
Non-Coagulant Spinning of High-Strength Fibers from Homopolymer Polyacrylonitrile Synthesized via Anionic Polymerisation

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Solutions
- -
- For solutions with a polymer concentration of 1–5 wt%, the mixture was stirred for 24 h at 50 °C using a magnetic stirrer. For concentrations below 1 wt%, the solution was sequentially diluted inside a Ubbelohde viscometer at 25 °C to determine the intrinsic viscosity.
- -
- Highly viscous solutions with a polymer content greater than 5 wt% were prepared using a paddle mixer with a J-shaped rotor. Mixing was conducted at 60 rpm for 24 h at 70 °C.
- -
- High-viscosity spinning solutions with concentrations above 20 wt% were prepared using a rotor speed of 10 rpm for 72 h at 70 °C.
2.2.2. Rheology of Solutions
2.2.3. Fiber Spinning
2.2.4. Fibers’ Mechanical Properties
2.2.5. Optical Analysis
2.2.6. Thermal Analysis
2.2.7. FT-IR Spectroscopy
3. Results and Discussion
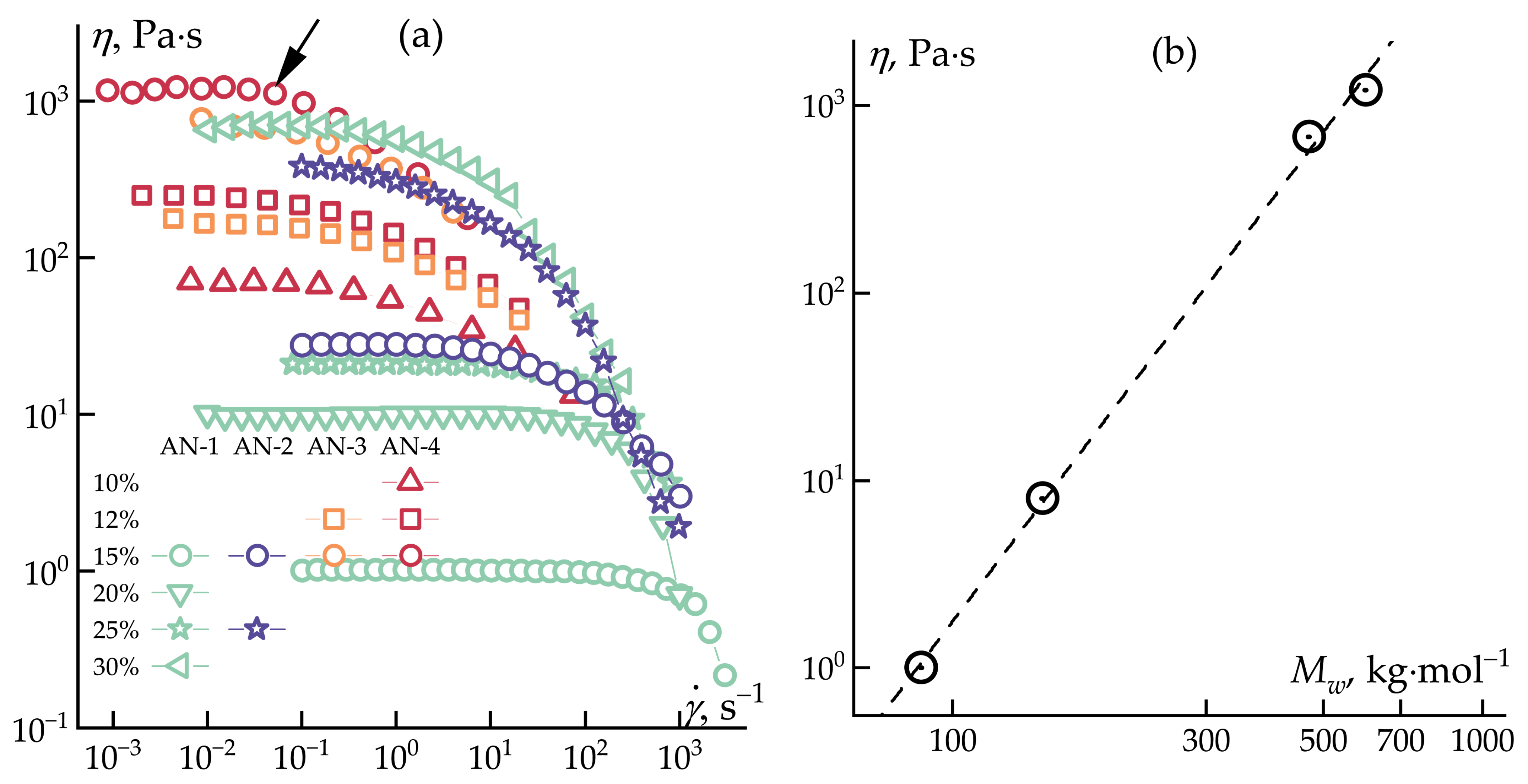
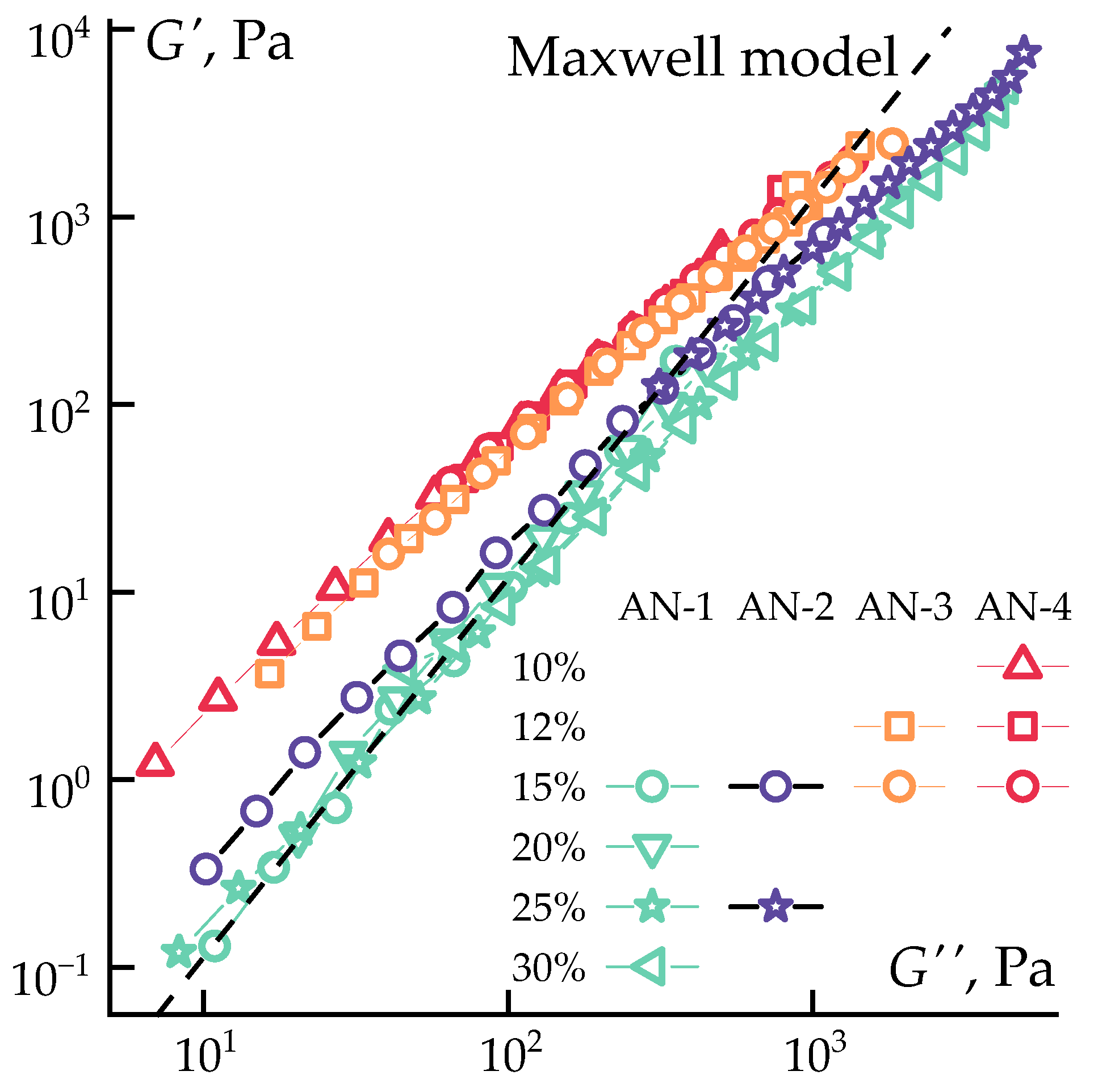
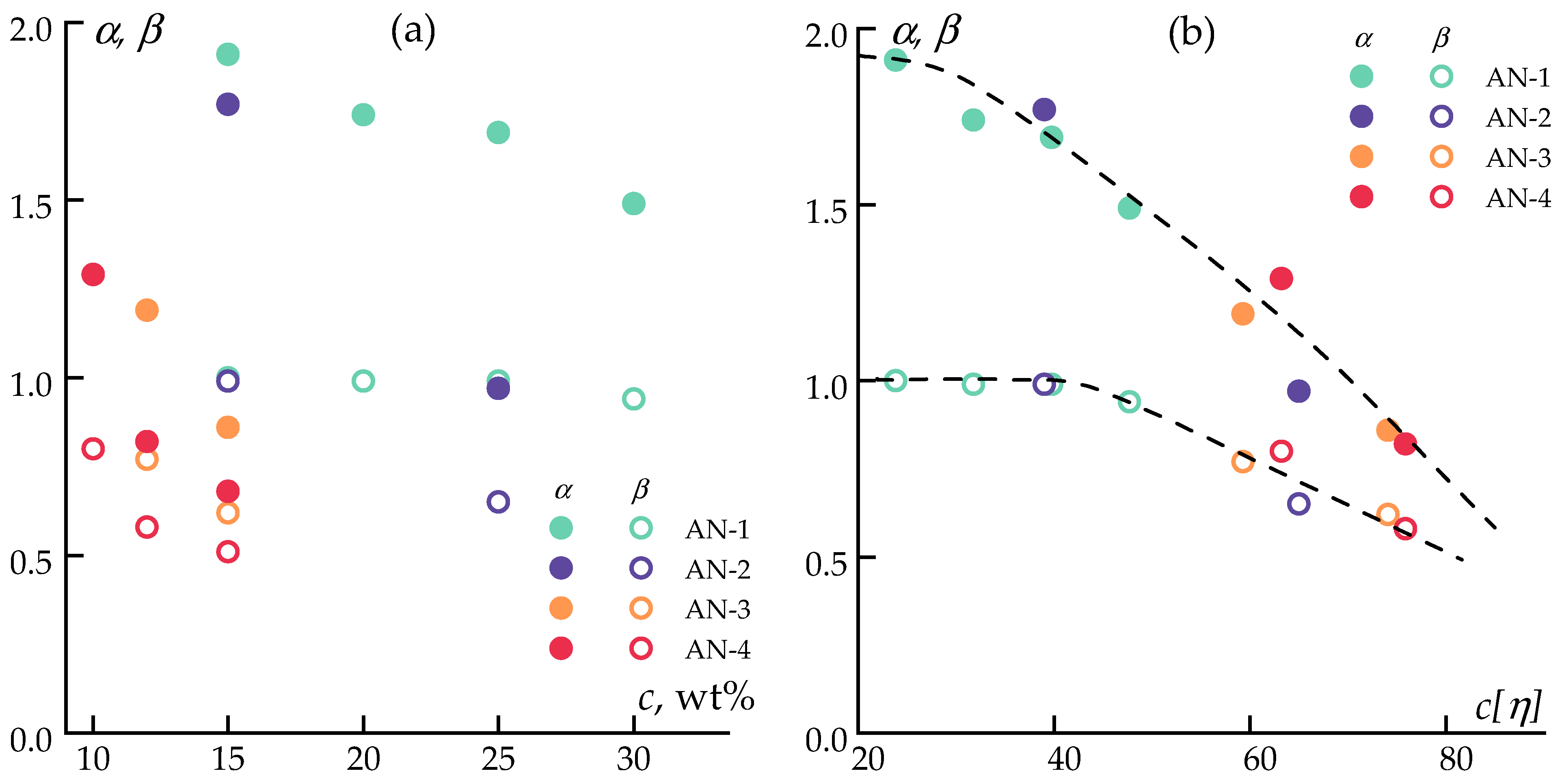
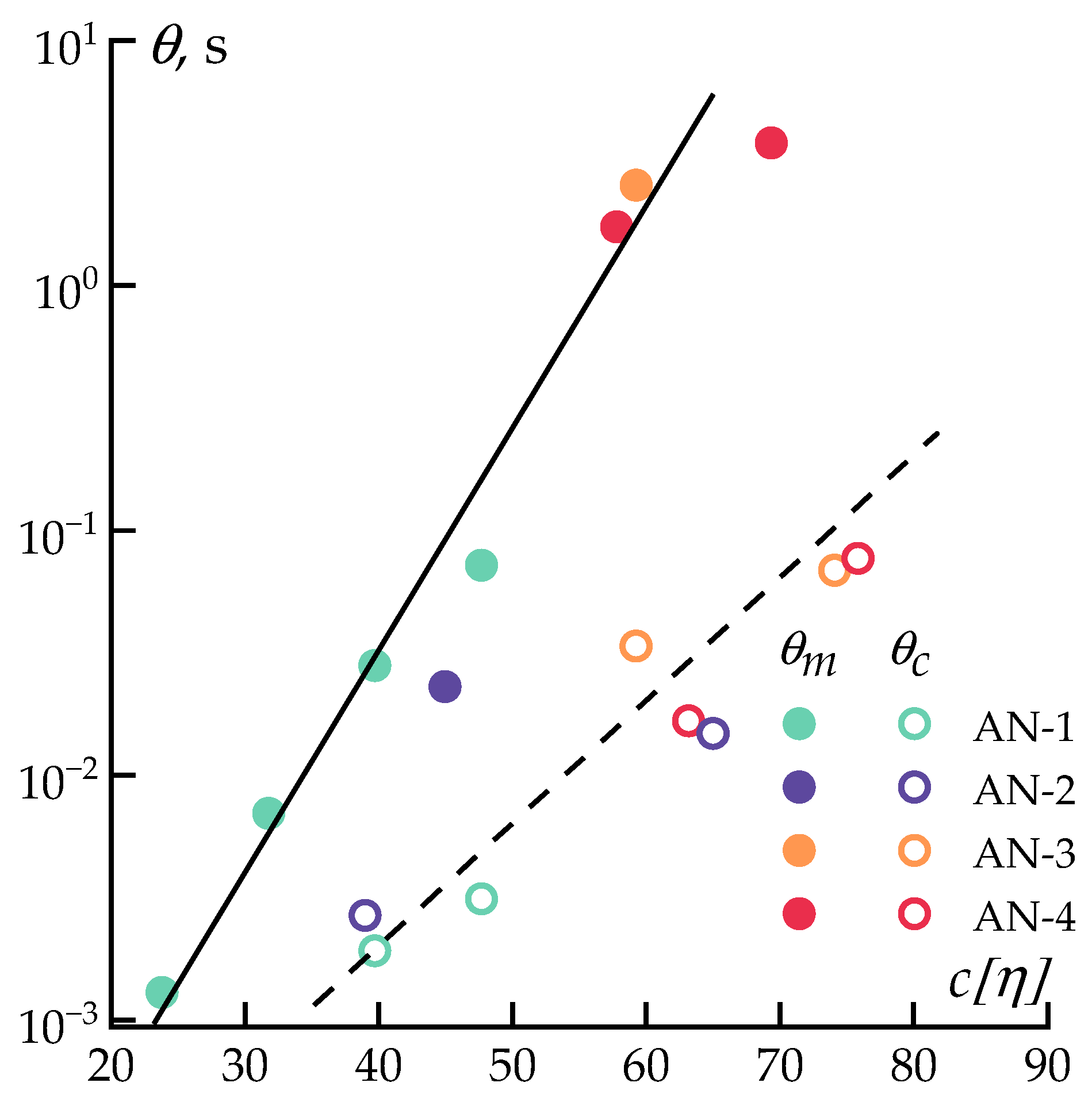
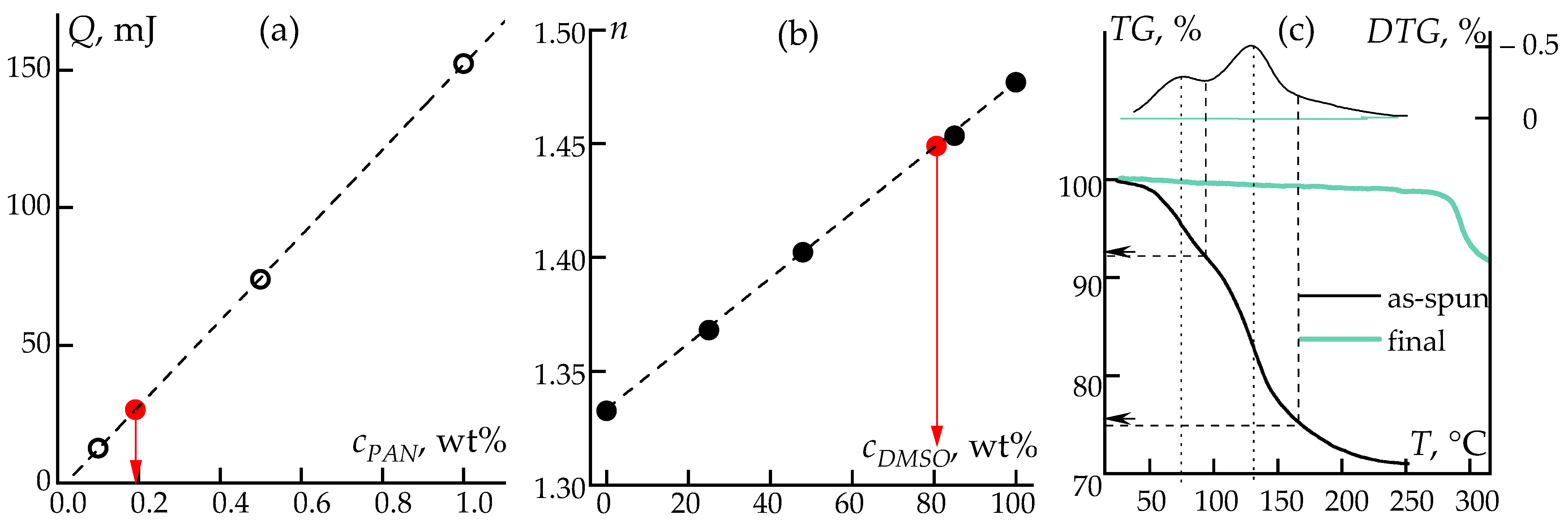
3.1. Solutions Rheology
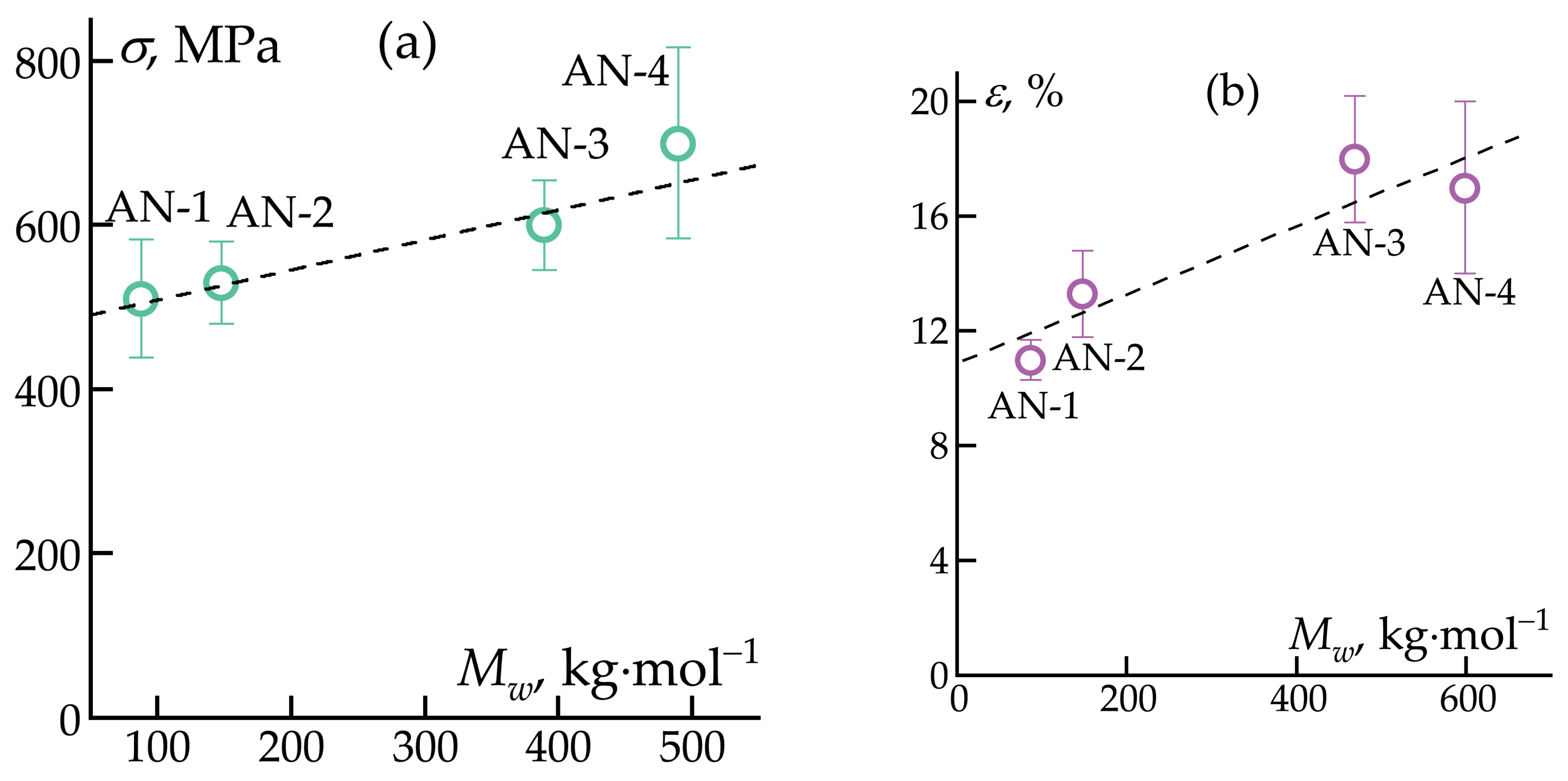
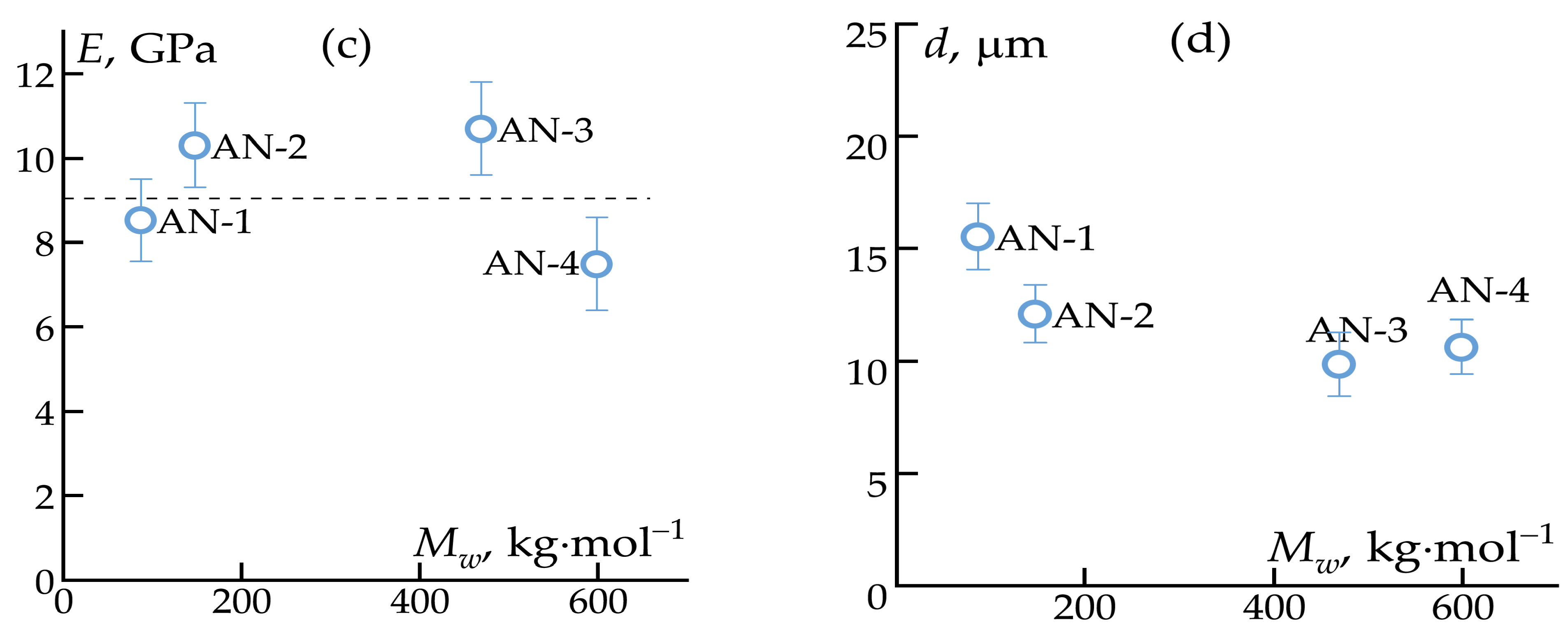
3.2. Fiber Spinning
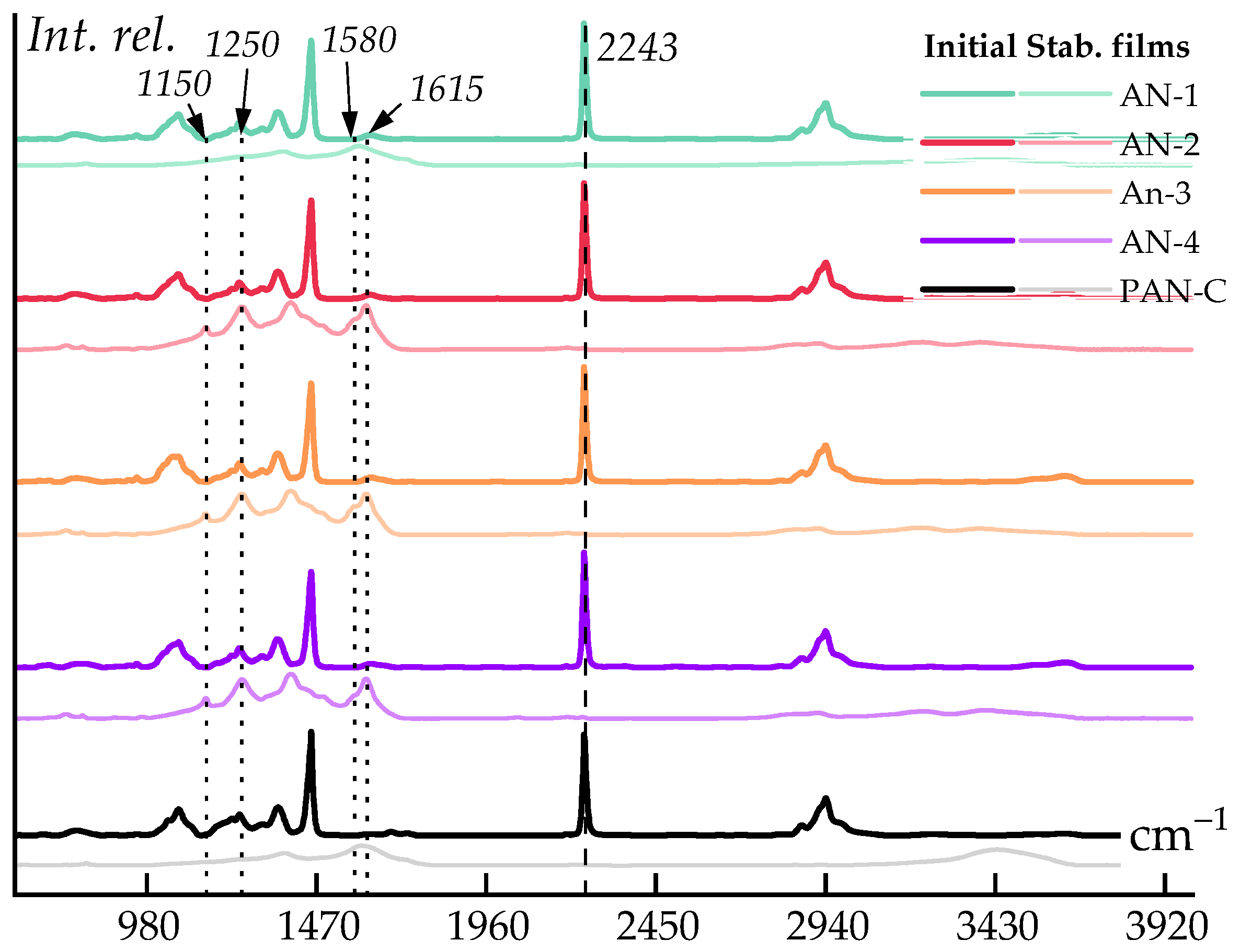
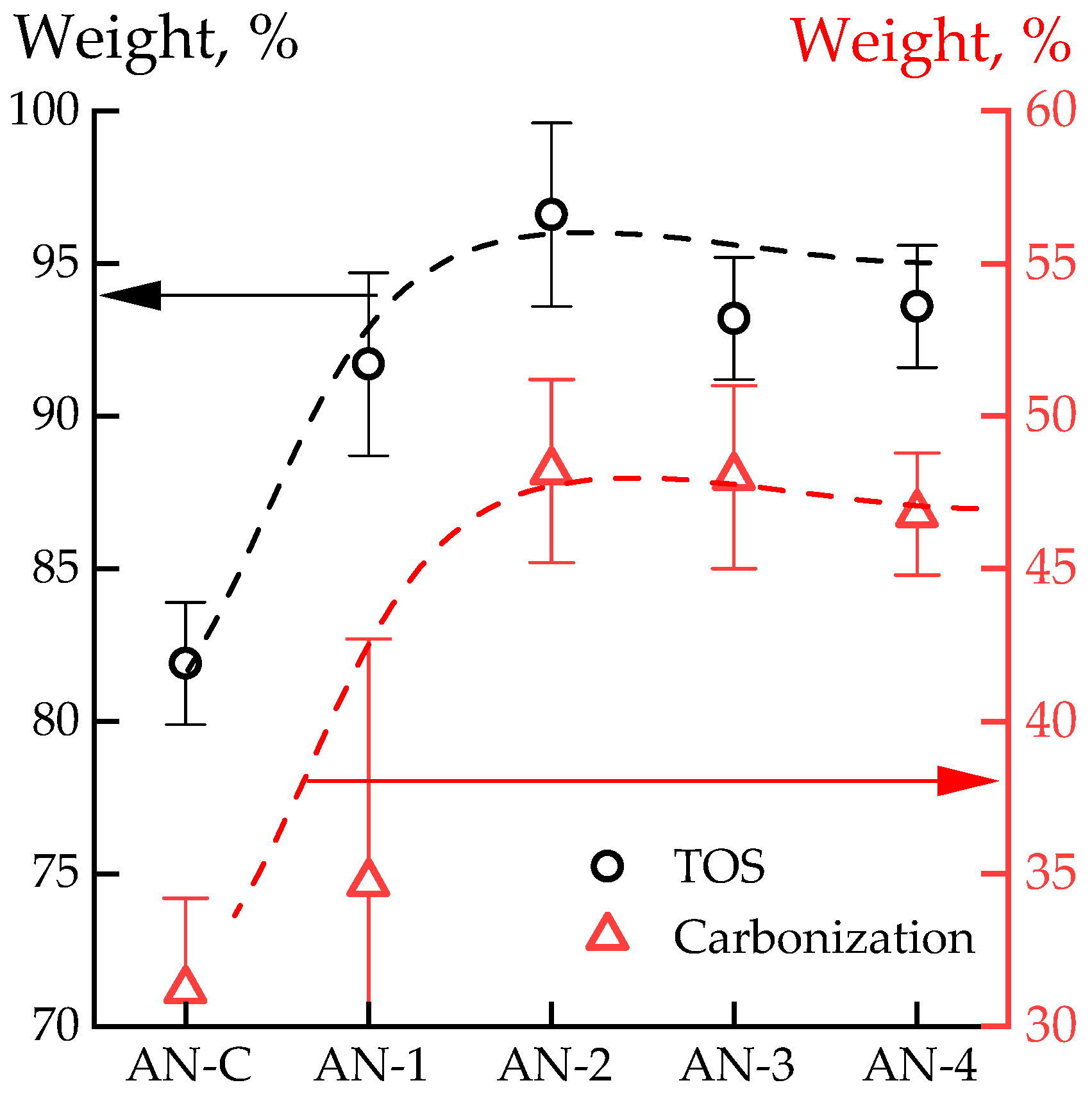
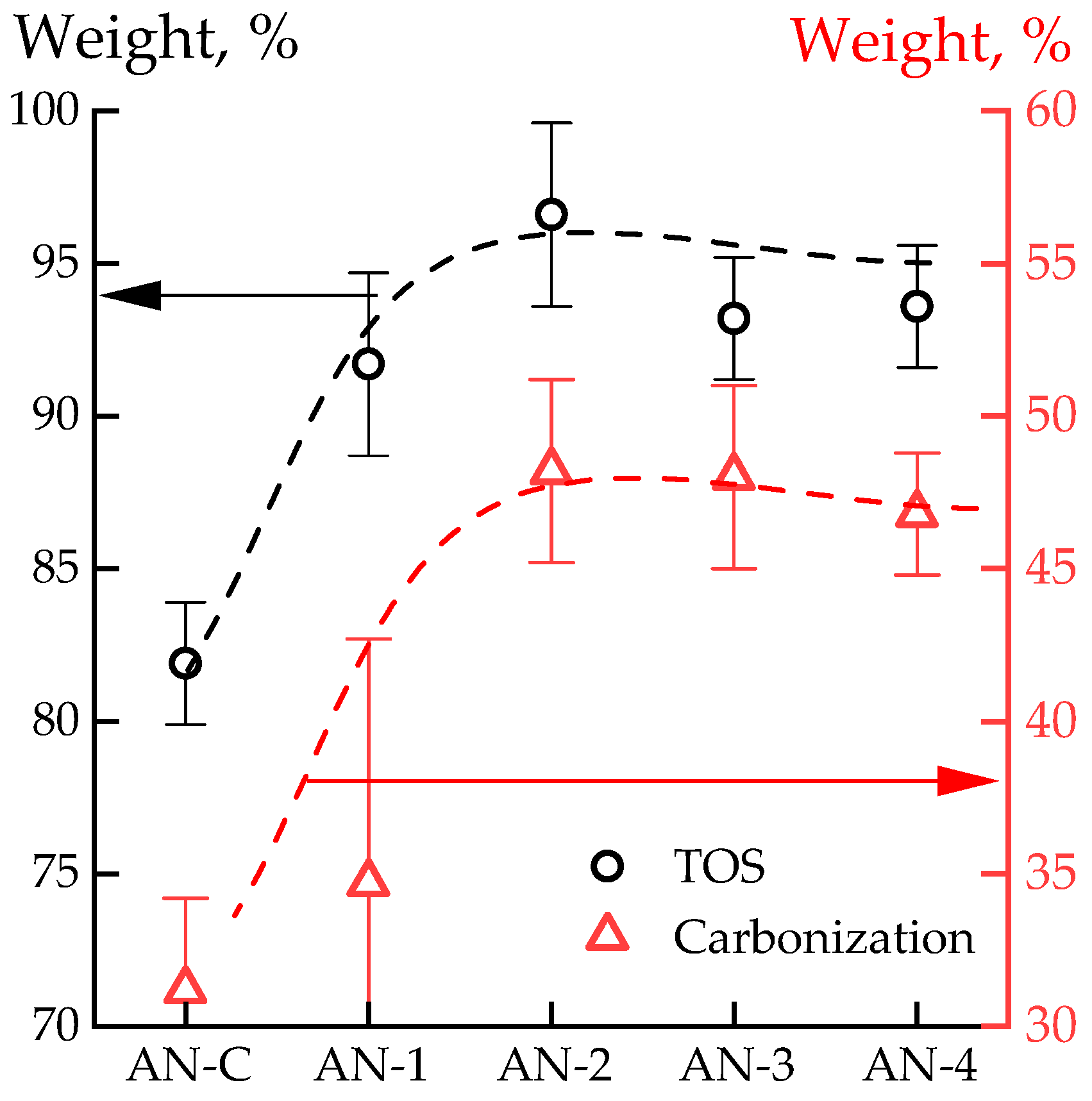
3.3. Thermal Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kausar, A. Polyacrylonitrile-Based Nanocomposite Fibers: A Review of Current Developments. J. Plast. Film Sheeting 2019, 35, 295–316. [Google Scholar] [CrossRef]
- Chernikova, E.; Toms, R.; Gervald, A.Y.; Prokopov, N. Fiber-Forming Acrylonitrile Copolymers: From Synthesis to Properties of Carbon Fiber Precursors and Prospects for Industrial Production. Polym. Sci. Ser. C 2020, 62, 17–50. [Google Scholar] [CrossRef]
- Meng, H.; Xu, T.; Gao, M.; Bai, J.; Li, C. An Oil-contamination-resistant PVP/PAN Electrospinning Membrane for High-efficient Oil–Water Mixture and Emulsion Separation. J. Appl. Polym. Sci. 2021, 138, 50043. [Google Scholar] [CrossRef]
- Scharnagl, N.; Buschatz, H. Polyacrylonitrile (PAN) Membranes for Ultra-and Microfiltration. Desalination 2001, 139, 191–198. [Google Scholar] [CrossRef]
- Maksimov, N.M.; Toms, R.V.; Balashov, M.S.; Gerval’d, A.Y.; Prokopov, N.I.; Plutalova, A.V.; Kuzin, M.S.; Skvortsov, I.Y.; Kulichikhin, V.G.; Chernikova, E.V. Novel Potential Precursor of Carbon Fiber Based on Copolymers of Acrylonitrile, Acrylamide, and Alkyl Acrylates. Polym. Sci. Ser. B 2022, 64, 670–687. [Google Scholar] [CrossRef]
- Zhang, C.; Gilbert, R.; Fornes, R. Preparation of Ultrahigh Molecular Weight Polyacrylonitrile and Its Terpolymers. J. Appl. Polym. Sci. 1995, 58, 2067–2075. [Google Scholar] [CrossRef]
- Thomas, W. Mechanism of Acrylonitrile Polymerization. In Fortschritte der Hochpolymeren-Forschung; Springer: Berlin/Heidelberg, Germany, 2006; pp. 401–441. [Google Scholar]
- Pan, X.; Lamson, M.; Yan, J.; Matyjaszewski, K. Photoinduced Metal-Free Atom Transfer Radical Polymerization of Acrylonitrile. ACS Macro Lett. 2015, 4, 192–196. [Google Scholar] [CrossRef]
- Kozhunova, E.Y.; Plutalova, A.V.; Chernikova, E.V. RAFT Copolymerization of Vinyl Acetate and Acrylic Acid in the Selective Solvent. Polymers 2022, 14, 555. [Google Scholar] [CrossRef]
- Basiouny, N.; Hefni, H.H.; Abouelmagd, W.; El-Rahman, A.; Esmail, E.; Youssif, M. Preparation of Polyacrylonitrile with Different Molecular Weights and High Conversion Yield in Aqueous Phase Polymerization. Egypt. J. Chem. 2022, 65, 1347–1353. [Google Scholar]
- Cundall, R.; Eley, D.; Worrall, J. The Anionic Polymerization of Acrylonitrile. J. Polym. Sci. 1962, 58, 869–880. [Google Scholar] [CrossRef]
- Ottolenghi, A.; Barzakay, S.; Zilkha, A. Anionic Polymerization of Acrylonitrile by Various Catalysts. J. Polym. Sci. Part A Gen. Pap. 1963, 1, 3643–3654. [Google Scholar] [CrossRef]
- Novoselova, A.; Shamanin, V.; Vinogradova, L. Synthesis of Ultra-High-Molecular-Weight Polyacrylonitrile by Anionic Polymerization. Polym. Sci. Ser. B 2009, 51, 205–211. [Google Scholar] [CrossRef]
- Shi, X.; Jiang, J. Anionic Polymerization Initiated by Lithium Amides for Preparing High Molecular Weight Polyacrylonitrile. Chin. Chem. Lett. 2019, 30, 473–476. [Google Scholar] [CrossRef]
- Kamide, K.; Ono, H.; Hisatani, K. Stereospecificity in the Polymerization of Acrylonitrile Using Anionic Initiators Including Dialkylmagnesium. Polym. J. 1992, 24, 917–930. [Google Scholar] [CrossRef]
- Eom, Y.; Kim, B.C. Solubility Parameter-Based Analysis of Polyacrylonitrile Solutions in N,N-Dimethyl Formamide and Dimethyl Sulfoxide. Polymer 2014, 55, 2570–2577. [Google Scholar] [CrossRef]
- Ratkanthwar, K.; Zhao, J.; Zhang, H.; Hadjichristidis, N.; Mays, J. Schlenk Techniques for Anionic Polymerization. In Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications; Springer: Tokyo, Japan, 2015; pp. 3–18. [Google Scholar]
- Liu, Q.; Wang, Y.; Niu, F.; Ma, L.; Qu, C.; Fu, S.; Chen, M. Spinnability of Polyacrylonitrile Gel Dope in the Mixed Solvent of Dimethyl Sulfoxide/Dimethylacetamide and Characterization of the Nascent Fibers. Polym. Sci. Ser. A 2018, 60, 638–646. [Google Scholar] [CrossRef]
- Estrin, Y.I.; Tarasov, A.E.; Grishchuk, A.A.; Chernyak, A.V.; Badamshina, E.R. Initiation of Anionic Polymerization of Acrylonitrile with Tertiary Amines and Ethylene or Propylene Oxide: Some Mechanistic Aspects. RSC Adv. 2016, 6, 106064–106073. [Google Scholar] [CrossRef]
- Estrin, Y.I.; Grishchuk, A.; Tarasov, A.; Perepelitsina, E.; Badamshina, E. Anionic Polymerization and Copolymerization of Acrylonitrile Initiated by Systems Based on Bicyclic Tertiary Amines and Ethylene Oxide. Polym. Sci. Ser. B 2016, 58, 19–26. [Google Scholar] [CrossRef]
- Tarasov, A.; Grishchuk, A.; Karpov, S.; Podval’naya, Y.V.; Chernyak, A.; Korchagina, S.; Badamshina, E. Anionic Copolymerization of Acrylonitrile with Methyl Acrylate under the Action of a Novel Initiating System Based on a Bicyclic Tertiary Amine and Ethylene Oxide. Russ. J. Appl. Chem. 2020, 93, 1009–1018. [Google Scholar] [CrossRef]
- Rui, G.; Lv, Q.; Lu, J.; Wu, T.; Zhao, S.; Huang, R.; Han, B.; Yang, W. A Metal-Free Method for Ultra-High Molecular Weight Polyacrylonitrile under Dimethyl Sulfoxide. Polymer 2021, 214, 123245. [Google Scholar] [CrossRef]
- Mironova, M.V.; Tarasov, A.E.; Kuzin, M.S.; Skvortsov, I.Y.; Arkharova, N.A.; Podval’naya, Y.V.; Grishchuk, A.A.; Badamshina, E.R.; Kulichikhin, V.G. Rheological and Relaxational Properties of Mixed Solutions Based on Linear and Highly Branched Polyacrylonitrile. Polym. Sci. Ser. A 2022, 64, 354–365. [Google Scholar] [CrossRef]
- Li, X.; Suo, X.; Liu, Y.; Li, Y. Effect of Gelation Time on the Microstructures, Mechanical Properties and Cyclization Reactions of Dry-Jet Gel-Spun Polyacrylonitrile Fibers. New Carbon Mater. 2019, 34, 9–18. [Google Scholar] [CrossRef]
- Toms, R.V.; Balashov, M.S.; Gervald, A.Y.; Prokopov, N.I.; Plutalova, A.V.; Chernikova, E.V. Influence of Monomer Sequence on the Cyclization Behavior of Poly(Acrylonitrile-Co-Acrylamide). Appl. Sci. 2023, 13, 3734. [Google Scholar] [CrossRef]
- Yusof, N.; Ismail, A. Post Spinning and Pyrolysis Processes of Polyacrylonitrile (PAN)-Based Carbon Fiber and Activated Carbon Fiber: A Review. J. Anal. Appl. Pyrolysis 2012, 93, 1–13. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A Review of Heat Treatment on Polyacrylonitrile Fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Ju, A.; Guang, S.; Xu, H. Effect of Comonomer Structure on the Stabilization and Spinnability of Polyacrylonitrile Copolymers. Carbon 2013, 54, 323–335. [Google Scholar] [CrossRef]
- Bajaj, P.; Sreekumar, T.V.; Sen, K. Structure Development during Dry–Jet–Wet Spinning of Acrylonitrile/Vinyl Acids and Acrylonitrile/Methyl Acrylate Copolymers. J. Appl. Polym. Sci. 2002, 86, 773–787. [Google Scholar] [CrossRef]
- Chernikova, E.; Kostina, Y.V.; Efimov, M.; Prokopov, N.; Gerval’d, A.Y.; Toms, R.; Nikolaev, A.Y.; Shkirev, M. Homo-and Copolymers of Acrylonitrile: Effect of the Reaction Medium on the Thermal Behavior in an Inert Atmosphere. Polym. Sci. Ser. B 2015, 57, 116–131. [Google Scholar] [CrossRef]
- Hajir Bahrami, S.; Bajaj, P.; Sen, K. Thermal Behavior of Acrylonitrile Carboxylic Acid Copolymers. J. Appl. Polym. Sci. 2003, 88, 685–698. [Google Scholar] [CrossRef]
- Szepcsik, B.; Pukánszky, B. The Mechanism of Thermal Stabilization of Polyacrylonitrile. Thermochim. Acta 2019, 671, 200–208. [Google Scholar] [CrossRef]
- Ge, Y.; Fu, Z.; Zhang, M.; Zhang, H. The Role of Structural Evolution of Polyacrylonitrile Fibers during Thermal Oxidative Stabilization on Mechanical Properties. J. Appl. Polym. Sci. 2021, 138, 49603. [Google Scholar] [CrossRef]
- Kulichikhin, V.; Skvortsov, I.; Subbotin, A.; Kotomin, S.; Malkin, A. A Novel Technique for Fiber Formation: Mechanotropic Spinning—Principle and Realization. Polymers 2018, 10, 856. [Google Scholar] [CrossRef]
- Skvortsov, I.Y.; Maksimov, N.M.; Kuzin, M.S.; Toms, R.V.; Varfolomeeva, L.A.; Chernikova, E.V.; Kulichikhin, V.G. Influence of Alkyl Acrylate Nature on Rheological Properties of Polyacrylonitrile Terpolymers Solutions, Spinnability and Mechanical Characteristics of Fibers. Materials 2022, 16, 107. [Google Scholar] [CrossRef]
- Podvalnaya, Y.V. Study of the Patterns of Anionic (Co)Polymerization of Acrylonitrile: From Linear to Hyperbranched Polymers; FRC PCP MC RAS: Chernogolovka, Russia, 2023. (In Russian) [Google Scholar]
- Podvalnaya, Y.V.; Tarasov, A.E.; Grishchuk, A.A.; Chernyayev, D.A.; Badamshina, E.R. Anionic Copolymerization of Acrylonitrile with Ethyl Acrylate under the Action of the Initiating System of 1, 4-Diazabicyclo [2.2. 2] Octane–Ethylene Oxide. Key Eng. Mater. 2021, 899, 226–231. [Google Scholar] [CrossRef]
- Eom, Y.; Kim, C.; Kim, B.C. Effects of Physical Association through Nitrile Groups on the MWD-Dependent Viscosity Behavior of Polyacrylonitrile Solutions. Macromol. Res. 2017, 25, 262–269. [Google Scholar] [CrossRef]
- Karacan, I.; Erdoğan, G. The Role of Thermal Stabilization on the Structure and Mechanical Properties of Polyacrylonitrile Precursor Fibers. Fibers Polym. 2012, 13, 855–863. [Google Scholar] [CrossRef]
- GOST 10213.0-2002; Staple Chemical Fibre and Tow. Rules of Acceptance and Method of Sampling. Russian Federation. 2002. Available online: https://internet-law.ru/gosts/gost/6362 (accessed on 1 April 2024).
- Subbotin, A.V.; Semenov, A.N. Phase Separation in Dilute Polymer Solutions at High-rate Extension. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1066–1073. [Google Scholar] [CrossRef]
- Subbotin, A.V.; Semenov, A.N. Dynamics of Dilute Polymer Solutions at the Final Stages of Capillary Thinning. Macromolecules 2022, 55, 2096–2108. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Semakov, A.V.; Skvortsov, I.Y.; Zatonskikh, P.; Kulichikhin, V.G.; Subbotin, A.V.; Semenov, A.N. Spinnability of Dilute Polymer Solutions. Macromolecules 2017, 50, 8231–8244. [Google Scholar] [CrossRef]
- Henrici-Olivé, G.; Olivé, S. Molecular Interactions and Macroscopic Properties of Polyacrylonitrile and Model Substances. In Chemistry; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1979; Volume 32, pp. 123–152. ISBN 978-3-540-09442-5. [Google Scholar]
- Beevers, R.B. The Effect of Solvent on Association in Polyacrylonitrile Solutions. Polymer 1969, 10, 791–799. [Google Scholar] [CrossRef]
- Skvortsov, I.Y.; Kuzin, M.S.; Gerasimenko, P.S.; Kulichikhin, V.G.; Malkin, A.Y. The Role of Small Amounts of Water in the Gelation of Polyacrylonitrile Solutions in Dimethyl Sulfoxide: Rheology, Kinetics and Mechanism. Macromolecules 2024, 57. [Google Scholar] [CrossRef]
- Subbotin, A.V.; Skvortsov, I.Y.; Kuzin, M.S.; Gerasimenko, P.S.; Kulichikhin, V.G.; Malkin, A.Y. The Shape of a Falling Jet Formed by Concentrated Polymer Solutions. Phys. Fluids 2021, 33, 083108. [Google Scholar] [CrossRef]
- Tan, L.; Liu, S.; Pan, D. Water Effect on the Gelation Behavior of Polyacrylonitrile/Dimethyl Sulfoxide Solution. Colloids Surf. A Physicochem. Eng. Asp. 2009, 340, 168–173. [Google Scholar] [CrossRef]
- Mezger, T.G. The Rheology Handbook, 4th ed.; European Coatings TECH FILES; Vincentz Network: Hannover, Germany, 2014; ISBN 978-3-86630-650-9. [Google Scholar]
- Larson, R.G. Instabilities in Viscoelastic Flows. Rheol. Acta 1992, 31, 213–263. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; International Series of Monographs on Physics; Clarendon Press: Oxford, UK, 1986; ISBN 978-0-19-852033-7. [Google Scholar]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1980; ISBN 0-471-04894-1. [Google Scholar]
- Skvortsov, I.Y.; Chernikova, E.V.; Kulichikhin, V.G.; Varfolomeeva, L.A.; Kuzin, M.S.; Toms, R.V.; Prokopov, N.I. The Effect of the Synthetic Procedure of Acrylonitrile–Acrylic Acid Copolymers on Rheological Properties of Solutions and Features of Fiber Spinning. Materials 2020, 13, 3454. [Google Scholar] [CrossRef]
- Skvortsov, I.Y.; Malkin, A.Y.; Kuzin, M.S.; Bondarenko, G.N.; Gerasimenko, P.S.; Litmanovich, E.A. Rheology and Molecular Interactions in Polyacrylonitrile Solutions: Role of a Solvent. J. Mol. Liq. 2022, 364, 119938. [Google Scholar] [CrossRef]
- Fischer, P.; Rehage, H. Rheological Master Curves of Viscoelastic Surfactant Solutions by Varying the Solvent Viscosity and Temperature. Langmuir 1997, 13, 7012–7020. [Google Scholar] [CrossRef]
- Brem, A. Solutions for Fiber Spinning: The Role of Solvent Quality in Gel-Spinning of Ultra-High-Molecular-Weight Polyethylene. Doctoral Dissertation, ETH Zurich, Zurich, Switzerland, 2019; p. 228. [Google Scholar]
- Larson, R.; Sridhar, T.; Leal, L.; McKinley, G.H.; Likhtman, A.; McLeish, T. Definitions of Entanglement Spacing and Time Constants in the Tube Model. J. Rheol. 2003, 47, 809–818. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Isayev, A.I. Rheology: Concepts, Methods, and Applications; Elsevier: Amsterdam, The Netherlands, 2022; ISBN 1-927885-94-9. [Google Scholar]
- Subbotin, A.V.; Semenov, A.N. Phase Separation in Polymer Solutions under Extension. Polym. Sci. Ser. C 2018, 60, 106–117. [Google Scholar] [CrossRef]
- Papkov, S.P. Jelly-like State of Polymers; Khimiya: Moscow, Russia, 1974. (In Russian) [Google Scholar]
- Schaller, R.; Feldman, K.; Smith, P.; Tervoort, T.A. High-Performance Polyethylene Fibers “Al Dente”: Improved Gel-Spinning of Ultrahigh Molecular Weight Polyethylene Using Vegetable Oils. Macromolecules 2015, 48, 8877–8884. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, B.; Sun, L.; Zhang, H. Study on the Thermal Oxidative Stabilization Reactions and the Formed Structures in Polyacrylonitrile during Thermal Treatment. Polym. Degrad. Stab. 2017, 140, 104–113. [Google Scholar] [CrossRef]
- Moskowitz, J.D.; Wiggins, J.S. Thermo-Oxidative Stabilization of Polyacrylonitrile and Its Copolymers: Effect of Molecular Weight, Dispersity, and Polymerization Pathway. Polym. Degrad. Stab. 2016, 125, 76–86. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn, kg·mol−1 | Mw, kg·mol−1 | Ð 1 | [η], dL·g−1 | Mη, kg·mol−1 |
|---|---|---|---|---|---|
| AN-1 | 40 | 87 | 2.2 | 1.56 | 83 |
| AN-2 | 82 | 148 | 1.8 | 2.7 | 171 |
| AN-3 | 217 | 469 | 2.2 | 5.06 | 434 |
| AN-4 | 344 | 599 | 1.7 | 6.32 | 599 |
| AN-C [38] | 88 | 150 | 1.7 | 1.75 | 88 |
| Sample | c, wt% | Ts 1, °C | V0 m·min−1 | Roller’s Velocity, m·min−1 | Drawing Ratio (V6/V1) | Total Drawing Ratio (V6/V0) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V1 | V2 | V3 | V4 | V5 | V6 | ||||||
| AN-1 | 30 | 25 | 0.16 | 1.3 | 1.4 | 2.4 | 6 | 6.3 | 12 | 9.2 | 75 |
| AN-2 | 25 | 90 | 0.04 | 2.2 | 2.6 | 3.7 | 5.5 | 5.6 | 10.3 | 4.7 | 258 |
| AN-3 | 13.5 | 70 | 0.1 | 4.2 | 4.8 | 16 | 16.8 | 16.8 | 24.5 | 5.8 | 245 |
| AN-4 | 12 | 25 | 0.08 | 4.4 | 5 | 12.9 | 16.3 | 16.6 | 44 | 10 | 550 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skvortsov, I.Y.; Kuzin, M.S.; Gerasimenko, P.S.; Mironova, M.V.; Golubev, Y.V.; Kulichikhin, V.G. Non-Coagulant Spinning of High-Strength Fibers from Homopolymer Polyacrylonitrile Synthesized via Anionic Polymerisation. Polymers 2024, 16, 1185. https://doi.org/10.3390/polym16091185
Skvortsov IY, Kuzin MS, Gerasimenko PS, Mironova MV, Golubev YV, Kulichikhin VG. Non-Coagulant Spinning of High-Strength Fibers from Homopolymer Polyacrylonitrile Synthesized via Anionic Polymerisation. Polymers. 2024; 16(9):1185. https://doi.org/10.3390/polym16091185
Chicago/Turabian StyleSkvortsov, Ivan Yu., Mikhail S. Kuzin, Pavel S. Gerasimenko, Maria V. Mironova, Yaroslav V. Golubev, and Valery G. Kulichikhin. 2024. "Non-Coagulant Spinning of High-Strength Fibers from Homopolymer Polyacrylonitrile Synthesized via Anionic Polymerisation" Polymers 16, no. 9: 1185. https://doi.org/10.3390/polym16091185
APA StyleSkvortsov, I. Y., Kuzin, M. S., Gerasimenko, P. S., Mironova, M. V., Golubev, Y. V., & Kulichikhin, V. G. (2024). Non-Coagulant Spinning of High-Strength Fibers from Homopolymer Polyacrylonitrile Synthesized via Anionic Polymerisation. Polymers, 16(9), 1185. https://doi.org/10.3390/polym16091185