. Storing of the Samples in Defined Conditions
2.4. Characterization Methods
2.4.1. Light and Electron Microscopy
2.4.2. Vibrational Spectroscopy
2.4.3. Wide-Angle X-Ray Scattering
2.4.4. Dynamic Mechanical Thermal Analysis
2.4.5. Rheology
2.4.6. Microindentation Hardness Testing
2.4.7. In Situ Measurements During Melt Mixing
3. Results and Discussion
3.1. Morphology and Homogeneity
3.1.1. Light and Electron Microscopy
3.1.2. Vibrational Spectroscopy
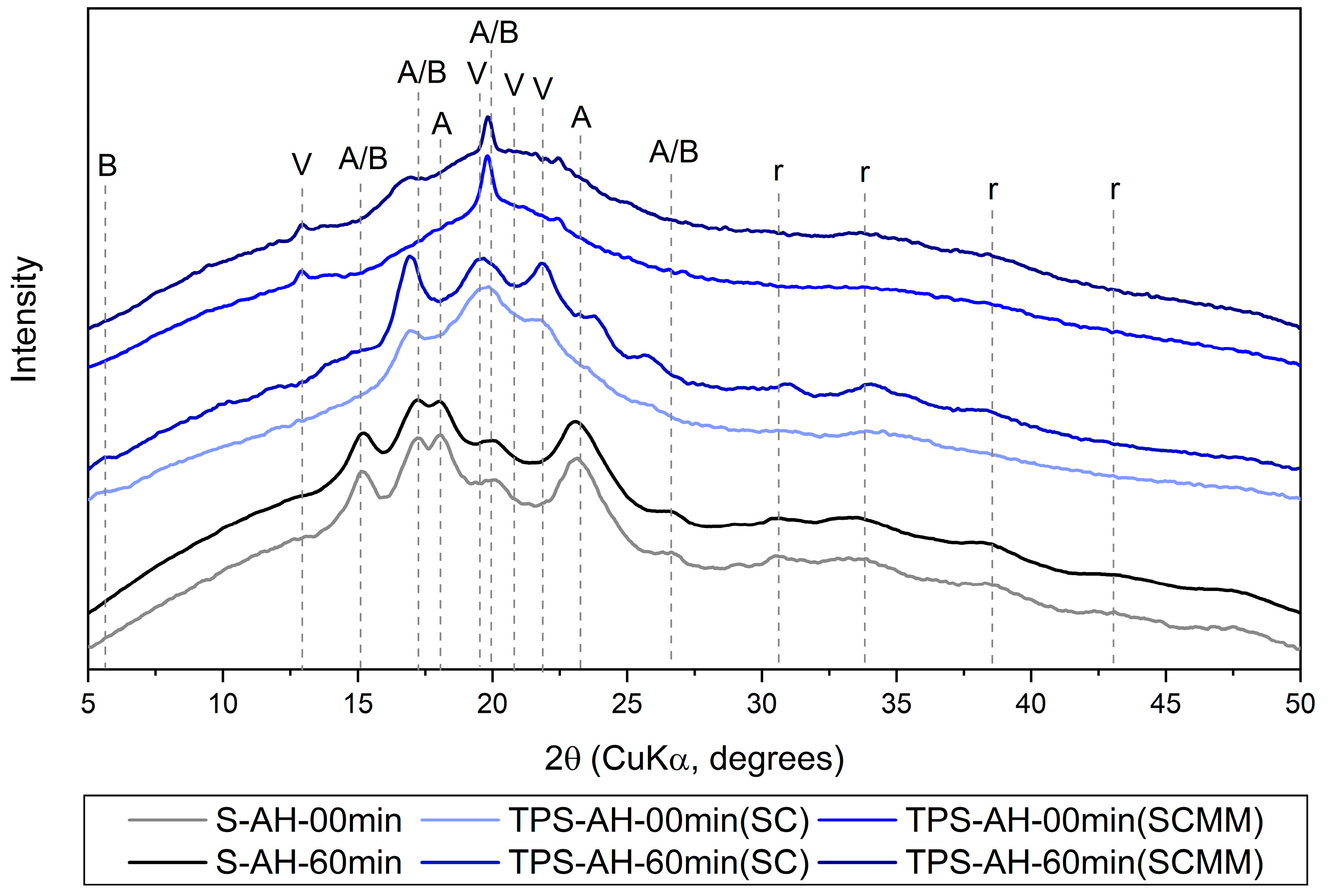
3.1.3. Wide-Angle X-Ray Scattering
3.2. Mechanical and Rheological Properties
3.2.1. Dynamic Mechanical Thermal Analysis
3.2.2. Rheological Properties
3.2.3. Micromechanical Properties
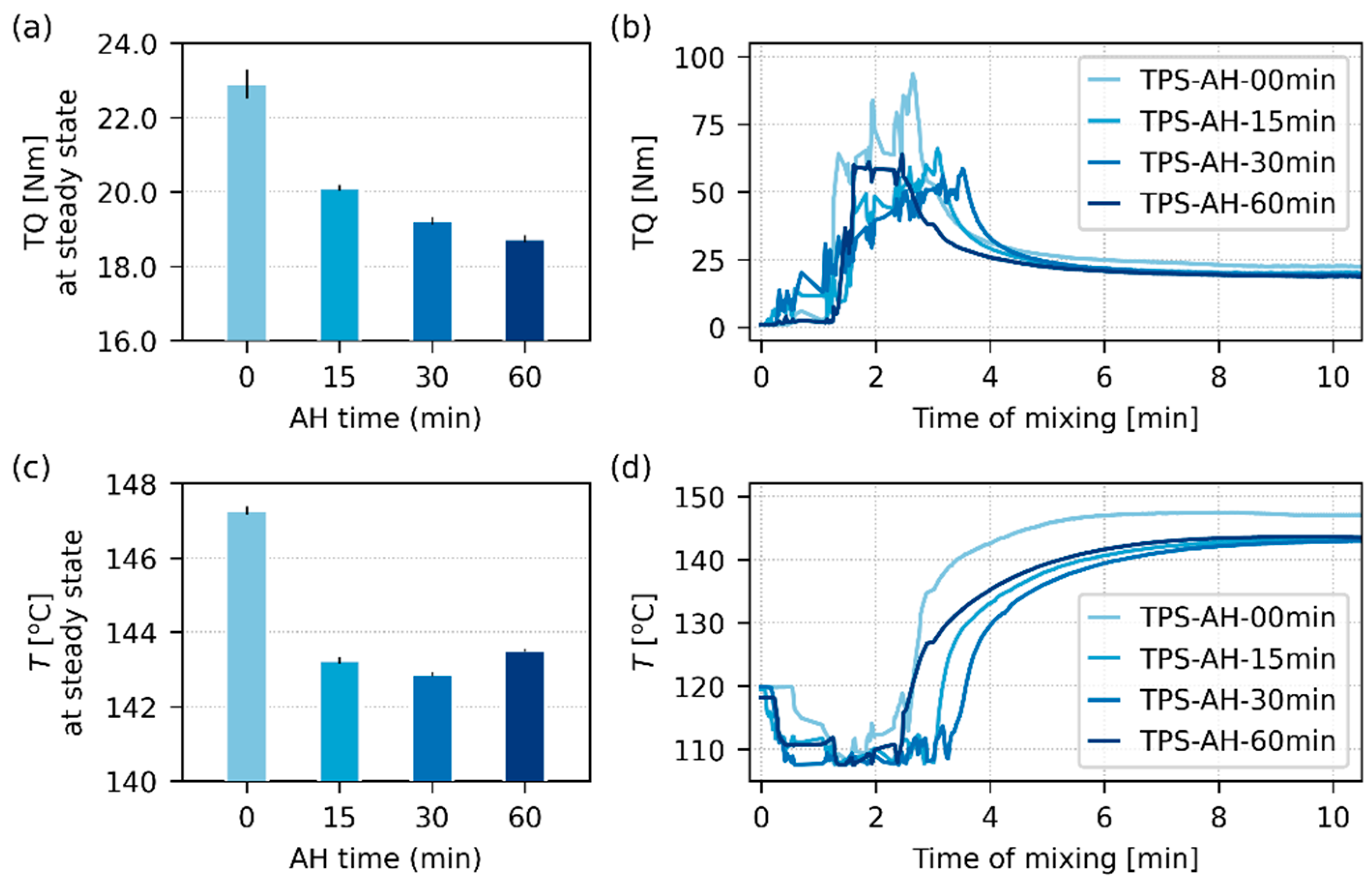
3.3. Processing of Acid-Hydrolyzed Starches
4. Conclusions
- The acid hydrolysis preferentially targeted the amorphous regions, which decreased the average molar mass and viscosity of the amorphous matrix. During the SC and MM, the lower-viscosity matrix in the AH-treated starches resulted in lower disruption of semicrystalline regions, a slightly coarser morphology (as observed by PLM), and higher crystallinity (as evaluated from WAXS).
- The fact that TPS after AH exhibited a less viscous, softer amorphous phase and higher fraction of stiffer crystalline phase resulted in similar thermomechanical, rheological, and micromechanical properties of all prepared systems (as evidenced by DMTA, rheometry, and MHI), because the two phenomena tended to cancel out.
- During the melt mixing, when the material was fully molten and forced to flow, the shorter molecules in all AH-treated TPSs started to act as a lubricant and decreased both torque (TQ) and processing temperature (T), as proven by in situ measurements in the kneading chamber during the MM. Even if the changes in TQ and T were quite moderate, we have demonstrated that AH is a feasible approach to save energy during TPS processing and/or to mitigate the negative effect of the MM on possible temperature-sensitive admixtures.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TPS | Thermoplastic starch |
| SC | Solution casting |
| MM | Melt mixing |
| SC+MM | Solution casting followed by melt mixing |
| AH | Acid hydrolysis |
| LM | Light microscopy |
| PLM | Polarized light microscopy |
| SEM | Scanning electron microscopy |
| FTIR | Fourier-transform infrared spectra |
| WAXS | Wide-angle X-ray scattering |
| DMTA | Dynamic mechanical thermal analysis |
| LVER | Linear viscoelasticity region |
| MHI | Microindentation hardness testing |
Appendix A. Additional TPS Characterization Results



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WAXS Diffraction | Observation of the Diffraction Peaks | Starch Structure Types | ||||
|---|---|---|---|---|---|---|
| 2θ (°) | Intensity 1 | S (Powder) | TPS (SC) | TPS (SC+MM) | Structure 2 | References |
| 5.3 | w | no | yes | no | B | [2] |
| 13.5 | m | no | no | yes | V | [50,53] |
| 15.1 | s | yes | no | no | A/B | [30] |
| 17.0 | s | yes | yes | no | A/B | [2,30] |
| 18.1 | s | yes | no | no | A | [30] |
| 19.7 | s | no | no | yes | V | [50,53] |
| 20.0 | s | yes | yes | no | A/B | [2,30] |
| 20.8 | w | no | no | yes | V | [50,53] |
| 21.8 | m | no | yes | no | V | [2] |
| 23.2 | w | yes | no | no | A | [30] |
| 26.6 | m | yes | yes | no | A/B | [30] |
| 30.6 | w | yes | yes | no | r | [29,66,67] |
| 33.4 | w | yes | yes | yes | r | [29,66,67] |
| 38.4 | w | yes | yes | yes | r | [29,66] |
| 43.1 | w | yes | no | no | r | [29,66] |
Appendix B. More Details About TPS Micromechanical Properties

References
- He, Z.; Chi, C.; Huang, S.; Li, X. A novel method for obtaining high amylose starch fractions from debranched starch. Curr. Res. Food Sci. 2023, 7, 100589. [Google Scholar] [CrossRef] [PubMed]
- Rana, L.; Kouka, S.; Gajdosova, V.; Strachota, B.; Konefał, M.; Pokorny, V.; Pavlova, E.; Stary, Z.; Lukes, J.; Patocka, M.; et al. Thermoplastic Starch with Maltodextrin: Preparation, Morphology, Rheology, and Mechanical Properties. Materials 2024, 17, 5474. [Google Scholar] [CrossRef] [PubMed]
- Buléon, A.; Colona, P.; Planchot, V.; Ball, S. Starch granules: Structure and biosynthesis. Int. J. Biol. Macromol. 1998, 23, 85–112. [Google Scholar] [CrossRef]
- Ai, Y.; Jane, J. Gelatinization and rheological properties of starch. Starch-Stärke 2015, 67, 213–224. [Google Scholar] [CrossRef]
- Suortti, T.; Gorenstein, M.V.; Roger, P. Determination of the molecular mass of amylose. J. Chromatogr. A 1998, 828, 515–521. [Google Scholar] [CrossRef]
- Cui, C.; Ji, N.; Wang, Y.; Xiong, L.; Sun, Q. Bioactive and intelligent starch-based films: A review. Trends Food Sci. Technol. 2021, 116, 854–869. [Google Scholar] [CrossRef]
- Mizuno, A.; Mitsuiki, M.; Motoki, M. Effect of Crystallinity on the Glass Transition Temperature of Starch. J. Agric. Food Chem. 1998, 46, 98–103. [Google Scholar] [CrossRef]
- Primo-Martín, C.; van Nieuwenhuijzen, N.H.; Hamer, R.J.; van Vliet, T. Crystallinity changes in wheat starch during the bread-making process: Starch crystallinity in the bread crust. J. Cereal Sci. 2007, 45, 219–226. [Google Scholar] [CrossRef]
- Temesgen, S.; Rennert, M.; Tesfaye, T.; Großmann, L.; Kuehnert, I.; Smolka, N.; Nase, M. Thermal, morphological, and structural characterization of starch-based bio-polymers for melt spinnability. e-Polymers 2024, 24, 20240025. [Google Scholar] [CrossRef]
- Ostafinska, A.; Fortelny, I.; Nevoralova, M.; Hodan, J.; Kredatusova, J.; Slouf, M. Synergistic effects in mechanical properties of PLA/PCL blends with optimized composition, processing, and morphology. RSC Adv. 2015, 5, 98971–98982. [Google Scholar] [CrossRef]
- Thakur, R.; Pristijono, P.; Scarlett, C.J.; Bowyer, M.; Singh, S.P.; Vuong, Q.V. Starch-based films: Major factors affecting their properties. Int. J. Biol. Macromol. 2019, 132, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, B.; Zhang, Y.; Xu, F.; Zhu, K.; Li, S.; Tan, L.; Wu, G.; Dong, W. Effect of degree of polymerization of amylopectin on the gelatinization properties of jackfruit seed starch. Food Chem. 2019, 289, 152–159. [Google Scholar] [CrossRef]
- Campos, A.D.; Sena Neto, A.R.D.; Rodrigues, V.B.; Luchesi, B.R.; Moreira, F.K.V.; Correa, A.C.; Mattoso, L.H.C.; Marconcini, J.M. Bionanocomposites produced from cassava starch and oil palm mesocarp cellulose nanowhiskers. Carbohydr. Polym. 2017, 175, 330–336. [Google Scholar] [CrossRef]
- Campos-Requena, V.H.; Rivas, B.L.; Pérez, M.A.; Figueroa, C.R.; Figueroa, N.E.; Sanfuentes, E.A. Thermoplastic starch/clay nanocomposites loaded with essential oil constituents as packaging for strawberries—In vivo antimicrobial synergy over Botrytis cinerea. Postharvest Biol. Technol. 2017, 129, 29–36. [Google Scholar] [CrossRef]
- Ostafińska, A.; Mikešová, J.; Krejčíková, S.; Nevoralová, M.; Šturcová, A.; Zhigunov, A.; Michálková, D.; Šlouf, M. Thermoplastic starch composites with TiO2 particles: Preparation, morphology, rheology and mechanical properties. Int. J. Biol. Macromol. 2017, 101, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Huneault, M.A.; Li, H. Preparation and properties of extruded thermoplastic starch/polymer blends. J. Appl. Polym. Sci. 2012, 126, E96–E108. [Google Scholar] [CrossRef]
- Mezger, T.G. Oscillatory tests. In The Rheology Handbook, 4th ed.; Vincentz Network: Hannover, Germany, 2014. [Google Scholar]
- Zeng, F.; Ma, F.; Kong, F.; Gao, Q.; Yu, S. Physicochemical properties and digestibility of hydrothermally treated waxy rice starch. Food Chem. 2015, 172, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Jambrak, A.R.; Herceg, Z.; Šubarić, D.; Babić, J.; Brnčić, M.; Brnčić, S.R.; Bosiljkov, T.; Čvek, D.; Tripalo, B.; Gelo, J. Ultrasound effect on physical properties of corn starch. Carbohydr. Polym. 2010, 79, 91–100. [Google Scholar] [CrossRef]
- Kumar, V.; Kumarasamy, V.; Bhatt, P.; Dixit, R.; Kumar, M.; Shukla, C.P.; Subramaniyan, V.; Kumar, S. Ultrasound assisted techniques for starch modification to develop novel drug delivery systems: A comprehensive study. J. Bioact. Compat. Polym. 2024, 39, 279–297. [Google Scholar] [CrossRef]
- Yan, X.; Wei, H.; Kou, L.; Ren, L.; Zhou, J. Acid hydrolysis of amylose granules and effect of molecular weight on properties of ethanol precipitated amylose nanoparticles. Carbohydr. Polym. 2021, 252, 117243. [Google Scholar] [CrossRef]
- Karma, V.; Gupta, A.D.; Yadav, D.K.; Singh, A.A.; Verma, M.; Singh, H. Recent Developments in Starch Modification by Organic Acids: A Review. Starch-Stärke 2022, 74, 2200025. [Google Scholar] [CrossRef]
- Wang, S.; Copeland, L. Effect of Acid Hydrolysis on Starch Structure and Functionality: A Review. Crit. Rev. Food Sci. Nutr. 2015, 55, 1081–1097. [Google Scholar] [CrossRef]
- Beninca, C.; Demiate, I.M.; Lacerda, L.G.; Filho, M.A.d.S.C.; Ionashiro, M.; Schnitzler, E. Thermal behavior of corn starch granules modified by acid treatment at 30 and 50 °C. Eclética Química 2008, 33, 13–17. [Google Scholar] [CrossRef]
- Xia, L.; Wenyuan, G.; Juan, W.; Qianqian, J.; Luqi, H. Comparison of the morphological, crystalline, and thermal properties of different crystalline types of starches after acid hydrolysis. Starch-Stärke 2010, 62, 686–696. [Google Scholar] [CrossRef]
- Sanchez de la Concha, B.B.; Agama-Acevedo, E.; Nuñez-Santiago, M.C.; Bello-Perez, L.A.; Garcia, H.S.; Alvarez-Ramirez, J. Acid hydrolysis of waxy starches with different granule size for nanocrystal production. J. Cereal Sci. 2018, 79, 193–200. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Truong, V.-D.; Wang, L. Structures and rheological properties of corn starch as affected by acid hydrolysis. Carbohydr. Polym. 2003, 52, 327–333. [Google Scholar] [CrossRef]
- Ulbrich, M.; Bai, Y.; Flöter, E. The supporting effect of ultrasound on the acid hydrolysis of granular potato starch. Carbohydr. Polym. 2020, 230, 115633. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Hong, Y.; Gu, Z.; Cheng, L.; Li, Z.; Li, C. Effects of acid hydrolysis intensity on the properties of starch/xanthan mixtures. Int. J. Biol. Macromol. 2018, 106, 320–329. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, H.; Liu, P.; Wang, W.; Dong, H. Effects of acid hydrolysis on the physicochemical properties of pea starch and its film forming capacity. Food Hydrocoll. 2019, 87, 173–179. [Google Scholar] [CrossRef]
- Gajdosova, V.; Strachota, B.; Strachota, A.; Michalkova, D.; Krejcikova, S.; Fulin, P.; Nyc, O.; Brinek, A.; Zemek, M.; Slouf, M. Biodegradable Thermoplastic Starch/Polycaprolactone Blends with Co-Continuous Morphology Suitable for Local Release of Antibiotics. Materials 2022, 15, 1101. [Google Scholar] [CrossRef]
- Atichokudomchai, N.; Shobsngob, S.; Varavinit, S. Morphological Properties of Acid-modified Tapioca Starch. Starch-Stärke 2000, 52, 283–289. [Google Scholar] [CrossRef]
- Ujcic, A.; Krejcikova, S.; Nevoralova, M.; Zhigunov, A.; Dybal, J.; Krulis, Z.; Fulin, P.; Nyc, O.; Slouf, M. Thermoplastic Starch Composites with Titanium Dioxide and Vancomycin Antibiotic: Preparation, Morphology, Thermomechanical Properties, and Antimicrobial Susceptibility Testing. Front. Mater. 2020, 7, 9. [Google Scholar] [CrossRef]
- Palacký, J.; Mojzeš, P.; Bok, J. SVD-based method for intensity normalization, background correction and solvent subtraction in Raman spectroscopy exploiting the properties of water stretching vibrations. J. Raman Spectrosc. 2011, 42, 1528–1539. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Oliver, W.C.; Pharr, G.M. Nanoindentation in materials research: Past, present, and future. MRS Bull. 2010, 35, 897–907. [Google Scholar] [CrossRef]
- Slouf, M.; Krajenta, J.; Gajdosova, V.; Pawlak, A. Macromechanical and micromechanical properties of polymers with reduced density of entanglements. Polym. Eng. Sci. 2021, 61, 1773–1790. [Google Scholar] [CrossRef]
- Fischer-Cripps, A.C. Nanoindentation. In Mechanical Engineering Series; Springer: New York, NY, USA, 2004. [Google Scholar] [CrossRef]
- Slouf, M.; Henning, S. Micromechanical Properties. In Encyclopedia of Polymer Science and Technology, 3rd ed.; Mark, H.F., Ed.; Wiley: New York, NY, USA, 2022; pp. 1–50. [Google Scholar]
- Slouf, M.; Strachota, B.; Strachota, A.; Gajdosova, V.; Bertschova, V.; Nohava, J. Macro-, Micro- and Nanomechanical Characterization of Crosslinked Polymers with Very Broad Range of Mechanical Properties. Polymers 2020, 12, 2951. [Google Scholar] [CrossRef]
- Slouf, M.; Arevalo, S.; Vlkova, H.; Gajdosova, V.; Kralik, V.; Pruitt, L. Comparison of macro-, micro- and nanomechanical properties of clinically-relevant UHMWPE formulations. J. Mech. Behav. Biomed. Mater. 2021, 120, 104205. [Google Scholar] [CrossRef]
- Gajdošová, V.; Špírková, M.; Aguilar Costumbre, Y.; Krejčíková, S.; Strachota, B.; Šlouf, M.; Strachota, A. Morphology, Micromechanical, and Macromechanical Properties of Novel Waterborne Poly(urethane-urea)/Silica Nanocomposites. Materials 2023, 16, 1767. [Google Scholar] [CrossRef]
- Hoover, R. Acid-Treated Starches. Food Rev. Int. 2000, 16, 369–392. [Google Scholar] [CrossRef]
- Fortelný, I.; Jůza, J. Description of the Droplet Size Evolution in Flowing Immiscible Polymer Blends. Polymers 2019, 11, 761. [Google Scholar] [CrossRef]
- Ostafinska, A.; Fortelný, I.; Hodan, J.; Krejčíková, S.; Nevoralová, M.; Kredatusová, J.; Kruliš, Z.; Kotek, J.; Šlouf, M. Strong synergistic effects in PLA/PCL blends: Impact of PLA matrix viscosity. J. Mech. Behav. Biomed. Mater. 2017, 69, 229–241. [Google Scholar] [CrossRef]
- Almeida, M.R.; Alves, R.S.; Nascimbem, L.B.L.R.; Stephani, R.; Poppi, R.J.; De Oliveira, L.F.C. Determination of amylose content in starch using Raman spectroscopy and multivariate calibration analysis. Anal. Bioanal. Chem. 2010, 397, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Basiak, E.; Lenart, A.; Debeaufort, F. How Glycerol and Water Contents Affect the Structural and Functional Properties of Starch-Based Edible Films. Polymers 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Basilio-Cortés, U.A.; González-Cruz, L.; Velazquez, G.; Teniente-Martínez, G.; Gómez-Aldapa, C.A.; Castro-Rosas, J.; Bernardino-Nicanor, A. Effect of Dual Modification on the Spectroscopic, Calorimetric, Viscosimetric and Morphological Characteristics of Corn Starch. Polymers 2019, 11, 333. [Google Scholar] [CrossRef]
- Chung, H.; Arnold, M.A. Near-Infrared Spectroscopy for Monitoring Starch Hydrolysis. Appl. Spectrosc. 2000, 54, 277–283. [Google Scholar] [CrossRef]
- Shi, R.; Liu, Q.; Ding, T.; Han, Y.; Zhang, L.; Chen, D.; Tian, W. Ageing of soft thermoplastic starch with high glycerol content. J. Appl. Polym. Sci. 2007, 103, 574–586. [Google Scholar] [CrossRef]
- Slouf, M.; Gajdosova, V.; Dybal, J.; Sticha, R.; Fulin, P.; Pokorny, D.; Mateo, J.; Panisello, J.J.; Canales, V.; Medel, F.; et al. European Database of Explanted UHMWPE Liners from Total Joint Replacements: Correlations among Polymer Modifications, Structure, Oxidation, Mechanical Properties and Lifetime In Vivo. Polymers 2023, 15, 568. [Google Scholar] [CrossRef]
- Slouf, M.; Synkova, H.; Baldrian, J.; Marek, A.; Kovarova, J.; Schmidt, P.; Dorschner, H.; Stephan, M.; Gohs, U. Structural changes of UHMWPE after e-beam irradiation and thermal treatment. J. Biomed. Mater. Res. 2008, 85B, 240–251. [Google Scholar] [CrossRef]
- Van Soest, J.J.G.; Hulleman, S.H.D.; De Wit, D.; Vliegenthart, J.F.G. Crystallinity in starch bioplastics. Ind. Crops Prod. 1996, 5, 11–22. [Google Scholar] [CrossRef]
- Schmitt, H.; Guidez, A.; Prashantha, K.; Soulestin, J.; Lacrampe, M.F.; Krawczak, P. Studies on the effect of storage time and plasticizers on the structural variations in thermoplastic starch. Carbohydr. Polym. 2015, 115, 364–372. [Google Scholar] [CrossRef]
- Van Soest, J.J.G.; Benes, K.; De Wit, D. The Influence of Acid Hydrolysis of Potato Starch on the Stress-Strain Propoerties of Thermoplastic Starch. Starch-Stärke 1995, 47, 429–434. [Google Scholar] [CrossRef]
- Angellier, H.; Putaux, J.; Molina-Boisseau, S.; Dupeyre, D.; Dufresne, A. Starch Nanocrystal Fillers in an Acrylic Polymer Matrix. Macromol. Symp. 2005, 221, 95–104. [Google Scholar] [CrossRef]
- Ujcic, A.; Nevoralova, M.; Dybal, J.; Zhigunov, A.; Kredatusova, J.; Krejcikova, S.; Fortelny, I.; Slouf, M. Thermoplastic Starch Composites Filled with Isometric and Elongated TiO2-Based Nanoparticles. Front. Mater. 2019, 6, 284. [Google Scholar] [CrossRef]
- Tabor, D. The Hardness of Metals; Clarendon Press: Oxford, UK; Oxford University Press: Oxford, UK, 1951. [Google Scholar]
- Struik, L.C.E. Some problems in the non-linear viscoelasticity of amorphous glassy polymers. J. Non-Cryst. Solids 1991, 131–133, 395–407. [Google Scholar] [CrossRef]
- Fulin, P.; Gajdosova, V.; Sloufova, I.; Hodan, J.; Pokorny, D.; Slouf, M. Comparison of various UHMWPE formulations from contemporary total knee replacements before and after accelerated aging. Mater. Des. 2025, 252, 113795. [Google Scholar] [CrossRef]
- Slouf, M.; Pavlova, E.; Krejcikova, S.; Ostafinska, A.; Zhigunov, A.; Krzyzanek, V.; Sowinski, P.; Piorkowska, E. Relations between morphology and micromechanical properties of alpha, beta and gamma phases of iPP. Polym. Test. 2018, 67, 522–532. [Google Scholar] [CrossRef]
- Slouf, M.; Steinhart, M.; Nemecek, P.; Gajdosova, V.; Hodan, J. Correlations between Microscale Indentation Creep and Macroscale Tensile Creep of Polymers. Materials 2023, 16, 834. [Google Scholar] [CrossRef]
- Flores, A.; Ania, F.; Baltá-Calleja, F.J. From the glassy state to ordered polymer structures: A microhardness study. Polymer 2009, 50, 729–746. [Google Scholar] [CrossRef]
- Zhu, Y.; Guo, F.; Li, J.; Wang, Z.; Liang, Z.; Yi, C. Development of a Novel Energy Saving and Environmentally Friendly Starch via a Graft Copolymerization Strategy for Efficient Warp Sizing and Easy Removal. Polymers 2024, 16, 182. [Google Scholar] [CrossRef]
- Jain, S.; Goossens, J.G.P.; Peters, G.W.M.; Van Duin, M.; Lemstra, P.J. Strong decrease in viscosity of nanoparticle-filled polymer melts through selective adsorption. Soft Matter 2008, 4, 1848. [Google Scholar] [CrossRef]
- Zuo, Y.; Gu, J.; Tan, H.; Qiao, Z.; Xie, Y.; Zhang, Y. The characterization of granule structural changes in acid-thinning starches by new methods and its effect on other properties. J. Adhes. Sci. Technol. 2014, 28, 479–489. [Google Scholar] [CrossRef]
- Pozo, C.; Rodríguez-Llamazares, S.; Bouza, R.; Barral, L.; Castaño, J.; Müller, N.; Restrepo, I. Study of the structural order of native starch granules using combined FTIR and XRD analysis. J. Polym. Res. 2018, 25, 266. [Google Scholar] [CrossRef]

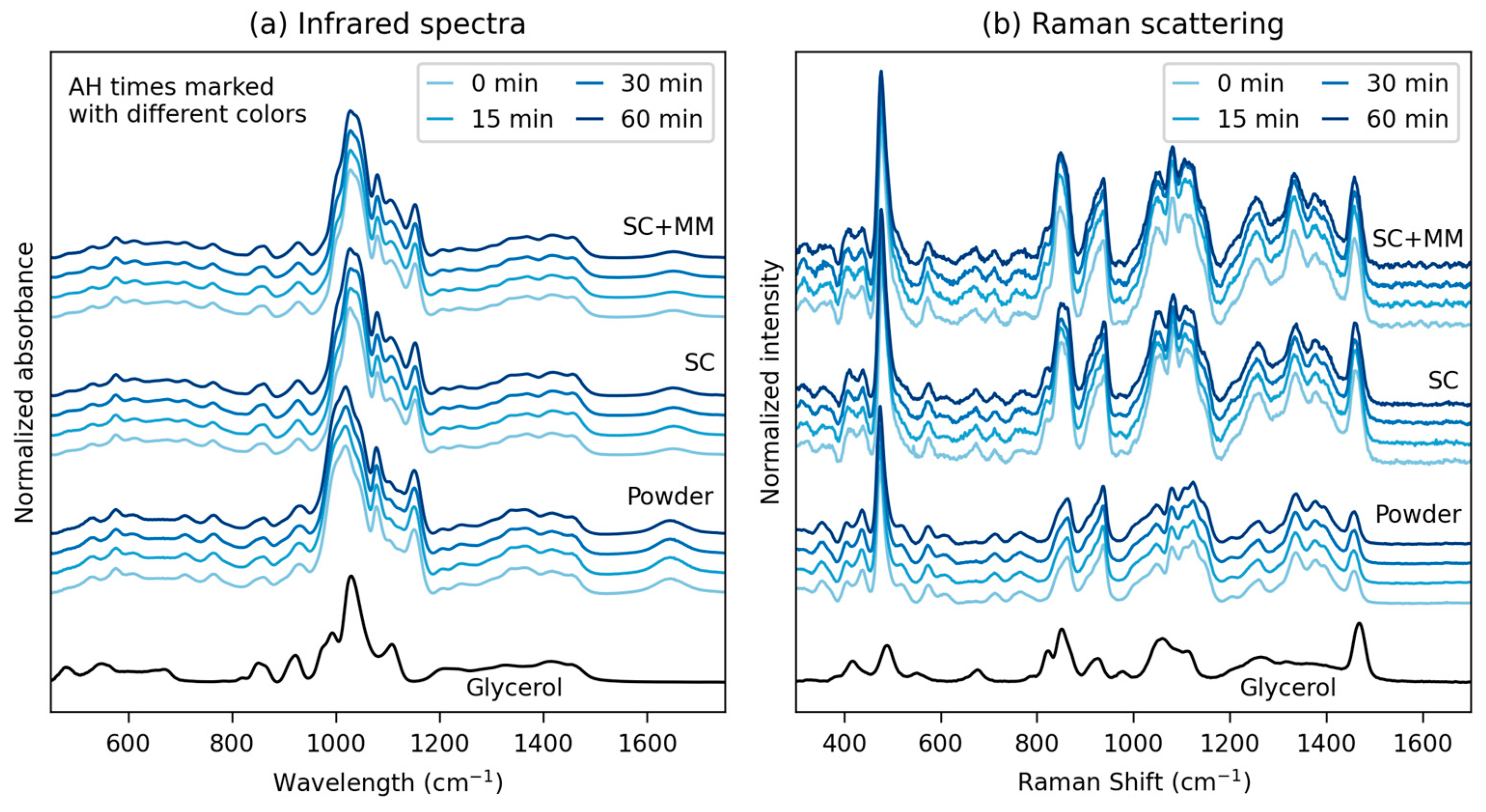


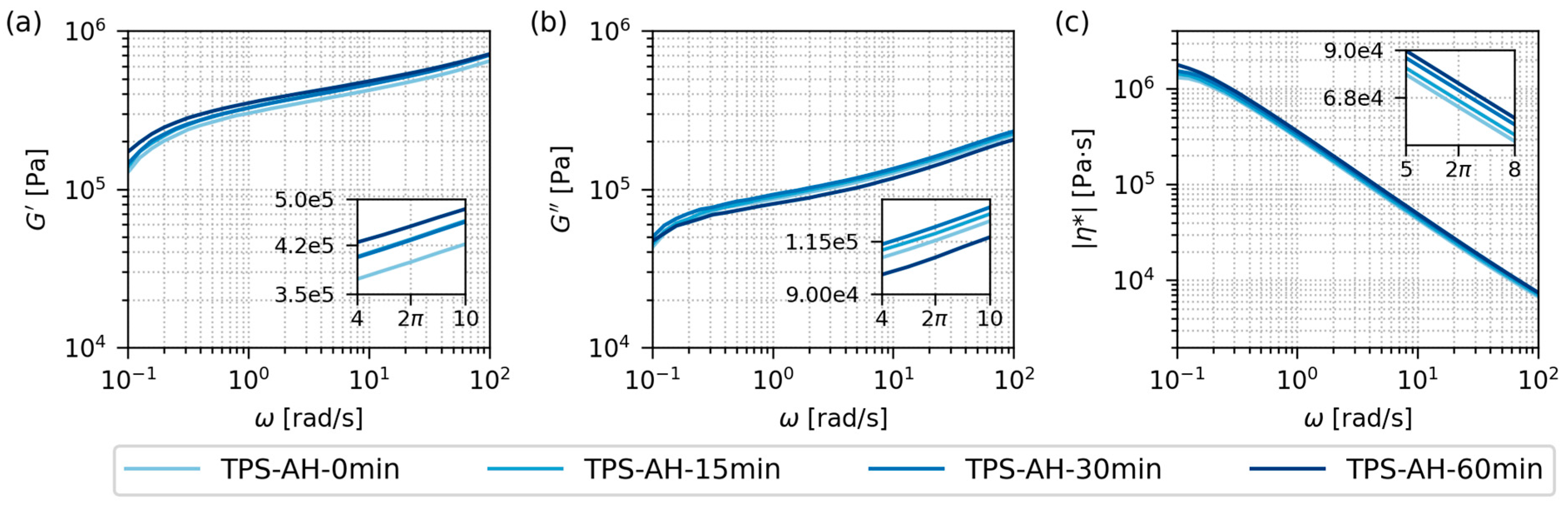
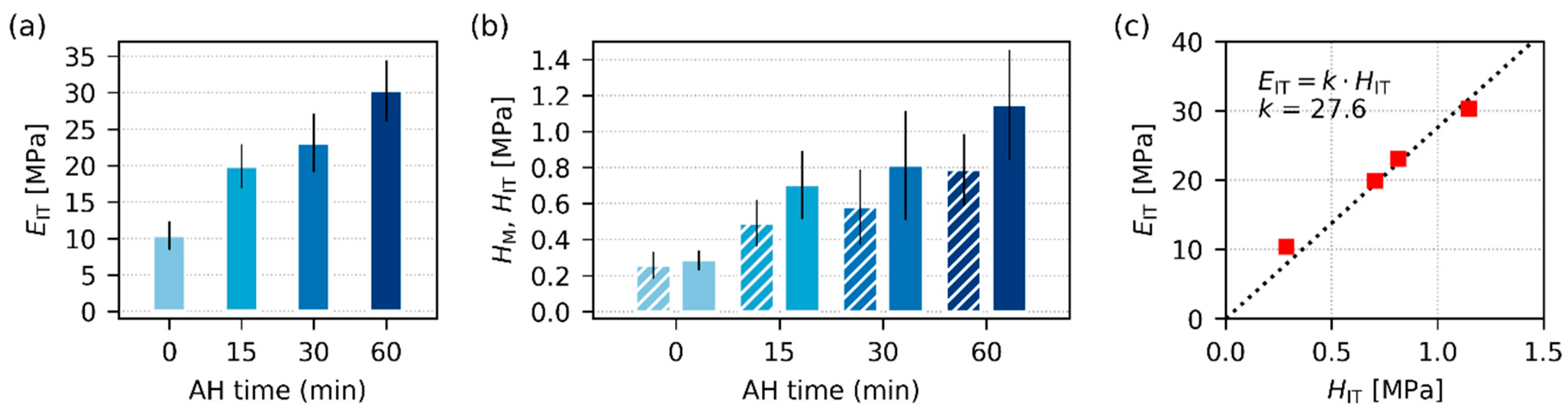

| Native Starches * | Thermoplastic Starches ** | AH Time (min) *** |
|---|---|---|
| S-AH-00min | TPS-AH-00min | 0 |
| S-AH-15min | TPS-AH-15min | 15 |
| S-AH-30min | TPS-AH-30min | 30 |
| S-AH-60min | TPS-AH-60min | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouka, S.; Gajdosova, V.; Strachota, B.; Sloufova, I.; Kuzel, R.; Stary, Z.; Slouf, M. Impact of Acid Hydrolysis on Morphology, Rheology, Mechanical Properties, and Processing of Thermoplastic Starch. Polymers 2025, 17, 1310. https://doi.org/10.3390/polym17101310
Kouka S, Gajdosova V, Strachota B, Sloufova I, Kuzel R, Stary Z, Slouf M. Impact of Acid Hydrolysis on Morphology, Rheology, Mechanical Properties, and Processing of Thermoplastic Starch. Polymers. 2025; 17(10):1310. https://doi.org/10.3390/polym17101310
Chicago/Turabian StyleKouka, Saffana, Veronika Gajdosova, Beata Strachota, Ivana Sloufova, Radomir Kuzel, Zdenek Stary, and Miroslav Slouf. 2025. "Impact of Acid Hydrolysis on Morphology, Rheology, Mechanical Properties, and Processing of Thermoplastic Starch" Polymers 17, no. 10: 1310. https://doi.org/10.3390/polym17101310
APA StyleKouka, S., Gajdosova, V., Strachota, B., Sloufova, I., Kuzel, R., Stary, Z., & Slouf, M. (2025). Impact of Acid Hydrolysis on Morphology, Rheology, Mechanical Properties, and Processing of Thermoplastic Starch. Polymers, 17(10), 1310. https://doi.org/10.3390/polym17101310