1. Introduction
In the last decades, environmental, economic, and safety challenges have focused the attention of scientists and companies to produce new biodegradable materials able to partially or totally substitute petrochemical-based plastics [
1]. In this context, one of the most promising biodegradable polymer in the market, also produced by renewable resources, is the poly(lactic acid) (PLA) [
2,
3], an aliphatic thermoplastic polyester with several advantages such as biodegradability, biocompatibility, and compostability which promote its use in many applications from commodities to engineering plastics [
4,
5,
6]. However, the mechanical response of PLA is low due to its high brittleness, low stretchability, and relatively poor impact strength which can limit its use in specific areas [
7,
8]. Many strategies have been explored in both academic and industrial fields to improve the mechanical properties of PLA and enhance its performances, such as copolymerization [
9], addition of additives [
10], blending with other polymers [
11] as well as by the incorporation of nanofillers [
12]. As expected, from the industrial point of view, polymer blending and the addition of additives can be considered as the most effective and cost-efficient methods to enhance PLA performances and use it in different applications where higher toughness is required [
4,
13]. Therefore, with the scope of preserving the sustainability of the final material, increasing attention has been given to the use of bio-based and renewable polymers and additives to obtain complete biodegradable and bio-based blends with improved mechanical and thermal properties with respect to the neat PLA.
In this regard, among bio-based polymers we can find starch, a biodegradable and biocompatible polysaccharide derived from renewable resources such as potatoes, peas, maize, etc., composed of the sequence α-D-glucose linked with each other through (1 → 4) glycosidic bonds. Two different structures are characteristic for starch, amylose which is linear and a branched one called amylopectin [
14]. Starch can be used as nanofillers in the form of starch nanocrystals, preserving its native crystalline form or as a matrix, in the form of thermoplastic matrix when processed with water and/or plasticizer, destructuring its granules [
15]. In the recent years, blends of PLA and thermoplastic starch (TPS) have been studied as environmentally friendly materials of great interest for food packaging and biomedical applications [
16,
17,
18]. However, the major drawback of processing PLA blends through simple physical blending is their immiscibility, poor compatibility, and the weak interfacial bonding of the blend components [
19,
20,
21]. In particular, PLA and TPS are immiscible due to the incompatibility between the hydrophobic nature of PLA and hydrophilic nature of starch. However, the addition of additives such as compatibilizer, plasticizer, coupling agent, etc., is used to reduce the interfacial tension and therefore improve the mechanical performances of the blends [
13]. Nowadays, the effect of compatibilizers and plasticizers on the morphology and mechanical properties of PLA/TPS blends using different additives have been studied demonstrating that more homogeneous morphology improved the mechanical properties and thermal stability of plasticized PLA/TPS blends [
22,
23,
24].
Between the additives, oligomeric lactic acid (OLA) is proposed as a renewable alternative to common plasticizers for PLA, taking advantage of their similar chemical structure that ensures high miscibility [
25]. In 2001, Martin and Averous [
26] reported that 20 wt.% of OLA with respect to neat PLA was enough to efficiently improve the mechanical properties of PLA and result in a decrease of about 20 °C in its glass transition temperature (T
g). Later, Burgos et al. [
27] found that amount of OLA higher than 20 wt.% can be incorporated into PLA obtaining more ductile homogeneous materials. However, they reported that PLA with 20 wt.% was the more stable formulation maintaining constant its thermal, mechanical, and oxygen barrier properties for at least 90 days at fixed conditions. Leones et al. [
28] reported that electrospun PLA-OLA fibers mats, with 20 wt.% OLA, show shape memory behavior at 40 °C and 45 °C, and Sonseca et al. [
29] confirmed that extruded PLA-OLA formulation with the same content of OLA also show shape-memory at a temperature close to the physiological one. This point is very important, taking into account that one of the most promising application fields of shape memory biopolymers is the biomedical sector [
30]. In particular, shape memory materials are smart materials able to respond to an external stimulus such as temperature, humidity, or light, recovering their original shape from a temporary shape previously programmed. These materials are composed of two different phases, a permanent one, responsible for recovering the original shape during the recovery stage and the switching one responsible for fixing the temporary shape during the programming stage [
30]. For thermally-activated shape memory polymers, the switching phase is activated at a specific transition temperature (T
trans) that generally is associated with its T
g or its melting temperature (T
m) [
30].
Nowadays, the use of PLA-based materials with shape memory properties in biomedical applications is still a challenge due to different reasons that limit the use of these materials in real applications [
31]. The shape memory effect in PLA is calculated for deformation of about 50% reaching maximum values of programming strains of about 150% [
32]. Moreover, the T
trans of PLA-based materials is related in general with its T
g that is around 60 °C. For this reason, when related to biomedical applications, it is highly desirable to have the T
trans slightly above body temperature to safely activate the shape memory capability avoiding the potential tissue damage due to a too high temperature. In this regard, in this work, with the main objective of increasing the flexibility of PLA without losing its shape memory effect at a temperature close to the physiological one, blends of plasticized PLA and TPS have been processed using a one-step method for potential application as resorbable implants. In particular, two lactic acid oligomers (OLA8), neat one (OLA8) and functionalized with maleic acid (mOLA8), were used to modulate physical and mechanical properties of PLA and its blends with 40% of TPS. An interesting approach is the use of an additive that could be used as both plasticizer and compatibilizer for the same blend. Mechanical, thermal, and morphological properties were studied. However, the attention was focused on their thermally-activated shape memory capability as well as on the compatibilizing effect of both plasticizers on the PLS/TPS blends, both being effects strongly correlated within themselves.
4. Characterization
Differential scanning calorimetry (DSC) experiments were carried out in a Mettler Toledo DSC822e instrument, under nitrogen flow (30 mL/min). The typical sample weight was around 10 mg. The cycle program consisted of a first heating stage from −90 to 180 °C at a rate of 10 °C min
−1, followed by cooling to −90 °C and subsequent heating up to 180 °C at 10 °C min
−1. The T
g was calculated from the first heating and was taken at the mid-point of heat capacity changes. The melting temperature (T
m) and cold crystallization temperature (T
cc) were obtained from the second heating, and the degree of crystallinity (χ
c) was determined by using Equation (1).
where ΔH
m is the enthalpy of fusion, ΔH
cc is the enthalpy of cool crystallization, is the enthalpy of fusion of a 100% crystalline PLA, taken as 93 J/g, and 1 − m
f is the weight fraction of PLA in the sample [
35].
Dynamic Mechanical Thermal Analysis (DMTA) of the samples was carried out using a DMA Q800 from TA Instrument in film tension mode with an amplitude of 5 μm, a frequency of 1 Hz, a force track of 125%, and a heating rate of 2 °C·min−1. Samples subjected to DMA were cut from compression-molded thin films into regular specimens of approximately 20 mm × 4 mm × 0.50 mm.
Thermogravimetric analysis (TGA) was carried out using a TA-TGA Q500 thermal analyzer. The different materials were analyzed by dynamic mode using about 10 milligrams of sample from room temperature to 800 °C at 10 °C min−1 under nitrogen atmosphere with a flow of 60 mL min−1. Temperatures at the maximum degradation rate (Tmax) were calculated from the first derivative of the TGA curves (DTG).
Mechanical properties were determined using an Instron Universal Testing Machine at a strain rate of 50 mm min−1. Tensile test measurements were performed on five dog-bone specimens with a width of 2 mm, thickness of 0.50 mm, and leaving an initial length between the clamps of 20 mm. From these experiments were obtained the Young Modulus, as the slope of the curve between 0% and 2% deformation, the elongation at break, and the maximum stress were reached.
SEM micrographs of the cryo-fracture surface of neat PLA and plasticized PLA were obtained by Scanning Electron Microscopy (SEM PHILIPS XL30 with a tungsten filament) in order to study their morphology. The polymer samples were frozen using liquid N2 and then cryo-fractured. All the samples were gold/palladium coated by an automatic sputter-coated Polaron SC7640. The investigated blends were analyzed also by Atomic Force Microscopy (AFM) operating in tapping mode with a scanning probe microscope (Icon form Bruker with Nanoscope V controller). All blends were cut using an ultra-microtome Leica Ultracut R with a diamond blade. Height and phase images were obtained under ambient conditions with typical scan speed of 0.5–1 line/s using a scan head with a maximum range of 16 μm × 16 μm. Surface roughness (the roughness average (Ra) and root mean square roughness (Rq)) of each investigated system was calculated using 5 μm × 5 μm AFM height image.
Samples for the thermally-activated shape memory studies were cut from compression-molded thin films into rectangular specimens of approximately 20 mm × 4 mm × 0.60 mm. All thermo-mechanical cycles were carried out using a stress-controlled DMA Q800 from TA Instruments in film tension mode. The samples were heated at the Ttrans (for neat PLA the Ttrans was 75 °C while for the rest of the materials, it was 45 °C) for 5 min and stretched until 50% by applying a constant deformation stress. They were then quenched at the fixing temperature, Tfix, (neat PLA, Tfix = 25 °C; rest of materials Tfix = 0 °C) under the same constant stress. The temporary shape, as characterized by an elongation of Ɛm, was recovered after releasing the stress, and the permanent shape, characterized by an elongation of Ɛp, was recovered upon heating (2 °C·min−1) to Ttrans.
Therefore, to get a quantitative estimation of the shape memory properties of the material, the strain fixity ratio (R
f), and the strain recovery ratio (R
r) have been calculated [
35]. In particular, R
r, the ability to recover the initial shape, was taken as the ratio of the recovered strain to the total strain, as given by the following equation:
R
f, the ability to fix the temporary shape, is the amplitude ratio of the fixed strain to the total strain, as presented by Equation (3):
where Ɛ
m is the deformed strain, Ɛ
u the fixed strain, Ɛ
p the recovered strain, and N is the number of cycles.
5. Results and Discussion
The effect of two different plasticizers on the thermal and mechanical properties of PLA-based blends was first studied to evaluate their miscibility and their effects as plasticizers for PLA, and as compatibilizers for PLA/TPS blends with the ultimate scope to study their thermally-activated shape memory response. Based on previous results, we fixed the amount of plasticizer at 20 wt.% with respect to PLA [
27,
36], considering that this amount is enough to decrease T
g of PLA to a temperature close to the physiological one, with the purpose of obtaining materials with a shape memory response suitable for biomedical applications avoiding migration phenomena [
27]. The preparation of blends with a composition of 40 wt.% of TPS was chosen following previous reports by Nazrin et al. [
34].
In order to understand the properties of the processed blends, it is necessary to study their morphology.
Figure 1 shows the SEM images of cryo-fractured surfaces of the samples corresponding to neat PLA, plasticized PLA with both OLA8 and mOLA8, and their respective blends obtained by adding 40 wt.% of TPS with respect to the plasticized PLA. Therefore, it is easy to note that PLA showed the typical brittle fracture with a smooth fracture surface due to its very low plastic deformation. In the case of oPLA and moPLA, no evidence of phase separation was detected. Indeed, plasticized PLAs/TPS blends showed different roughness with respect to neat PLA, also confirmed by further AFM analysis (
Figure 2). This fact can be due to the difference in the hydrophilic nature of PLA and TPS phases. However, from the SEM analysis, moPLA/TPS seems to present a better dispersion of the TPS phase obtaining a more homogeneous material. This is possibly due to the improvement in interfacial adhesion between the two phases thanks to the compatibilizer effect of the plasticizer mOLA8.
However, the effect of the addition of both OLA8 and mOLA8 on the morphology of PLA/TPS blend was deeply studied by AFM. The obtained AFM height and phase images are shown in
Figure 2. The cross-section AFM phase image of PLA indicated its homogeneous morphology, which also confirmed the very low surface roughness of PLA. The addition of OLA8 and mOLA8 into PLA resulted in changes in the morphology observed for PLA. In the case of oPLA, the regular crystalline-like structure with separated domains of around 200 nm was detected while moPLA showed a different regular structure with small spherulites of the size of 400 ± 50 nm. The surface roughness of oPLA was eight times higher than the surface roughness of PLA and moPLA was four times higher, suggesting that both OLA8 and mOLA8 are good plasticizers for PLA.
In the case of PLA/TPS blends, the addition of OLA8 led to the regular structure which was very similar to the morphology of PLA. The only difference was the presence of small-in-size (~20 nm) spherical domains. The moPLA/TPS blend demonstrates still very regular spherulites-like structures well detected for moPLA with clearly visible small-in-size (~35 nm) spherical domains.
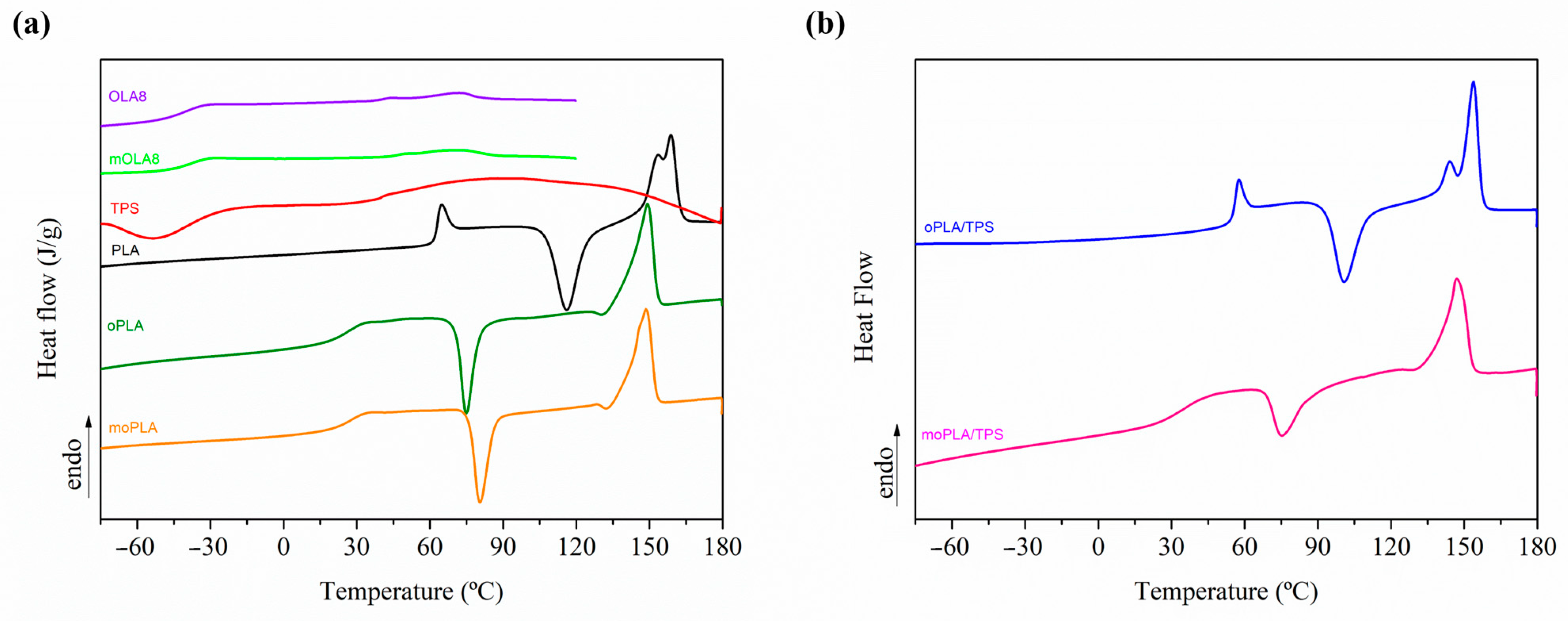
Figure 3 shows the DSC thermograms reporting the first heating scan for the neat materials and plasticized PLA on the left, and plasticized PLA/TPS blends on the right. The first heating scan is taken into account for the thermally activated shape memory study, considering that no further thermal treatments are applied to the samples before being tested by thermo-mechanical cycles. Therefore, from the DSC analysis, we can point out that both plasticized PLA-based materials, with OLA8 and mOLA8, showed a single T
g, indicating the absence of macroscopic phase separation, confirming the morphological analysis, and a consequent good miscibility between the components. As expected, the incorporation of the plasticizer induced an increment in the free volume between PLA chains, consequently increasing the chain mobility which resulted in a reduction in T
g values for plasticized PLA-based materials in comparison with neat PLA. In particular, for plasticized PLA-based materials a T
g of about 25 °C is obtained, an increase of about 10 °C, when mixed with TPS. Moreover, only oPLA shows a quite high degree of crystallinity (28%) and moPLA presents a quite small degree of crystallinity (6%), the other samples being almost amorphous. However, when mixed with TPS, the degree of crystallinity of oPLA/TPS decreases up to 5%, a decrease of about 80%. For moPLA, the degree of crystallinity is quite low (6%) however, when mixed with TPS, the degree of crystallinity increases up to 20%, confirming the crystalline structure obtained by AFM.
The behavior observed when mOLA8 was used as plasticizer could be attributed to the presence of a better dispersed and compatibilized TPS phase into the PLA matrix.
Moreover, the thermal stability of the materials was studied by TGA.
Figure 4 shows the weight loss and the first derivate (DTG) for all the samples, while the main results are summarized in
Table 1. Neat PLA decomposed in a single-step process with a maximum degradation rate (T
max) at 375 °C, in agreement with values previously reported [
27,
37].
As expected, the maximum degradation temperature for both plasticized PLA is much smaller than neat PLA, being, however, higher than the temperature shown for the processing of these materials, which is 160 °C. These results demonstrate that both plasticizers can be processed at the same melt processing window of neat PLA avoiding their thermal degradation. The same occurs for TPS, with a T
max, of about 300 °C. Moreover, from
Figure 4, it is possible to note the different stages of the thermal decomposition mechanism of TPS. In particular, the first stage is related to the physical dehydration of water, volatile compounds, and glycerol decomposition while the second stage is related to the chemical dehydration of bonded water and the thermal decomposition of starch [
38]. Analyzing the thermal stability of the plasticized PLA/TPS blends, it is easy to note that it is composed of a two-step degradation and that they presented intermediate thermal stability compared with both neat polymers. In particular, the DTG curves show a main peak of degradation related to PLA decomposition, and a small shoulder at lower temperatures related to the TPS thermal decomposition. Regarding the blends, moPLA/TPS showed the highest thermal stability in terms of T
max, about 340 °C, ten degrees higher than oPLA/TPS T
max. These results indicate that mOLA8 is able to increase the compatibility of plasticized PLA/TPS blends, as previously observed in the morphology analysis.
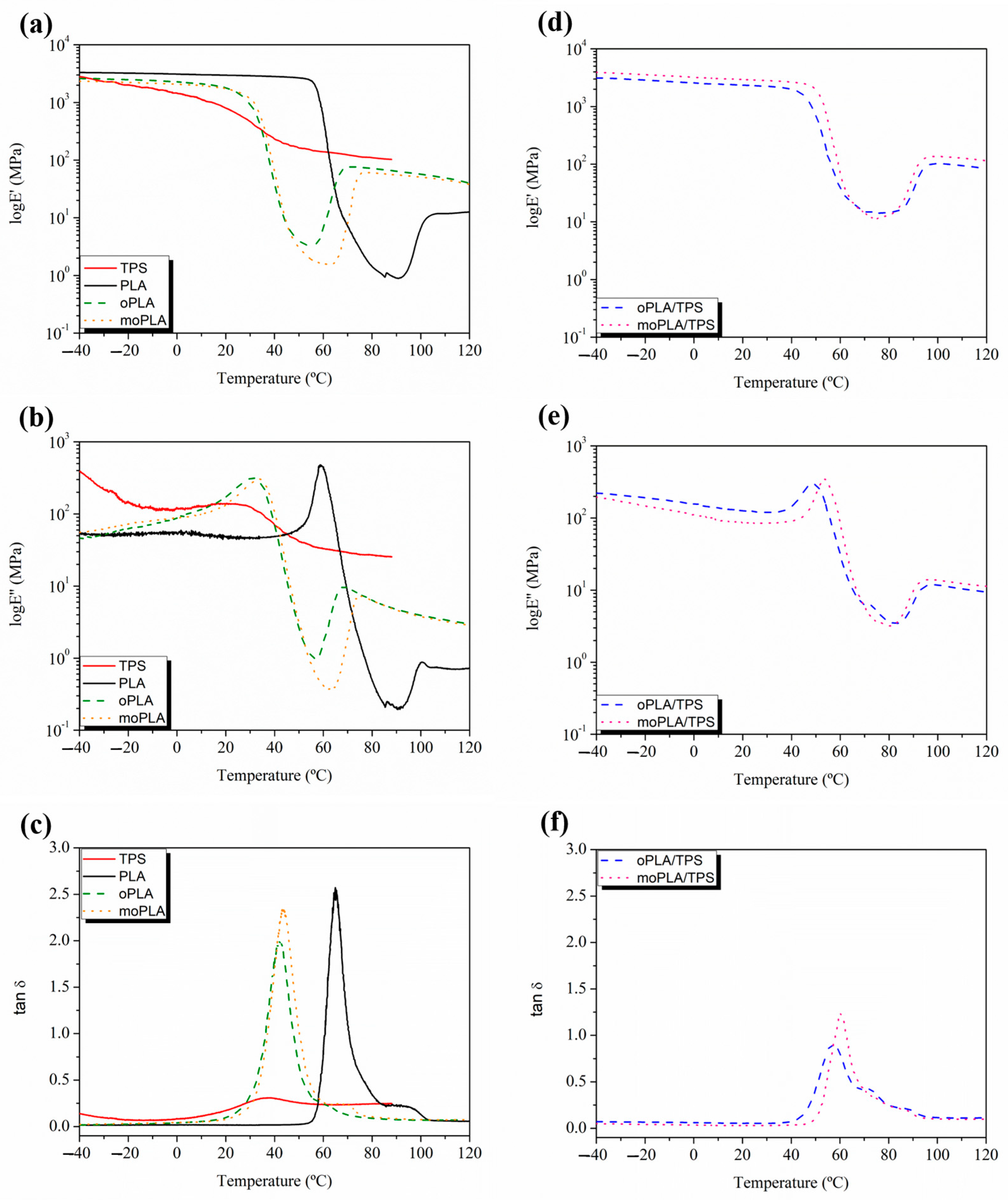
Figure 5 shows the DMTA curves for neat PLA and TPS, plasticized PLAs, on the left and their blends, on the right. The variation in the E’ values of neat PLA (
Figure 5a) showed a dramatic drop between 55 °C and 80 °C, which is representative of the relaxation process of the PLA chains due to the glass transition temperature. A second transition is observed at around 110 °C that reflects the cold crystallization of PLA. Adding both plasticizers, the E’ curves and consequently their drop, shifted to lower temperatures indicating a decrease in T
g and the T
cc, as previously observed by DSC analysis. Furthermore, the addition of both plasticizers leads to a broadening of the T
g peak (
Figure 5b,c) indicating a wide range of relaxation times. Moreover, the E’ values decreased with the addition of both plasticizers, suggesting a decrease in rigidity and an increased ductility of the materials. More accurate T
g values can be obtained by determining the peak maximum of tanδ and loss modulus (
Figure 5b,c). Observing the tanδ curve, neat PLA showed a T
g of 65 °C and the plasticized PLA reached the values of 44 °C and 42 °C for moPLA and oPLA, respectively. These results are in agreement with the results obtained by the DSC characterization.
Moreover, according to the literature [
39], TPS shows two relaxations due to a phase-separated system. The first one below −40 °C, termed β-relaxation, which is ascribed to a starch-poor phase rich in glycerol–water content while the relaxation at around 25 °C is termed α-relaxation and is ascribed to a starch-rich phase. Both TPS relaxations are highly dependent on the amount of bonded and non-bonded water enclosed in the starch chains, which can act as a plasticizer decreasing their temperatures.
Regarding the blends based on plasticized PLA and TPS, it is easy to note that the characteristic relaxations of PLA are shifted to higher temperatures compared with those of plasticized PLA. In this case, the plasticizers are acting partly as plasticizer for PLA and as compatibilizer for the blends, interacting with both PLA and TPS. The typical interactions in these systems are hydrogen bonding between the hydroxyl groups from PLA or starch and hydroxyl, as well as oxygen, from the OLA units, which caused the broadening of the PLA glass transition peak, as previously reported [
40]. Moreover, the E’ values of the blends at 25 °C significantly increased compared with those of plasticized PLAs, reaching values close to that of neat PLA.
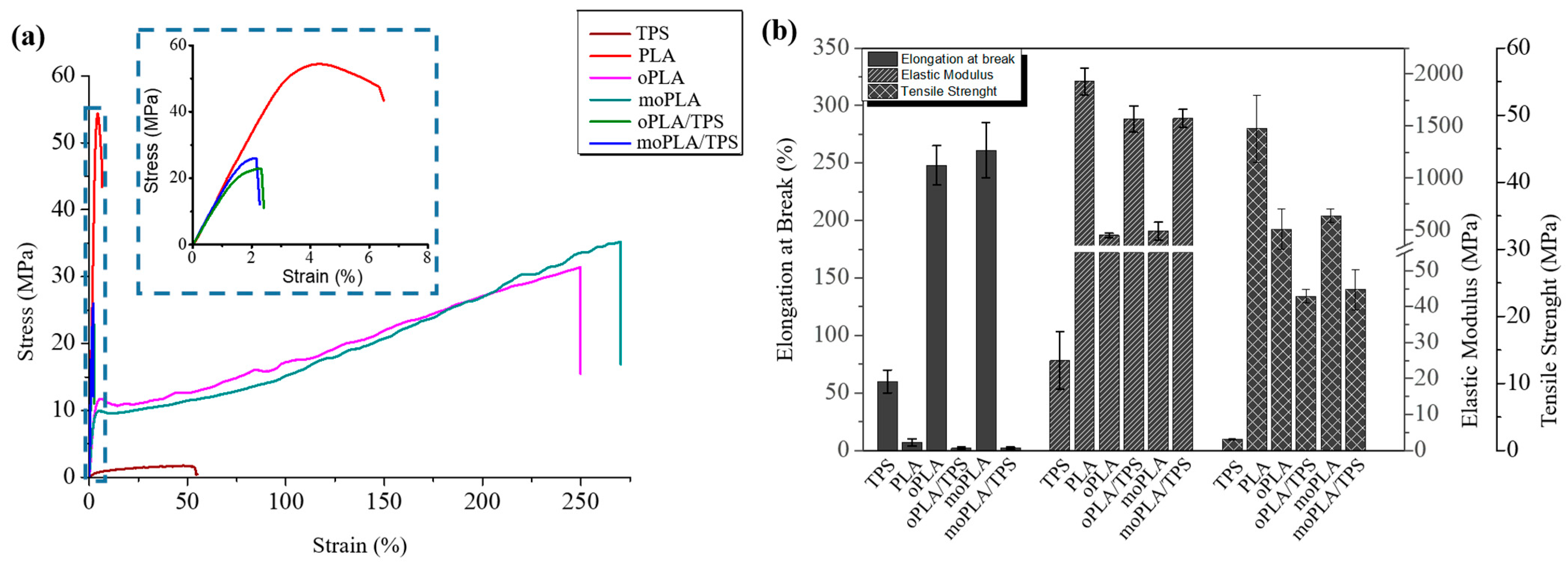
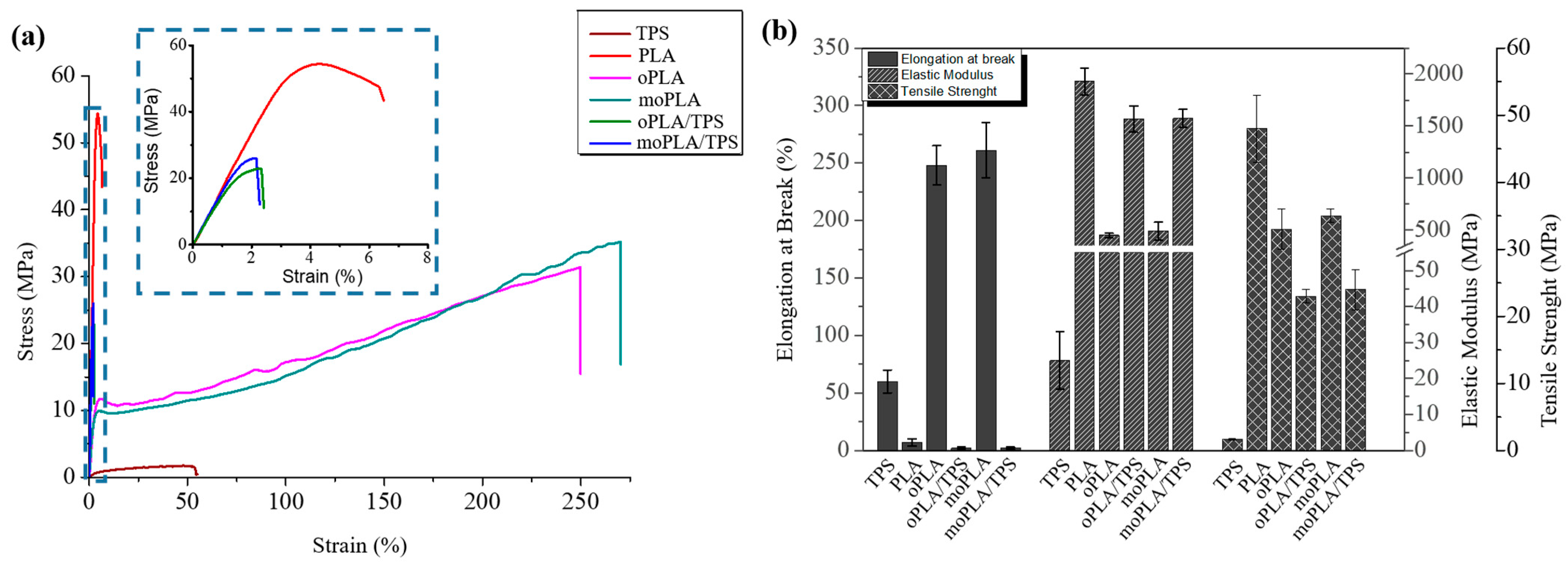
Mechanical properties of neat PLA, plasticized PLAs, and their blends with 40 wt.% of TPS were measured by tensile test. The tensile mechanical behavior of all the materials can be observed in
Figure 6a. When looking at the mechanical properties, plasticizers increase the ductility of the PLA-based materials leading to a higher elongation at break and reducing their stiffness and brittleness. As mentioned before, the mechanism involved in the plasticization process can be described by the interaction of the plasticizer molecules with the PLA chains, reducing the intermolecular forces, which results in an increase in free volume and chain mobility. The results of tensile tests are resumed in
Table 1.
It is easy to notice that plasticized PLA-based materials are more flexible than neat PLA. At the same time, a high decrease in the elastic modulus as well as in the tensile strength is observed compared with neat PLA. Therefore, the use of both plasticizers could lead to plasticized PLA-based materials with better performance for applications where high ductility is required compared with other plasticizers [
27]. Therefore, both plasticizers showed similar effects on tensile properties giving comparable values of tensile strength, elastic modulus, and elongation at break.
Moreover, when the plasticized PLA-based materials are blended with TPS, they are able to considerably increase the elastic modulus, slightly decrease their tensile strength, and dramatically drop down the elongation at break reaching values similar to that of neat PLA. Finally, the effect of the PLA plasticization and the compatibilization of its blends with TPS on the thermally-activated shape memory properties were studied in order to evaluate if these materials can overcome the PLA limitation and open up its potential application in biomedical field [
31]. Therefore, knowing the morphology of the materials as well as their mechanical and thermal properties, and considering that in general a T
trans ≈ T
g + 15 °C [
31,
32] is shown to study the thermally-activated shape memory response, we chose two different transition temperatures, one for neat PLA and one for the plasticized systems, closer to the human body temperature. In particular, the shape memory of neat PLA was thermally-activated at 75 °C while that of plasticized PLA-based materials and their blends with TPS was activated at 45 °C for comparison. A visualization test,
Figure 7, of the thermally-activated shape memory properties of neat PLA was done by bending the sample at the T
trans in the temporary shape then fixed at room temperature and recovered at the same temperature. The mechanism proposed involved a permanent network, composed of PLA chains physically entangled or linked thanks to the PLA crystals, able to memorize the permanent shape and store the driving energy needed to recover it. On the other hand, the switching phase, characterized by the T
trans, is formed by the amorphous PLA chains which can be frozen below their T
g in order to fix the temporary shape and reheated above their T
g to gain high mobility and activate the recovery of the permanent shape.
The thermo-mechanical cycles were performed at 50% elongation. The programming step was designed with a uniaxial stretching at the selected Ttrans (75 °C for PLA and 45 °C for the rest of materials), followed by a fast quenching of the stretched state at the Tfix, in particular 25 °C for PLA and 0 °C for the rest of materials. For PLA, it is not necessary bring down the fix temperature to 0 °C. However, for plasticized systems, taking into account that the transition temperature is only 45 °C, we have to choose a fix temperature lower than 25 °C in order to fix the temporary shape, choosing a difference of about 50 °C between both temperatures, in both cases, therefore a Tfix of 0 °C for the plasticized samples.
The stretched state was maintained after quenching and subsequent removal of the stress at the same temperature. Finally, the recovery of the permanent shape was activated at the T
trans.
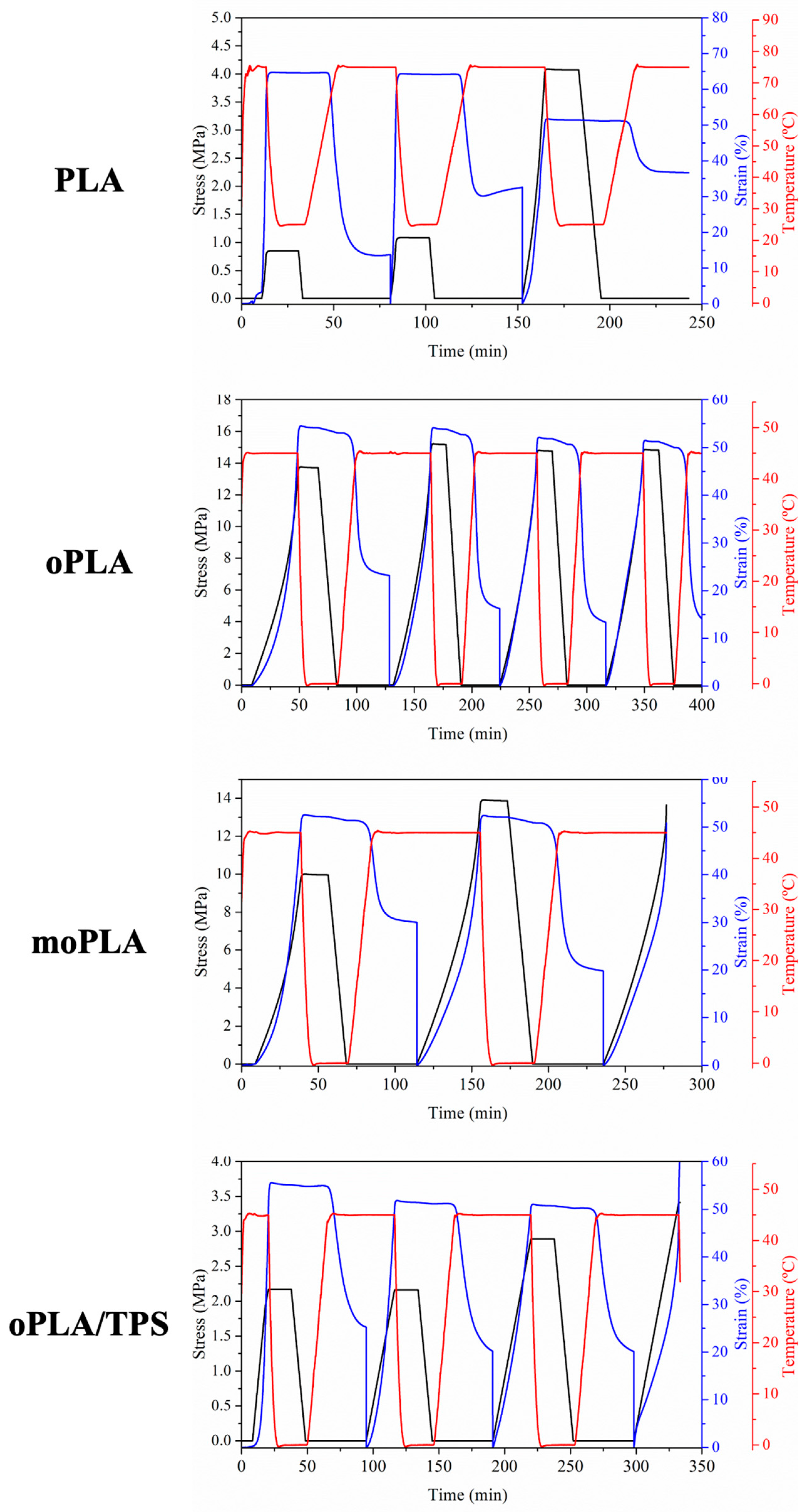
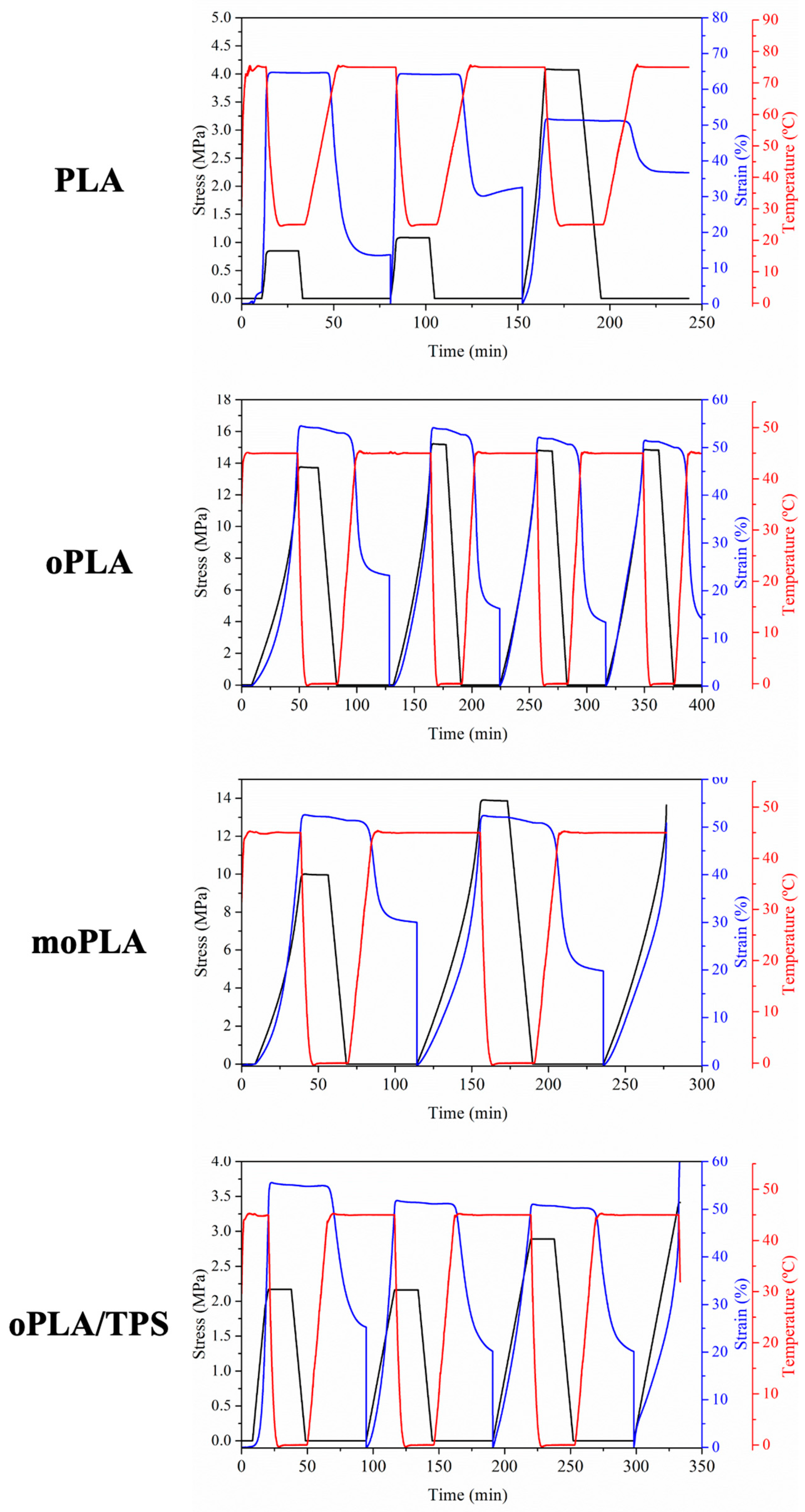
Figure 8 presents the evolution of strain, stress, and temperature in function of time during the dual-shape memory programming step for all the materials. The results related to the sample moPLA/TPS at 45 °C are not shown because it was not possible to perform the thermo-mechanical cycle due to the breaking of the sample.
It is worth noting that PLA showed an increase in the applied stress increasing the number of cycles, probably due to the induced crystallization as a consequence of the annealing process triggered at the T
trans during the recovery stage of the thermo-mechanical cycle. The effect of the annealing process was already reported in the literature for PLA/PCL blends demonstrating that the increase in the degree of crystallinity can increase the mechanical properties [
32]. The effect of PLA annealing is also visible in the values of R
r obtained for PLA by the thermo-mechanical cycles. In
Table 2, the results of the shape memory properties characterization, in terms of R
r and R
f, are summarized for all the samples. As it is easy to notice, PLA showed an excellent ability to fix the temporary shape, showing maximum values as high as 100%. On the other hand, the R
r values of PLA decreased, increasing the number of cycles and reaching minimum values of 28%. As mentioned above, this was probably due to the induced crystallization at the T
trans used, which is largely above the T
g of PLA. Thus, the mobility of the chains is increased and during the recovery step they might have the time to organize themselves in ordered crystals avoiding complete recovery. This behavior is due to the lack of stiffness of the permanent network that compose the PLA sample which is characterized mainly by physical entanglement that at the T
trans might not store all the driving energy needed to recover the permanent shape in the following step. Recently, Leones et al. [
28] reported the shape memory ability of neat electrospun PLA fibers thermally-activated at 60 °C showing excellent values of R
r and R
f. In their case, a part to adapt the T
trans used to be equal to the T
g of neat PLA, avoiding in this way the relaxation of the permanent network, and the neat PLA electrospun fibers showed a 5% of degree of crystallinity, being enough to increase the stiffness of the physical network of the sample at the T
trans.
Therefore, in our systems, oPLA showed excellent values of Rf and high values of Rr. This improvement is due to the fact that the permanent network of this sample is composed by physical entanglement together with crystals that increase the stiffness of the network that has to store the stretching energy during the programming step.
Looking at the shape memory ability of moPLA, we can easily notice that it was not possible to complete more than 2 consecutive cycles with the same sample. The applied stress needed to reach the elongation of 50% at the Ttrans was higher compared with oPLA. Evaluating the cycles obtained, it was possible to observe that the sample showed a higher Rr value in the second cycle while excellent Rf values were obtained for both cycles. Moreover, oPLA/TPS showed excellent Rf values and good Rr values. Compared with its counterpart without TPS, we can state that the thermally-activated shape memory ability depending on the PLA-based phase was almost totally preserved even while using 40 wt.% of TPS. Finally, as we mentioned above, it was not possible to perform the thermo-mechanical cycles with the compatibilized blend, moPLA/TPS. The high degree of crystallinity of the blend (20%) probably designed a stiff and brittle permanent network to be stretched at the Ttrans used, leading to the breaking of the sample. We can conclude that plasticized PLA/TPS blends show good thermally-activated shape memory response in the case of oPLA/TPS but the improvement of their compatibility in the moPLA/TPS leads to the loss of this property.



 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}