
Simultaneous Formation of Polyhydroxyurethanes and Multicomponent Semi-IPN Hydrogels

 , , , ,
, , , ,  , and
, and 
Abstract

1. Introduction
2. Experimental Section
2.1. Materials and General Methods
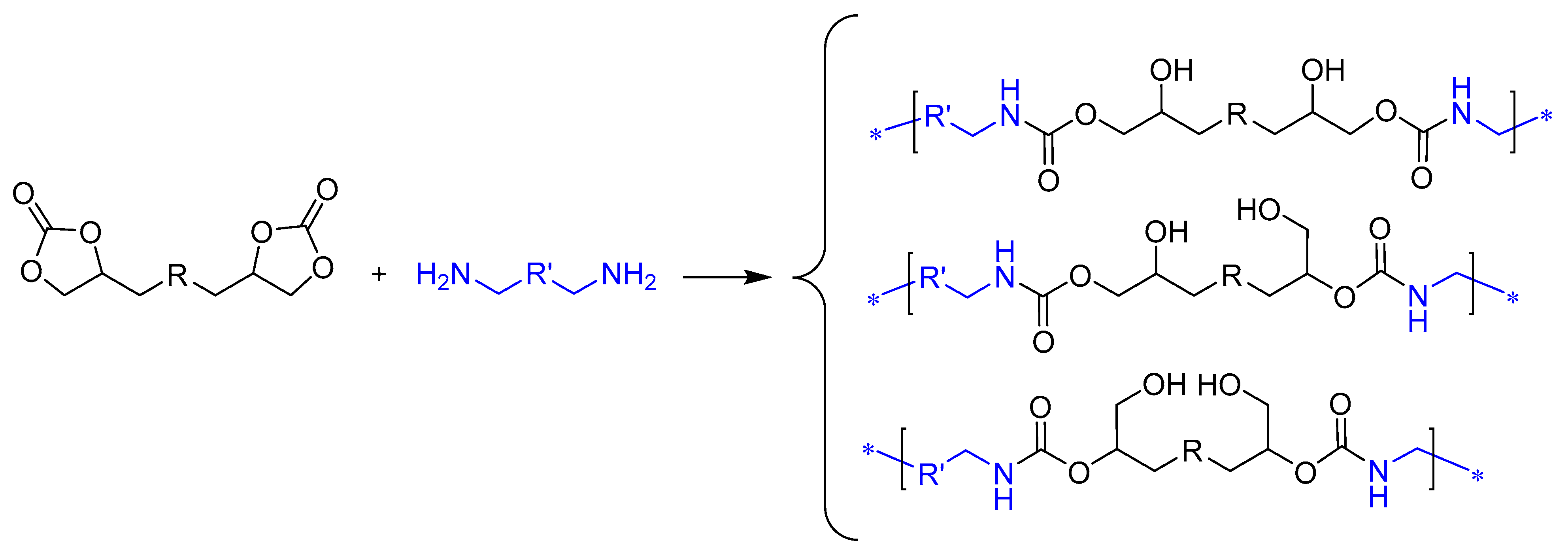
2.2. Preparation of PHU by Means of Non-Isocyanate Polyurethane (NIPU) Methodology
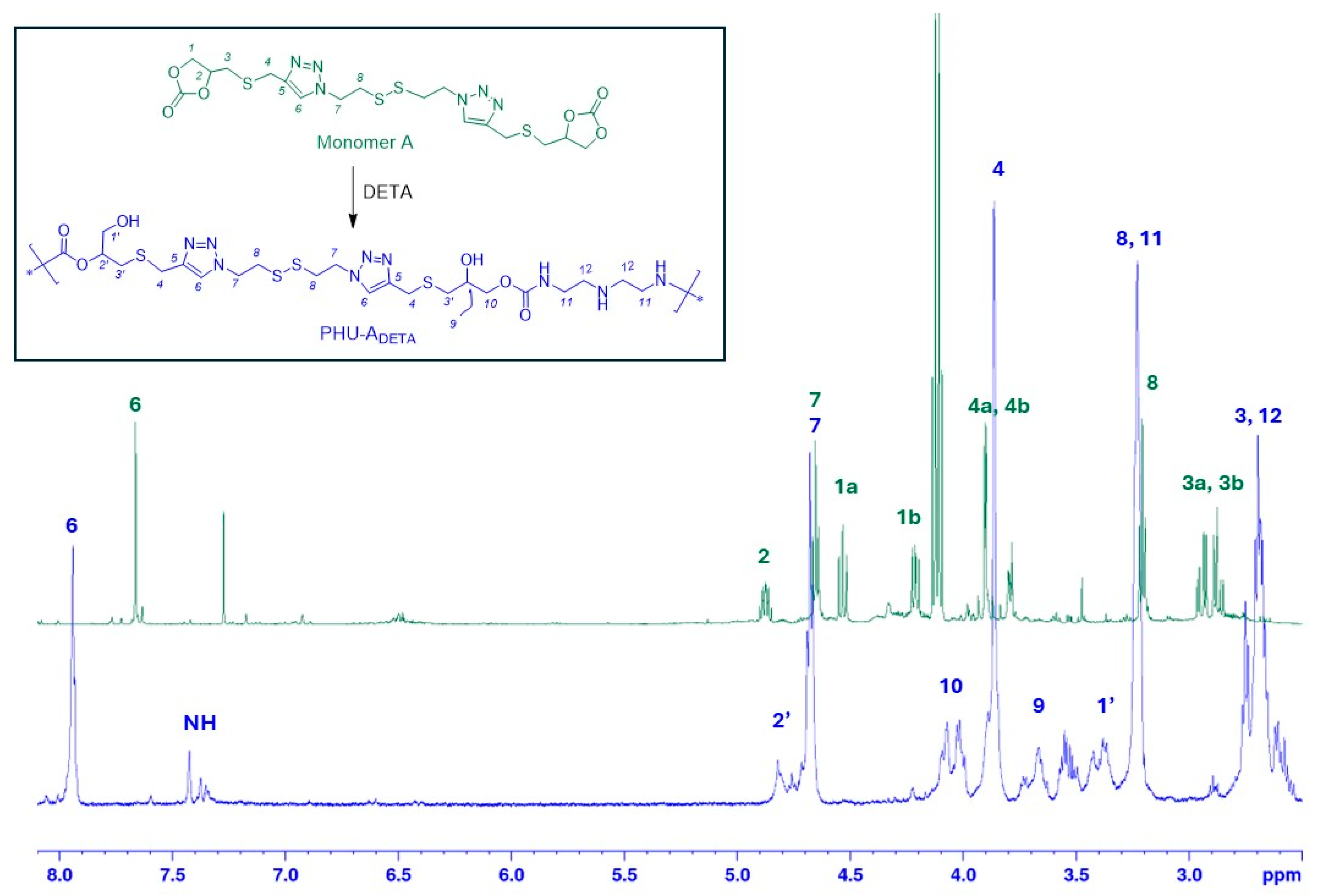
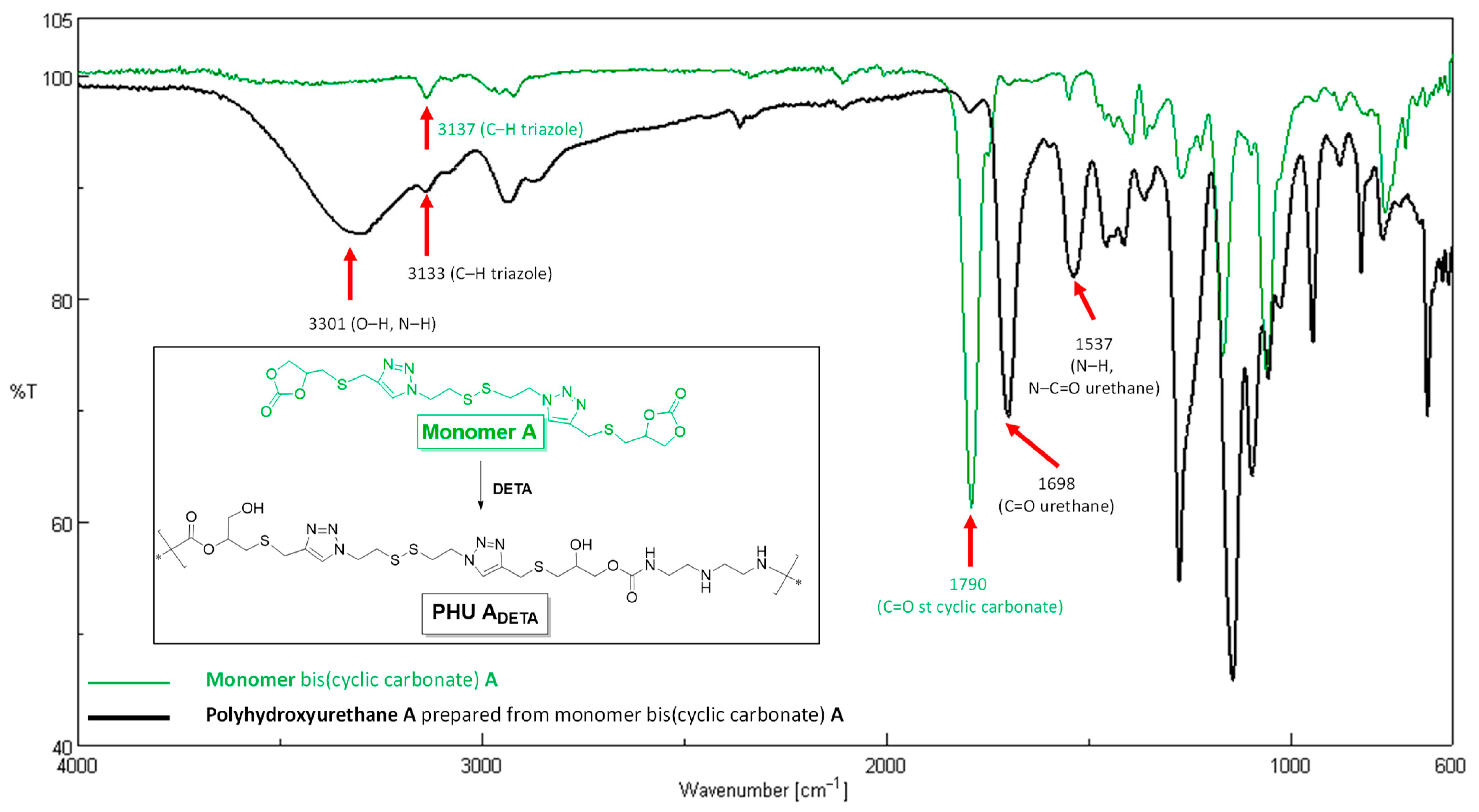
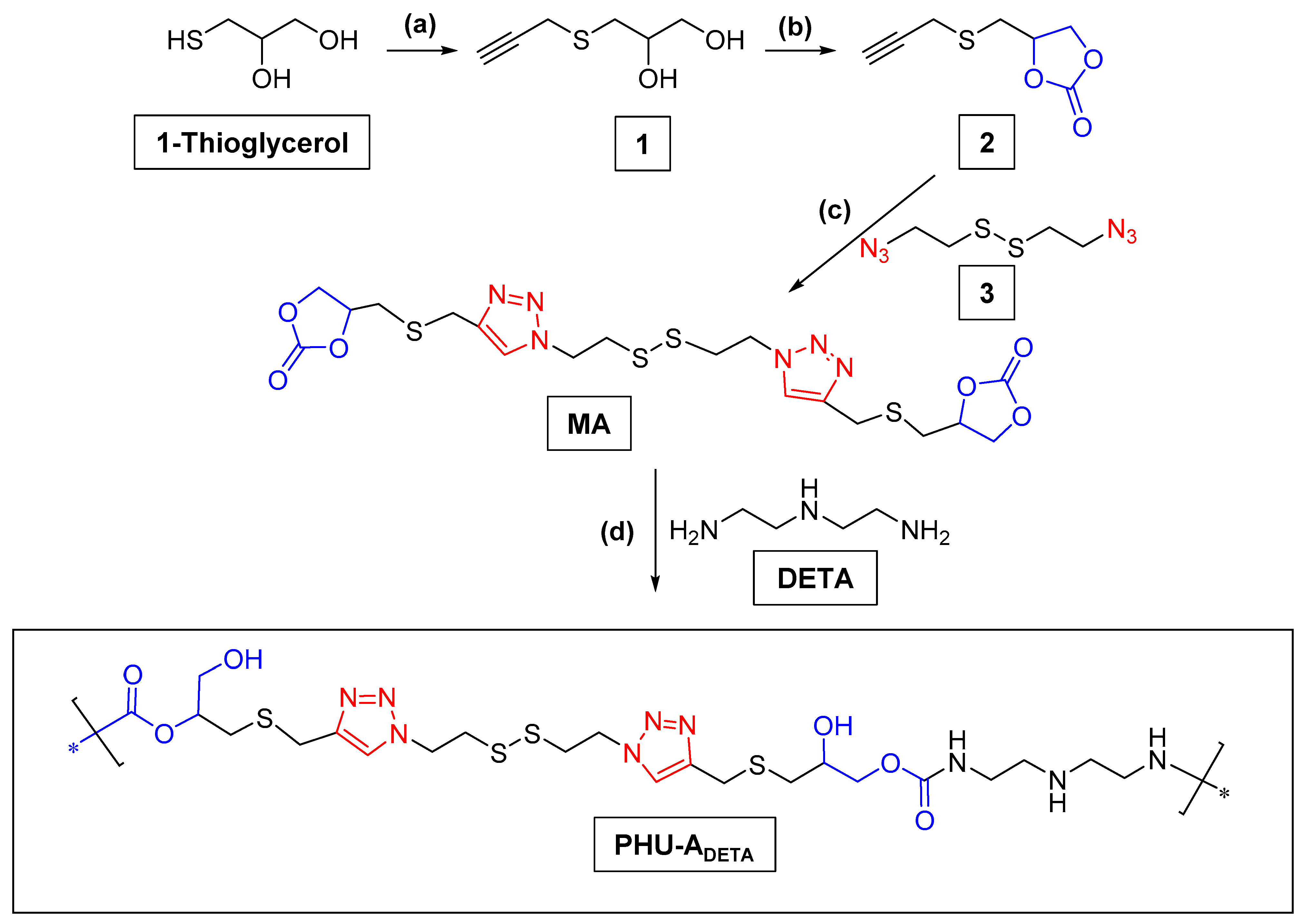
2.2.1. Preparation of PHU-ADETA from Monomer A and DETA
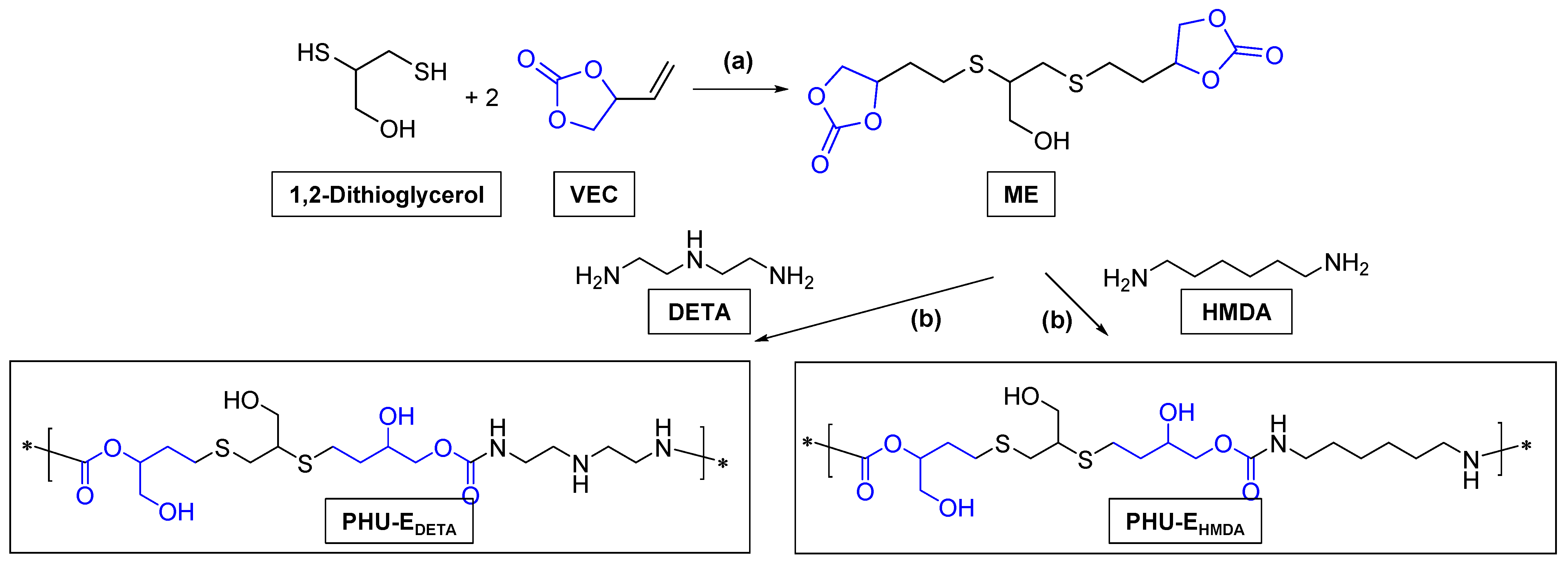
2.2.2. Preparation of PHU-EDETA and PHU-EHMDA
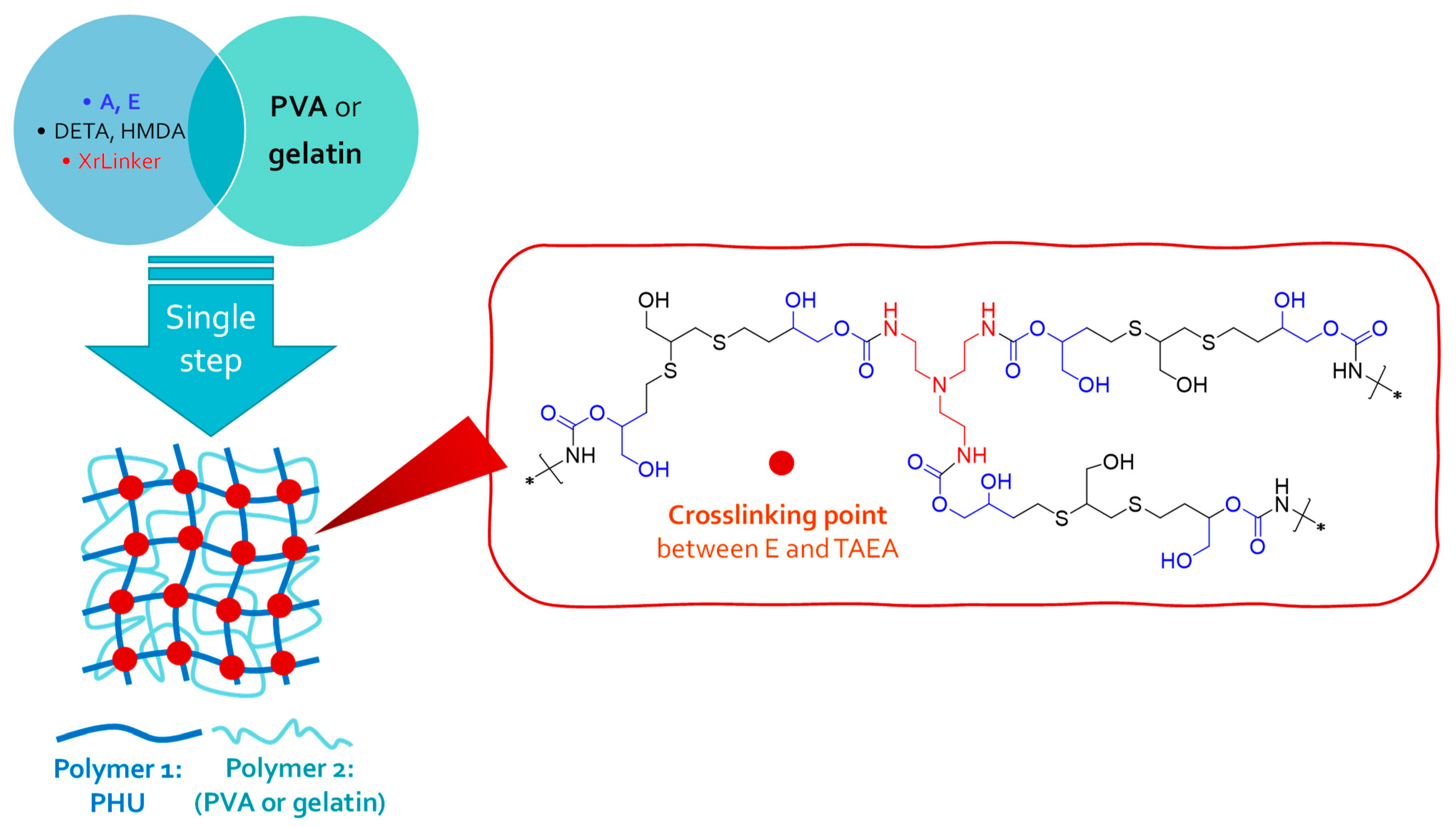
2.3. One-Step Procedure for the Manufacture of SIPN-Based Hydrogels. Characterization of the New Multicomponent Hydrogels
3. Results and Discussion
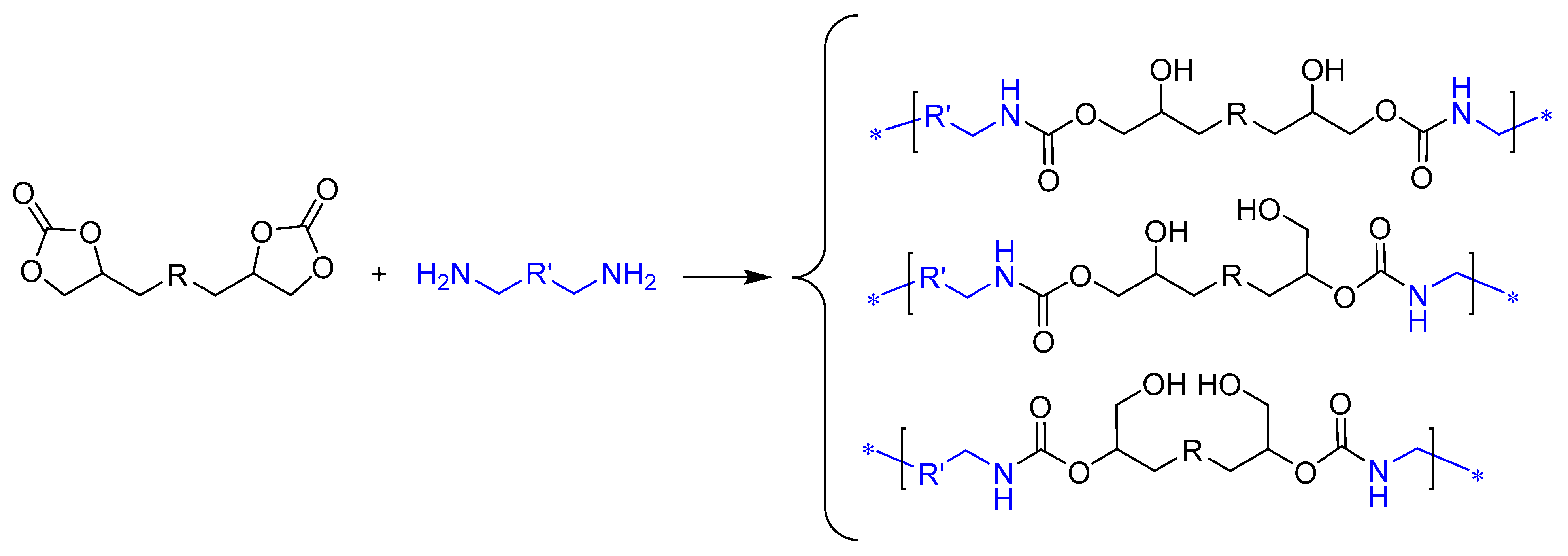
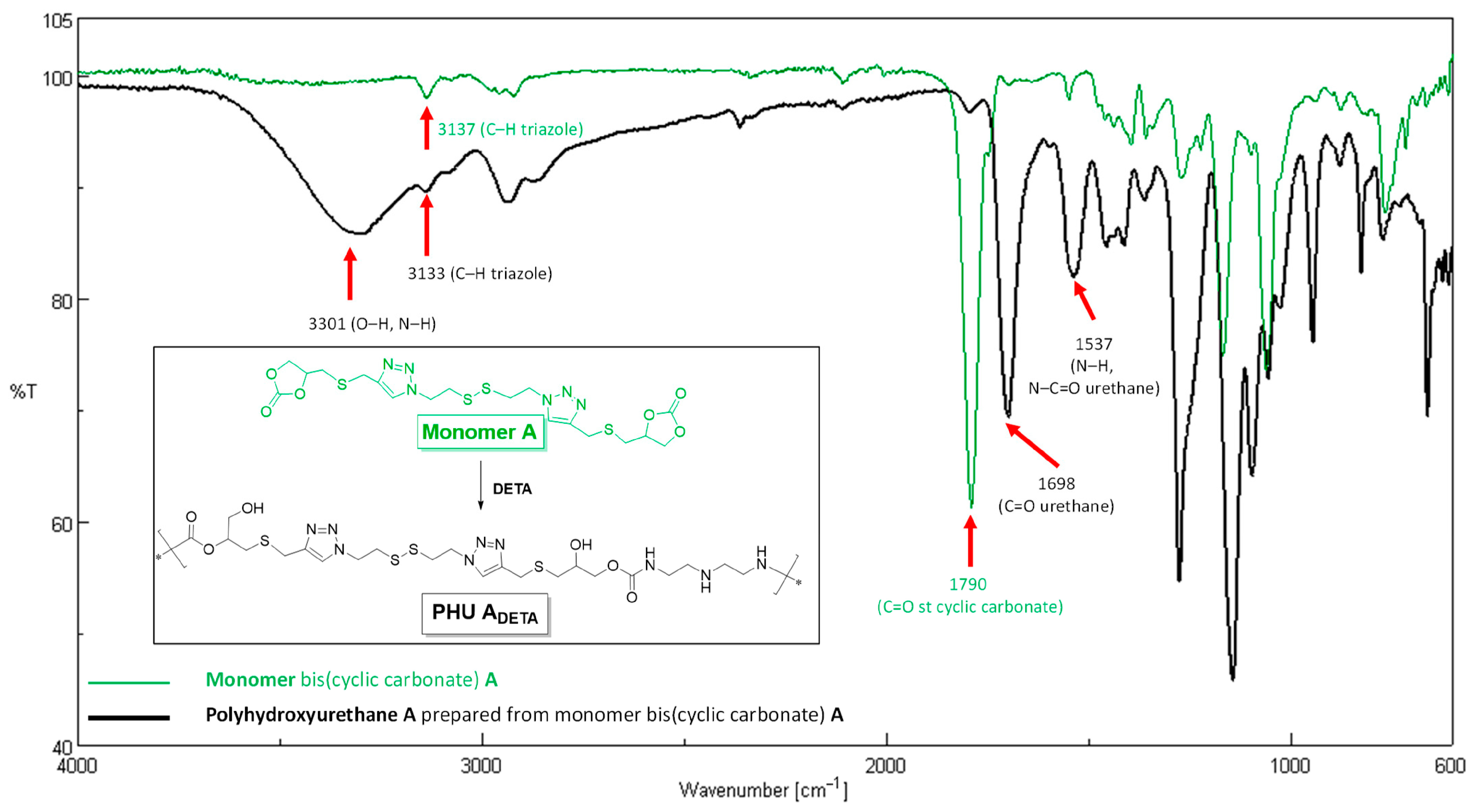
3.1. Synthesis of Monomer A and Preliminary Studies on PHU-ADETA Formation
3.2. Optimization of Polymerization Conditions for PHU-ADETA Formation
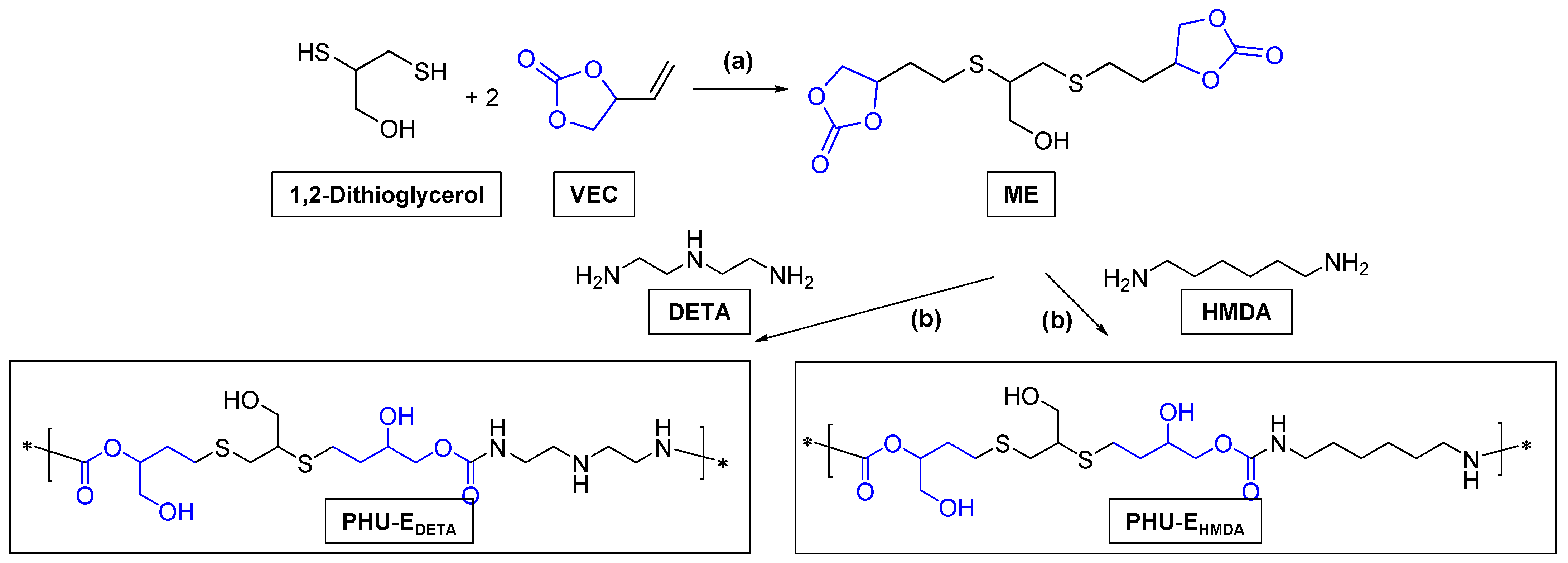
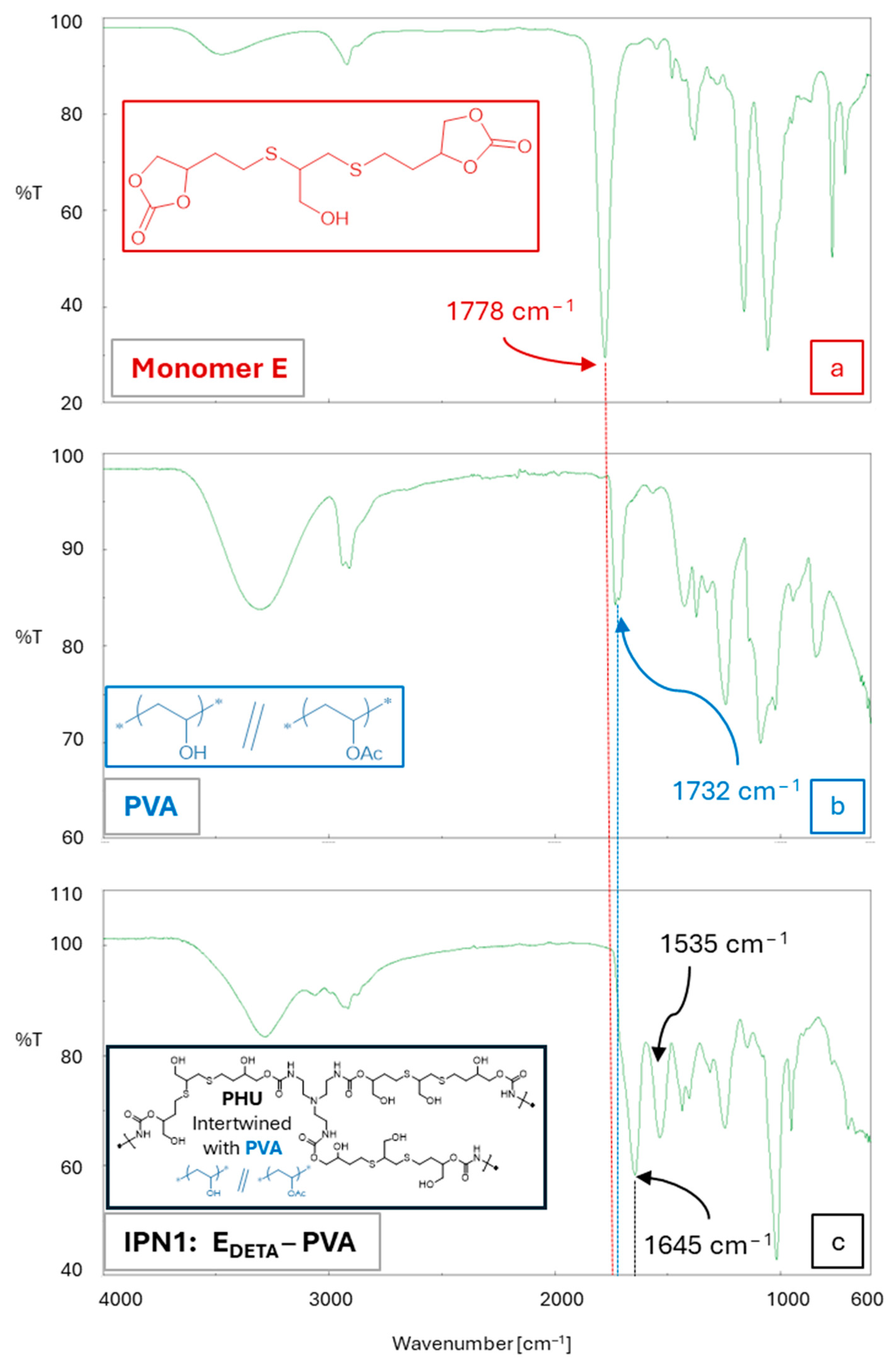
3.3. Synthesis of bis(Cyclic Carbonate) Monomer E and PHU-EDETA and PHU-EHMDA
3.4. One-Step Procedure for the Manufacture of PHU-Based Multicomponent Hydrogels
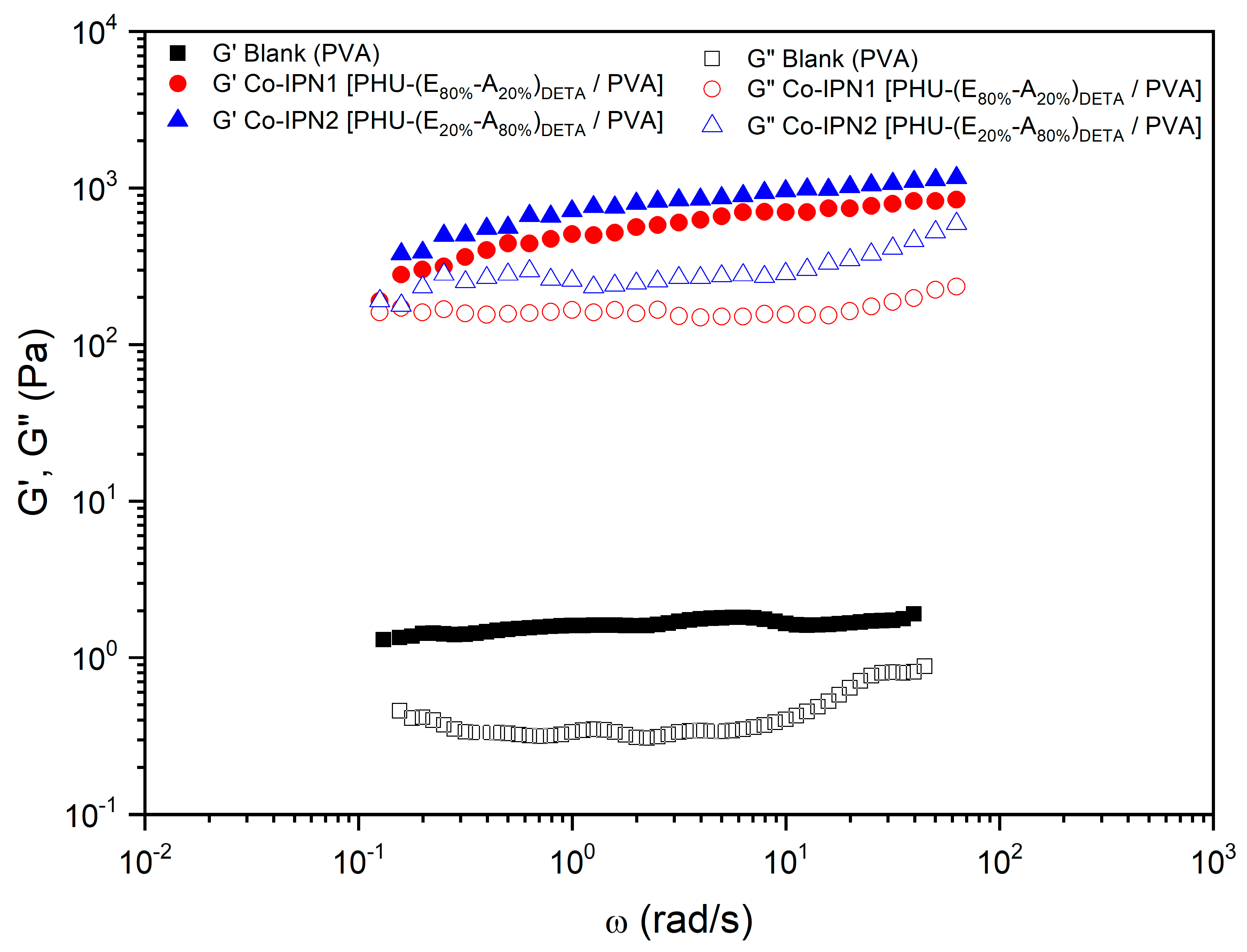
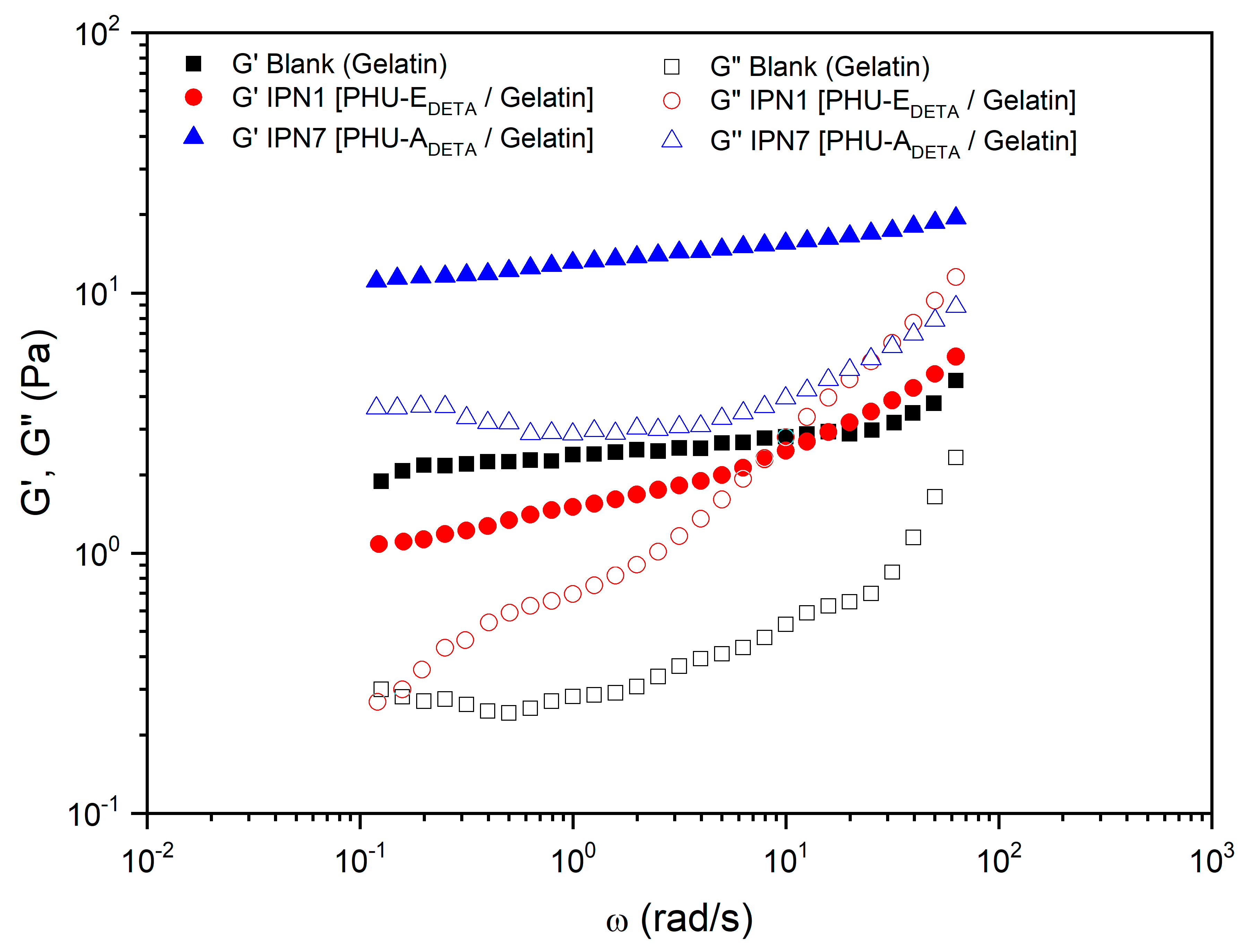
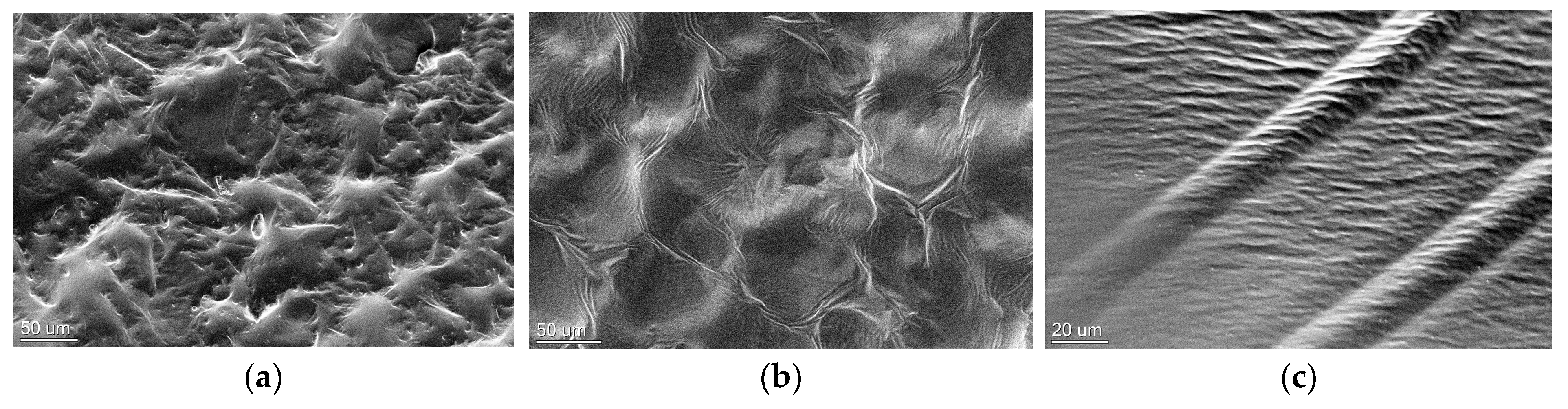
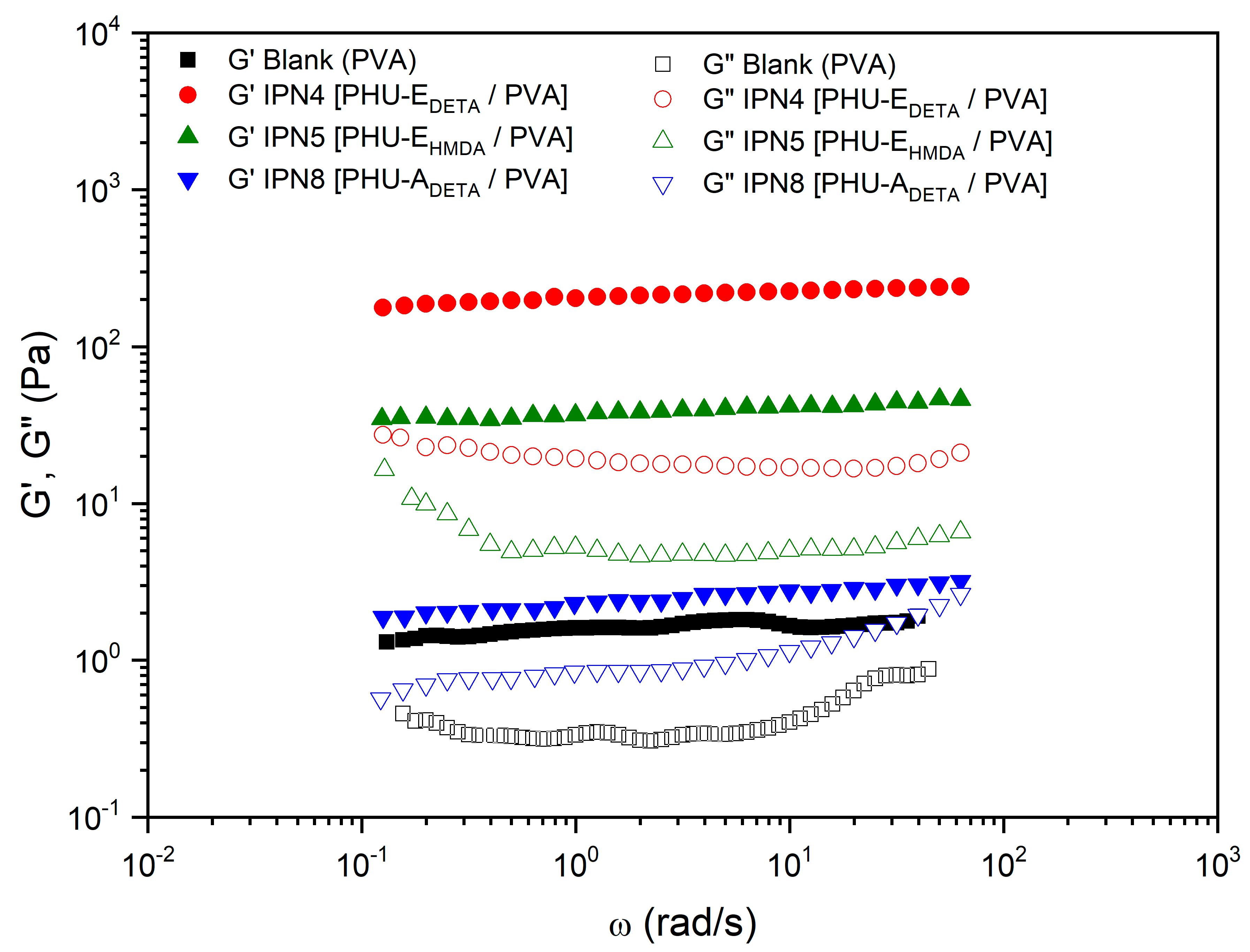
3.5. Characterization of Multicomponent Hydrogels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, H.K.; Kiick, K.L. Opportunities for multicomponent hybrid hydrogels in biomedical applications. Biomacromolecules 2015, 16, 28–42. [Google Scholar] [CrossRef]
- Jia, X.; Kiick, K.L. Hybrid multicomponent hydrogels for tissue engineering. Macromol. Biosci. 2009, 9, 140–156. [Google Scholar] [CrossRef]
- Derkus, B.; Okesola, B.O.; Barrett, D.W.; D’Este, M.; Chowdhury, T.T.; Eglin, D.; Mata, A. Multicomponent hydrogels for the formation of vascularized bone-like constructs in vitro. Acta Biomater. 2020, 109, 82–94. [Google Scholar] [CrossRef]
- Tanaka, W.; Shigemitsu, H.; Fujisaku, T.; Kubota, R.; Minami, S.; Urayama, K.; Hamachi, I. Post-assembly Fabrication of a Functional Multicomponent Supramolecular Hydrogel Based on a Self-Sorting Double Network. J. Am. Chem. Soc. 2019, 141, 4997–5004. [Google Scholar] [CrossRef]
- Galbis, J.A.; García-Martín, M.G.; De Paz, M.V.; Galbis, E. Synthetic Polymers from Sugar-Based Monomers. Chem. Rev. 2016, 116, 1600–1636. [Google Scholar] [CrossRef]
- Galbis, J.A.; García-Martín, M.G.; De-Paz, M.-V.; Galbis, E. Bio-based Polyurethanes from Carbohydrate Monomers. In Aspects of Polyurethanes; Yilmaz, F., Ed.; Intech: Rijeka, Croatia, 2017; pp. 155–192. ISBN 978-953-51-3545-6. [Google Scholar]
- Santerre, J.P.; Woodhouse, K.; Laroche, G.; Labow, R.S. Understanding the biodegradation of polyurethanes: From classical implants to tissue engineering materials. Biomaterials 2005, 26, 7457–7470. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.; de-Paz, M.V.; Aguilar-de-Leyva, A.; Caraballo, I.; Galbis, J.A. Reduction-sensitive functionalized copolyurethanes for biomedical applications. Polym. Chem. 2014, 5, 2370–2381. [Google Scholar] [CrossRef]
- Benito, E.; Romero-Azogil, L.; Galbis, E.; De-Paz, M.V.; García-Martín, M.G. Structurally simple redox polymersomes for doxorubicin delivery. Eur. Polym. J. 2020, 137, 109952. [Google Scholar] [CrossRef]
- de-Paz, M.V.; Marin, R.; Zamora, F.; Hakkou, K.; Alla, A.; Galbis, J.A.; Munoz-Guerra, S. Linear polyurethanes derived from alditols and diisocyanates. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4109–4117. [Google Scholar] [CrossRef]
- Mehta, R.; Kumar, V.; Bhunia, H.; Upadhyay, S.N. Synthesis of Poly(Lactic Acid): A Review. J. Macromol. Sci. Part C 2005, 45, 325–349. [Google Scholar] [CrossRef]
- Robert, J.L.; Aubrecht, K.B. Ring-Opening Polymerization of Lactide to Form a Biodegradable Polymer. J. Chem. Educ. 2008, 85, 258. [Google Scholar] [CrossRef]
- Valette, V.; Kébir, N.; Tiavarison, F.B.; Burel, F.; Lecamp, L. Preparation of flexible biobased non-isocyanate polyurethane (NIPU) foams using the transurethanization approach. React. Funct. Polym. 2022, 181, 105416. [Google Scholar] [CrossRef]
- Wołosz, D.; Parzuchowski, P.G.; Świderska, A. Synthesis and characterization of the non-isocyanate poly(carbonate-urethane)s obtained via polycondensation route. Eur. Polym. J. 2021, 155, 110574. [Google Scholar] [CrossRef]
- He, X.; Xu, X.; Wan, Q.; Bo, G.; Yan, Y. Solvent- and catalyst-free synthesis, hybridization and characterization of biobased nonisocyanate polyurethane (NIPU). Polymers 2019, 11, 1026. [Google Scholar] [CrossRef] [PubMed]
- Monie, F.; Grignard, B.; Thomassin, J.M.; Mereau, R.; Tassaing, T.; Jerome, C.; Detrembleur, C. 01.- Chemo- and Regioselective Additions of Nucleophiles to Cyclic Carbonates for the Preparation of Self-Blowing Non-Isocyanate Polyurethane Foams. Angew. Chem. Int. Ed. 2020, 59, 17033–17041. [Google Scholar] [CrossRef]
- Mao, H.I.; Chen, C.W.; Yan, H.C.; Rwei, S.P. Synthesis and characteristics of nonisocyanate polyurethane composed of bio-based dimer diamine for supercritical CO2 foaming applications. J. Appl. Polym. Sci. 2022, 139, e52841. [Google Scholar] [CrossRef]
- Ke, J.; Li, X.; Jiang, S.; Liang, C.; Wang, J.; Kang, M.; Li, Q.; Zhao, Y. Promising approaches to improve the performances of hybrid non-isocyanate polyurethane. Polym. Int. 2019, 68, 651–660. [Google Scholar] [CrossRef]
- Cornille, A.; Auvergne, R.; Figovsky, O.; Boutevin, B.; Caillol, S. A perspective approach to sustainable routes for non-isocyanate polyurethanes. Eur. Polym. J. 2017, 87, 535–552. [Google Scholar] [CrossRef]
- Grosso, R.; Benito, E.; Carbajo-Gordillo, A.I.; García-Martín, M.G.; Perez-Puyana, V.; Sánchez-Cid, P.; De-Paz, M.-V. Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept. Int. J. Mol. Sci. 2023, 24, 2281. [Google Scholar] [CrossRef] [PubMed]
- Matricardi, P.; Di Meo, C.; Coviello, T.; Hennink, W.E.; Alhaique, F. Interpenetrating polymer networks polysaccharide hydrogels for drug delivery and tissue engineering. Adv. Drug Deliv. Rev. 2013, 65, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cid, P.; Romero, A.; Díaz, M.J.; De-Paz, M.V.; Perez-Puyana, V. Chitosan-based hydrogels obtained via photoinitiated click polymer IPN reaction. J. Mol. Liq. 2023, 379, 121735. [Google Scholar] [CrossRef]
- Heo, J.Y.; Noh, J.H.; Park, S.H.; Ji, Y.B.; Ju, H.J.; Kim, D.Y.; Lee, B.; Kim, M.S. An injectable click-crosslinked hydrogel that prolongs dexamethasone release from dexamethasone-loaded microspheres. Pharmaceutics 2019, 11, 438. [Google Scholar] [CrossRef]
- León-Campos, M.I.; Claudio-Rizo, J.A.; Rodriguez-Fuentes, N.; Cabrera-Munguía, D.A.; Becerra-Rodriguez, J.J.; Herrera-Guerrero, A.; Soriano-Corral, F. Biocompatible interpenetrating polymeric networks in hydrogel state comprised from jellyfish collagen and polyurethane. J. Polym. Res. 2021, 28, 291. [Google Scholar] [CrossRef]
- Mushtaq, F.; Raza, Z.A.; Batool, S.R.; Zahid, M.; Onder, O.C.; Rafique, A.; Nazeer, M.A. Preparation, properties, and applications of gelatin-based hydrogels (GHs) in the environmental, technological, and biomedical sectors. Int. J. Biol. Macromol. 2022, 218, 601–633. [Google Scholar] [CrossRef] [PubMed]
- Bray, D. Critical Point Drying of Biological Specimens for Scanning Electron Microscopy. In Supercritical Fluid Methods and Protocols; Methods in Biotechnology; Williams, J.R., Clifford, A.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 235–243. [Google Scholar] [CrossRef]
- Carbajo-Gordillo, A.I.; Benito, E.; Galbis, E.; Grosso, R.; Iglesias, N.; García-Martín, M.-G.; De-Paz, M.-V. Spectra and GPC Data for Polyhydroxyurethanes Formation from Bis(cyclic carbonate) Monomers in Multicomponent Semi-IPN Hydrogels Fabrication [Dataset]; idUS (Depósito de Investigación de la Universidad de Sevilla): Sevilla, Spain, 2024. [Google Scholar] [CrossRef]
- Liu, C.; He, J.; van Ruymbeke, E.; Keunings, R.; Bailly, C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. [Google Scholar] [CrossRef]
- Baumgaertel, M.; Winter, H.H. Interrelation between continious and discrete time spectra. J. Non-Newton. Fluid Mech. 1992, 44, 15–36. [Google Scholar] [CrossRef]
- Besse, V.; Foyer, G.; Auvergne, R.; Caillol, S.; Boutevin, B.; Burk, R.M.; Roof, M.B. A safe and efficient method for conversion of 1,2- and 1,3-diols to cyclic carbonates utilizing triphosgene. Tetrahedron Lett. 1993, 51, 3284–3296. [Google Scholar] [CrossRef]
- Blain, M.; Yau, H.; Jean-Gérard, L.; Auvergne, R.; Benazet, D.; Schreiner, P.R.; Caillol, S.; Andrioletti, B. Urea-and thiourea-catalyzed aminolysis of carbonates. ChemSusChem 2016, 9, 2269–2272. [Google Scholar] [CrossRef]
- Haniffa, M.A.C.M.; Munawar, K.; Ching, Y.C.; Illias, H.A.; Chuah, C.H. Bio-based Poly(hydroxy urethane)s: Synthesis and Pre/Post-Functionalization. Chem. Asian J. 2021, 16, 1281–1297. [Google Scholar] [CrossRef]
- Blattmann, H.; Fleischer, M.; Bähr, M.; Mülhaupt, R. Isocyanate- and phosgene-free routes to polyfunctional cyclic carbonates and green polyurethanes by fixation of carbon dioxide. Macromol. Rapid Commun. 2014, 35, 1238–1254. [Google Scholar] [CrossRef]
- Schmidt, S.; Gatti, F.J.; Luitz, M.; Ritter, B.S.; Bruchmann, B.; Mülhaupt, R. Erythritol dicarbonate as intermediate for solvent-and isocyanate-free tailoring of bio-based polyhydroxyurethane thermoplastics and thermoplastic elastomers. Macromolecules 2017, 50, 2296–2303. [Google Scholar] [CrossRef]
- Blain, M.; Cornille, A.; Boutevin, B.; Auvergne, R.; Benazet, D.; Andrioletti, B.; Caillol, S. Hydrogen bonds prevent obtaining high molar mass PHUs. J. Appl. Polym. Sci. 2017, 134, 44958. [Google Scholar] [CrossRef]
- Cornille, A.; Blain, M.; Auvergne, R.; Andrioletti, B.; Boutevin, B.; Caillol, S. A study of cyclic carbonate aminolysis at room temperature: Effect of cyclic carbonate structures and solvents on polyhydroxyurethane synthesis. Polym. Chem. 2017, 8, 592–604. [Google Scholar] [CrossRef]
- Cornille, A.; Guillet, C.; Benyahya, S.; Negrell, C.; Boutevin, B.; Caillol, S. Room temperature flexible isocyanate-free polyurethane foams. Eur. Polym. J. 2016, 84, 873–888. [Google Scholar] [CrossRef]
- Guzmán Agudelo, A.F.; Pérez-Sena, W.Y.; Kebir, N.; Salmi, T.; Ríos, L.A.; Leveneur, S. Influence of steric effects on the kinetics of cyclic-carbonate vegetable oils aminolysis. Chem. Eng. Sci. 2020, 228, 115954. [Google Scholar] [CrossRef]
- Mikhailov, O.V. Molecular structure design and soft template synthesis of aza-, oxaaza- and thiaazamacrocyclic metal chelates in the gelatin matrix. Arab. J. Chem. 2017, 10, 47–67. [Google Scholar] [CrossRef]
- Tijing, L.D.; Woo, Y.C.; Yao, M.; Ren, J.; Shon, H.K. 1.16 Electrospinning for Membrane Fabrication: Strategies and Applications. In Comprehensive Membrane Science and Engineering; Drioli, E., Giorno, L., Fontananova, E., Eds.; Elsevier: Oxford, UK, 2017; pp. 418–444. ISBN 978-0-444-63796-3. [Google Scholar]
- Almdal, K.; Dyre, J.; Hvidt, S.; Kramer, O. Towards a phenomenological definition of the term “gel”. Polym. Gels Netw. 1993, 1, 5–17. [Google Scholar] [CrossRef]
- Lu, L.; Liu, X.; Tong, Z. Critical exponents for sol-gel transition in aqueous alginate solutions induced by cupric cations. Carbohydr. Polym. 2006, 65, 544–551. [Google Scholar] [CrossRef]
- Sachlos, E.; Wahl, D.A.; Triffitt, J.T.; Czernuszka, J.T. The impact of critical point drying with liquid carbon dioxide on collagen-hydroxyapatite composite scaffolds. Acta Biomater. 2008, 4, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, R.; Xu, N.; Du, F.S.; Wang, Y.L.; Tan, Y.X.; Ji, S.P.; Liang, D.H.; Li, Z.C. Reduction-degradable linear cationic polymers as gene carriers prepared by Cu(I)-catalyzed azide-alkyne cycloaddition. Biomacromolecules 2011, 12, 66–74. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Hydrogels Prepared and Blanks | Polymer 1 (PHU) Conc.: 10% w/v | Polymer 2 (Polymer Scaffold) Conc.: 10% w/v | Solvents | Catalyst | (Pa) | tan (δ)1 (b) | |
|---|---|---|---|---|---|---|---|---|
| Monomer 1 (bisCC) | Monomer 2 (Diamine) | |||||||
| 1 | Blank: Gelatin | - | - | Gelatin | DMSO-H2O 1:1 | 2.24 | 0.10 | |
| 2 | Blank: Gelatin | - | - | Gelatin | EtOH-H2O 1:1 | 1240.46 | 0.12 | |
| 3 | Blank: PVA | - | - | PVA | DMSO-H2O 1:1 | 1.86 | 0.21 | |
| 4 | Blank: PVA | - | - | PVA | EtOH-H2O 1:1 | 11.9 | 0.35 | |
| 5 | IPN1 [PHU-EDETA/Gelatin] | E | DETA | Gelatin | DMSO-H2O 1:1 | DBU | 1.45 | 0.44 |
| 6 | IPN2 [PHU-EHMDA/Gelatin] | E | HMDA | Gelatin | DMSO-H2O 1:1 | DBU | 4.29 | 0.20 |
| 7 | IPN3 [PHU-EDETA/Gelatin] | E | DETA | Gelatin | EtOH-H2O 1:1 | TU | 5.19 | 0.81 |
| 8 | IPN4 [PHU-EDETA/PVA] | E | DETA | PVA | DMSO-H2O 1:1 | DBU | 225.44 | 0.09 |
| 9 | IPN5 [PHU-EHMDA/PVA] | E | HMDA | PVA | DMSO-H2O 1:1 | DBU | 40.97 | 0.14 |
| 10 | IPN6 [PHU-EDETA/PVA] | E | DETA | PVA | EtOH-H2O 1:1 | TU | 8.28 | 0.19 |
| 11 | IPN7 [PHU-ADETA/Gelatin] | A | DETA | Gelatin | DMSO-H2O 1:1 | DBU | 14.34 | 0.22 |
| 12 | IPN8 [PHU-ADETA/PVA] | A | DETA | PVA | DMSO-H2O 1:1 | DBU | 2.39 | 0.38 |
| 13 | co-IPN1 [PHU-(E80%-A20%)DETA/PVA] | E80%-A20% | DETA | PVA | DMSO-H2O 1:1 | DBU | 742.12 | 0.32 |
| 14 | co-IPN2 [PHU-(E20%-A80%)DETA/PVA] | E20%-A80% | DETA | PVA | DMSO-H2O 1:1 | DBU | 958.39 | 0.35 |
| Sample | BisCC | Diamine | [Monomer] (mol/L) | Solvent | Temp. (°C) | Catalyst | (kDa) | (b) (°C) | (c) (°C) |
|---|---|---|---|---|---|---|---|---|---|
| PHU-ADETA (d) | A | DETA | 1.8 mol/L | TFE | 25 | TU | 16.4 | −3.0 | -- |
| PHU-EDETA (e) | E | DETA | 1.8 mol/L | EtOH | 50 | TU | 29.6 | −10.3 | -- |
| PHU-EHMDA (f) | E | HMDA | 1.8 mol/L | DMSO | 50 | DBU | 34.1 | 14.2 | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbajo-Gordillo, A.I.; Benito, E.; Galbis, E.; Grosso, R.; Iglesias, N.; Valencia, C.; Lucas, R.; García-Martín, M.-G.; de-Paz, M.-V. Simultaneous Formation of Polyhydroxyurethanes and Multicomponent Semi-IPN Hydrogels. Polymers 2024, 16, 880. https://doi.org/10.3390/polym16070880
Carbajo-Gordillo AI, Benito E, Galbis E, Grosso R, Iglesias N, Valencia C, Lucas R, García-Martín M-G, de-Paz M-V. Simultaneous Formation of Polyhydroxyurethanes and Multicomponent Semi-IPN Hydrogels. Polymers. 2024; 16(7):880. https://doi.org/10.3390/polym16070880
Chicago/Turabian StyleCarbajo-Gordillo, Ana I., Elena Benito, Elsa Galbis, Roberto Grosso, Nieves Iglesias, Concepción Valencia, Ricardo Lucas, M.-Gracia García-Martín, and M.-Violante de-Paz. 2024. "Simultaneous Formation of Polyhydroxyurethanes and Multicomponent Semi-IPN Hydrogels" Polymers 16, no. 7: 880. https://doi.org/10.3390/polym16070880
APA StyleCarbajo-Gordillo, A. I., Benito, E., Galbis, E., Grosso, R., Iglesias, N., Valencia, C., Lucas, R., García-Martín, M.-G., & de-Paz, M.-V. (2024). Simultaneous Formation of Polyhydroxyurethanes and Multicomponent Semi-IPN Hydrogels. Polymers, 16(7), 880. https://doi.org/10.3390/polym16070880