


Improved Approach for ab Initio Calculations of Rate Coefficients for Secondary Reactions in Acrylate Free-Radical Polymerization


Abstract

1. Introduction
2. Computational Details
2.1. Level of Theory
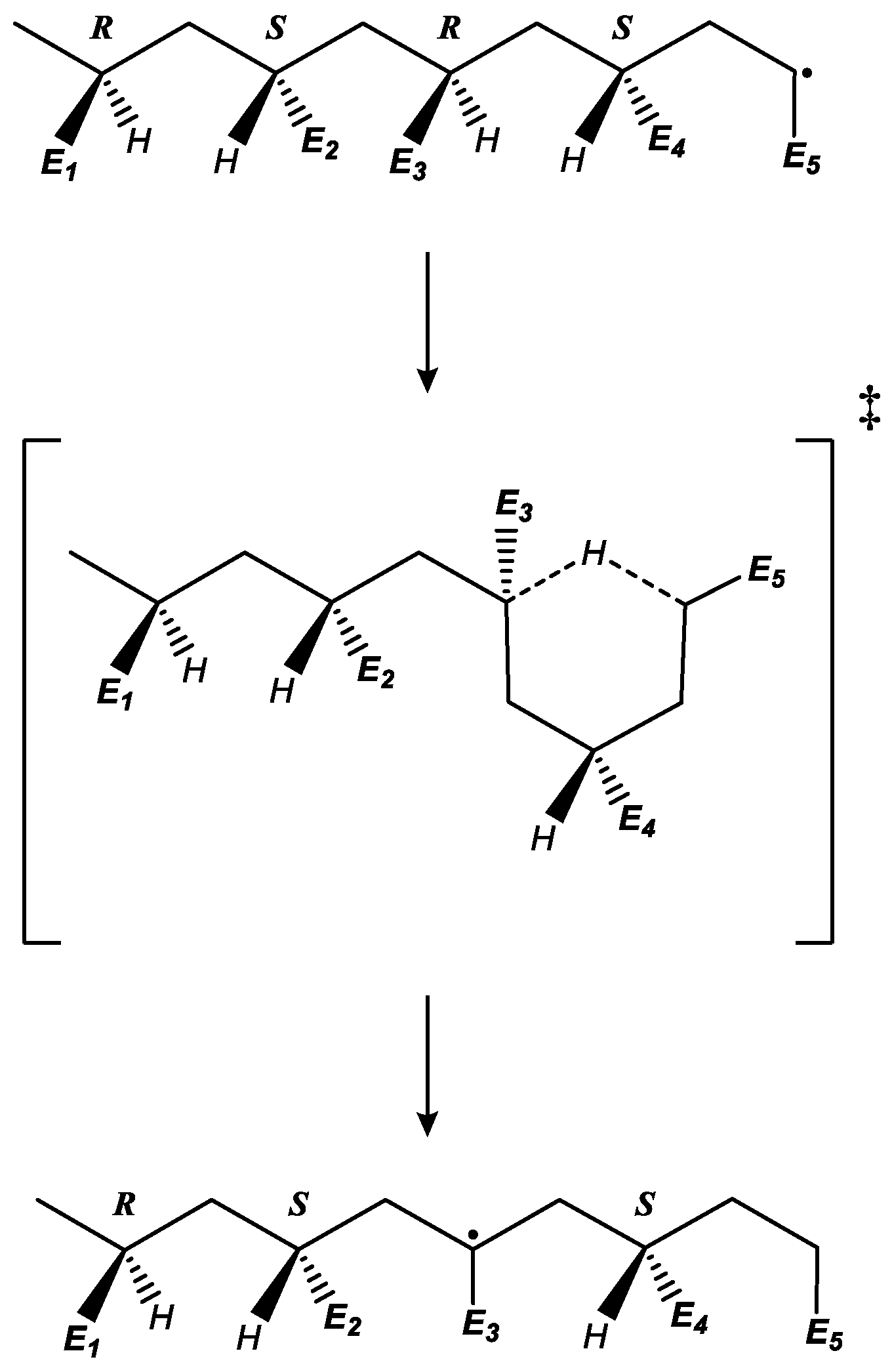
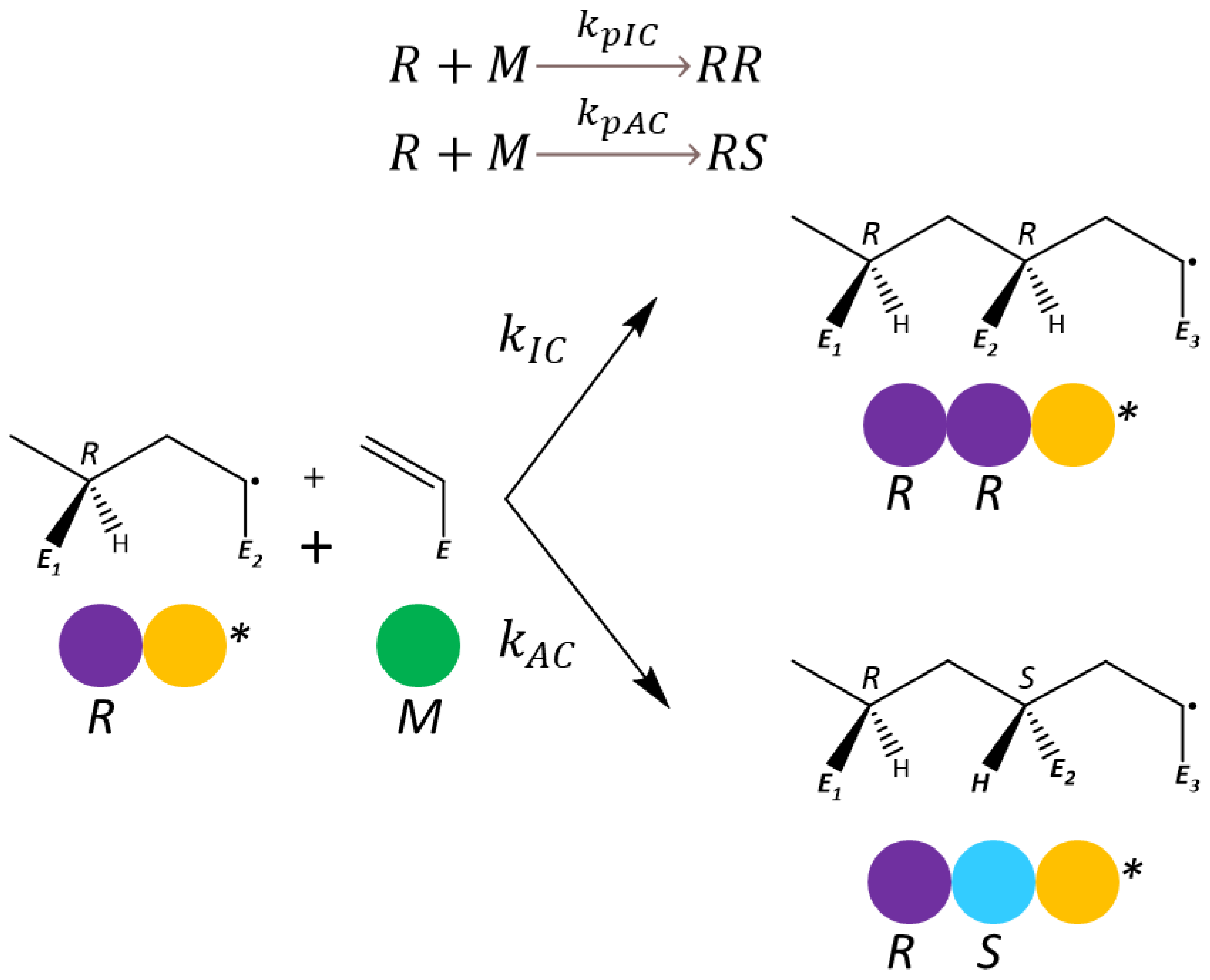
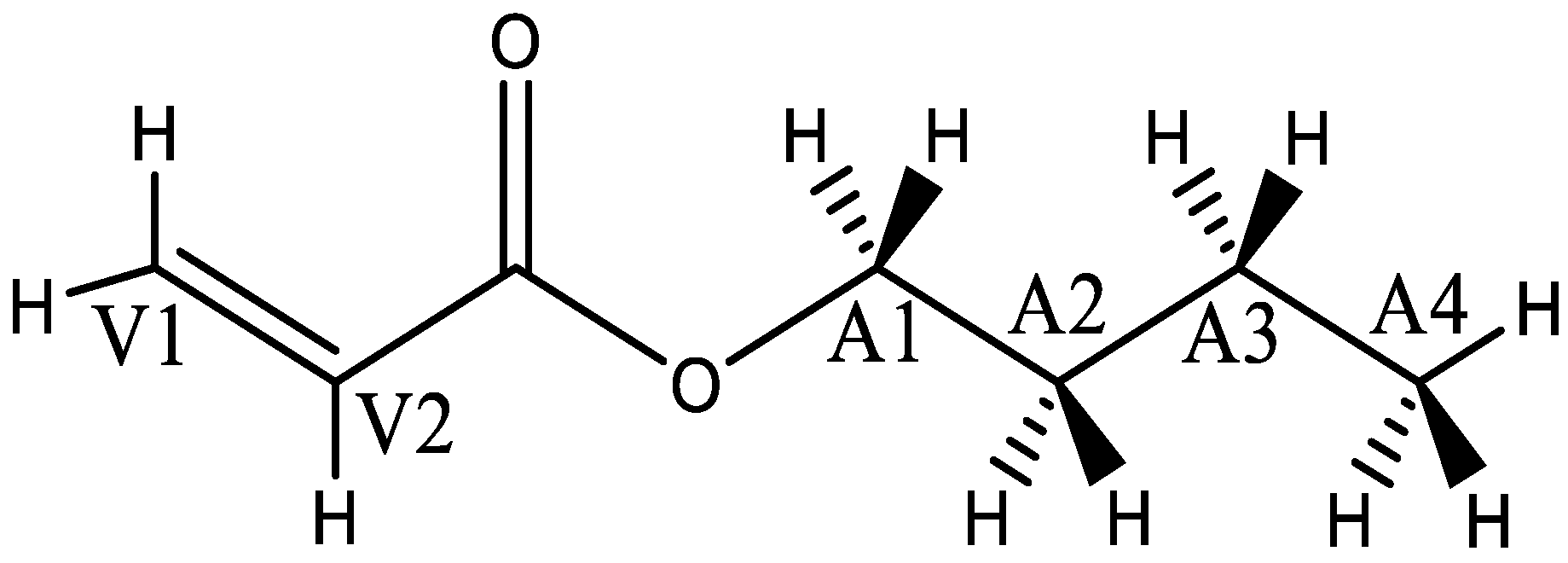
2.2. Molecular-Model Construction Considering Chiral Effects
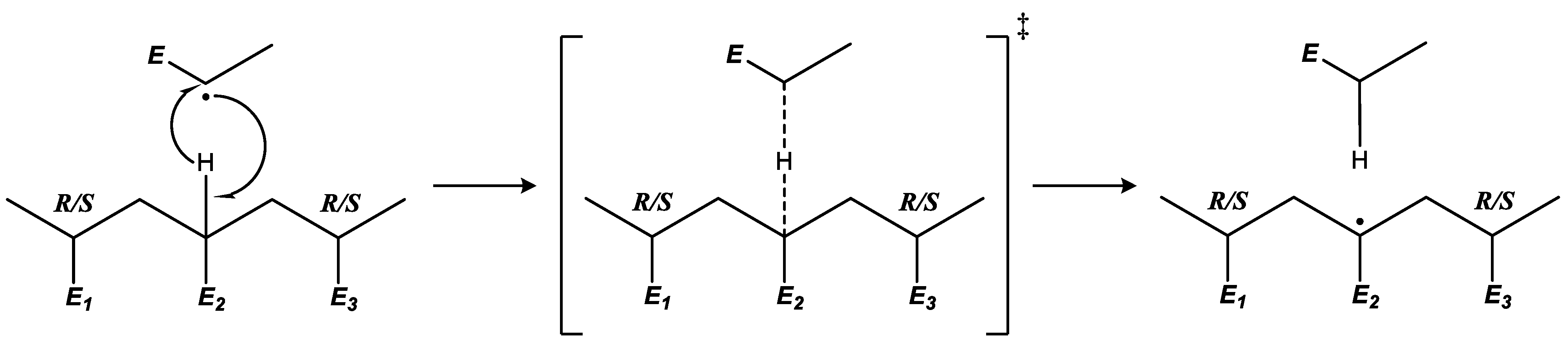
2.3. Reduced-Flexibility Approach
2.4. Simulation Details
3. Results and Discussion
3.1. Backbiting in Atactic Polymer Chains
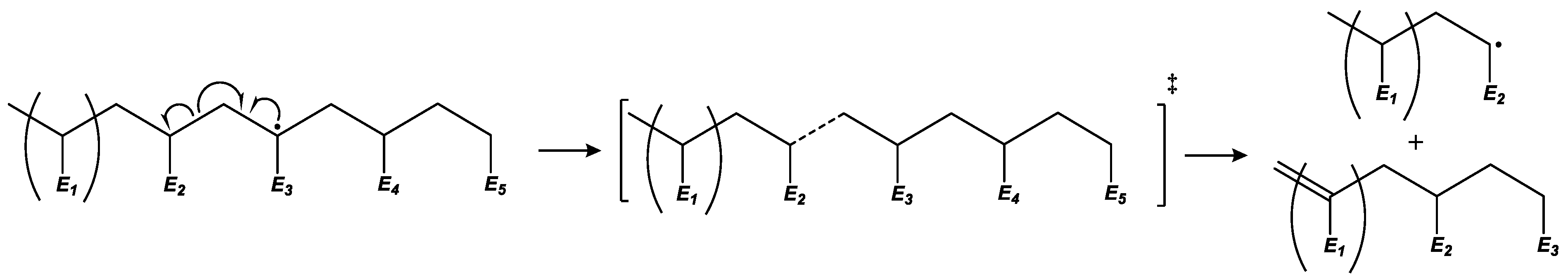
3.2. β-Scission
3.3. Migration
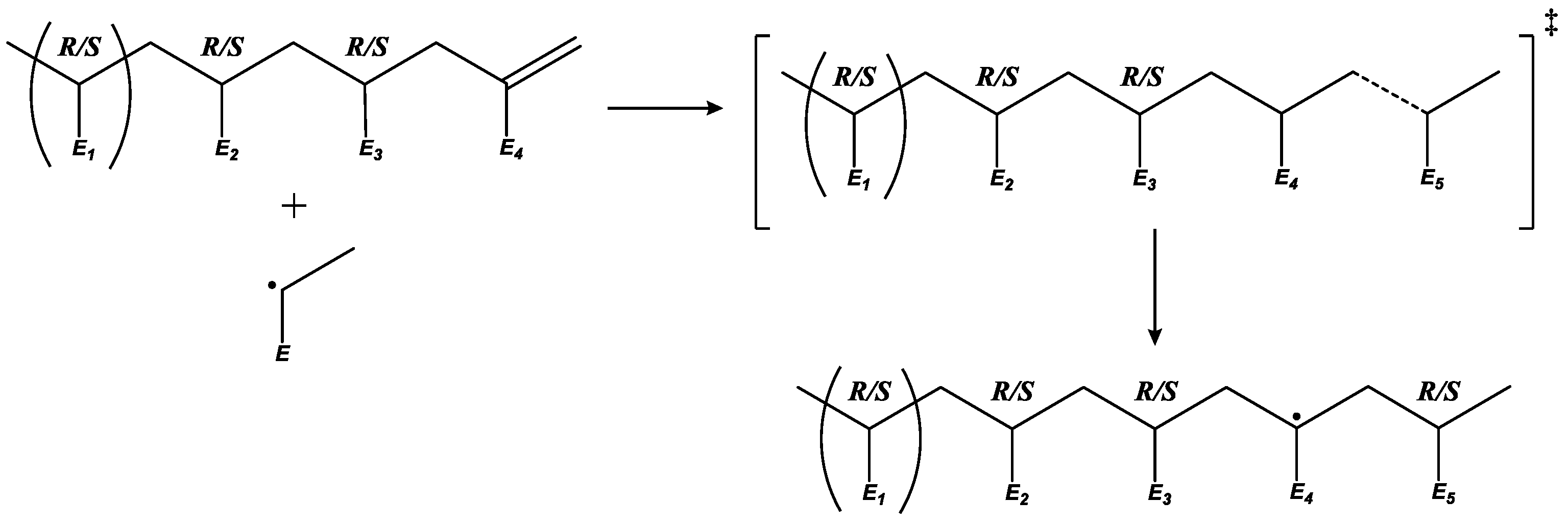
3.4. Macromonomer Propagation
3.5. MCR Propagation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acrylate | k @ 298.15K [L mol−1 s−1] | k @ 413.15K [L mol−1 s−1] | A [L mol−1 s−1] | Ea [kJ mol−1] | Source |
|---|---|---|---|---|---|
| methyl R-MCR-R | 3.11 × 101 | 6.78 × 102 | 6.5 × 106 | 30.0 | This work |
| R-MCR-S | 3.66 × 101 | 1.06 × 103 | 2.0 × 106 | 27.5 | |
| WA | 3.36 × 101 | 8.49 × 102 | 3.7 × 106 | 28.8 | |
| methyl | 1.79 × 101 | 3.64 × 102 | 8.9 ± 0.5 × 105 | 26.8 ± 1.5 | [60] |
| n-butyl | 1.31 × 101 | 3.37 × 102 | 1.52 ± 0.14 × 106 | 28.9 ± 3.2 | [58] |
| n-butyl | 1.05 × 101 | 2.51 × 102 | 9.2 × 105 | 28.3 | [71] |
| n-butyl | 1.05 × 101 | 3.10 × 102 | 1.98 × 106 * | 30.1 ± 9.7 | [50] |
| t-butyl | 2.87 × 100 | 3.21 × 101 | 1.68 × 104 ** | 21.5 ± 3.6 | [69] |
| dodecyl | 4.57 × 100 | 1.12 × 102 | 4.5 ± 0.8 × 105 | 28.5 ± 1.4 | [60] |
3.6. Chain Transfer to Monomer
3.7. Chain Transfer to Polymer
3.8. kMC Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Serrano-Aroca, Á.; Deb, S. Acrylate Polymers for Advanced Applications; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar]
- Agboluaje, M.; Refai, I.; Manston, H.H.; Hutchinson, R.A.; Dušička, E.; Urbanová, A.; Lacík, I. A comparison of the solution radical propagation kinetics of partially water-miscible non-functional acrylates to acrylic acid. Polym. Chem. 2020, 11, 7104–7114. [Google Scholar] [CrossRef]
- Penzel, E.; Ballard, N.; Asua, J.M. Polyacrylates. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley Online Library: Wiley-VCH, Weinheim, 2018; pp. 1–20. [Google Scholar]
- Gómez, P.G. Development of Electrochemical (bio) Sensors and Microanalytical Systems: Application to the Wine Industry. Ph.D. Thesis, Universitat Autònoma de Barcelona, Barcelona, Spain, 2017. [Google Scholar]
- Vandenbergh, T.J.J.; Olabisi, O.; Adewale, K. Handbook of Thermoplastics; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Barner-Kowollik, C.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Coote, M.L.; Hutchinson, R.A.; Junkers, T.; Lacík, I.; Russell, G.T.; et al. Critically evaluated rate coefficients in radical polymerization—7. Secondary-radical propagation rate coefficients for methyl acrylate in the bulk. Polym. Chem. 2014, 5, 204–212. [Google Scholar] [CrossRef]
- Bakhshi, H.; Kuang, G.; Wieland, F.; Meyer, W. Photo-Curing Kinetics of 3D-Printing Photo-Inks Based on Urethane-Acrylates. Polymers 2022, 14, 2974. [Google Scholar] [CrossRef]
- Konuray, O.; Morancho, J.M.; Fernández-Francos, X.; García-Alvarez, M.; Ramis, X. Curing kinetics of dually-processed acrylate-epoxy 3D printing resins. Thermochim. Acta 2021, 701, 178963. [Google Scholar] [CrossRef]
- Ballard, N.; Asua, J.M. Radical polymerization of acrylic monomers: An overview. Prog. Polym. Sci. 2018, 79, 40–60. [Google Scholar] [CrossRef]
- Pirman, T.; Ocepek, M.; Likozar, B. Radical Polymerization of Acrylates, Methacrylates, and Styrene: Biobased Approaches, Mechanism, Kinetics, Secondary Reactions, and Modeling. Ind. Eng. Chem. Res. 2021, 60, 9347–9367. [Google Scholar] [CrossRef]
- Miasnikova, A.; Laschewsky, A. Influencing the phase transition temperature of poly(methoxy diethylene glycol acrylate) by molar mass, end groups, and polymer architecture. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3313–3323. [Google Scholar] [CrossRef]
- Márquez, I.; Alarcia, F.; Velasco, J.I. Synthesis and Properties of Water-Based Acrylic Adhesives with a Variable Ratio of 2-Ethylhexyl Acrylate and n-Butyl Acrylate for Application in Glass Bottle Labels. Polymers 2020, 12, 428. [Google Scholar] [CrossRef]
- Edeleva, M.; Marien, Y.W.; Van Steenberge, P.H.M.; D’Hooge, D.R. Impact of side reactions on molar mass distribution, unsaturation level and branching density in solution free radical polymerization of n-butyl acrylate under well-defined lab-scale reactor conditions. Polym. Chem. 2021, 12, 2095–2114. [Google Scholar] [CrossRef]
- Olaj, O.F.; Bitai, I. The laser flash-initiated polymerization as a tool of evaluating (individual) kinetic constants of free radical polymerization, 3. Information from degrees of polymerization. Die Angew. Makromol. Chem. 1987, 155, 177–190. [Google Scholar] [CrossRef]
- Davis, T.P.; O’Driscoll, K.F.; Piton, M.C.; Winnik, M.A. Determination of propagation rate constants using a pulsed laser technique. Macromolecules 1989, 22, 2785–2788. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Russell, G.T.; Hill, D.J.T.; Moad, G.; O’Driscoll, K.F.; Shen, J.; Winnik, M.A. Consistent values of rate parameters in free radical polymerization systems. II. Outstanding dilemmas and recommendations. J. Polym. Sci. Part A Polym. Chem. 1992, 30, 851–863. [Google Scholar] [CrossRef]
- Deady, M.; Mau, A.W.H.; Moad, G.; Spurling, T.H. Evaluation of the kinetic parameters for styrene polymerization and their chain length dependence by kinetic simulation and pulsed laser photolysis. Die Makromol. Chem. 1993, 194, 1691–1705. [Google Scholar] [CrossRef]
- O’Driscoll, K.F.; Kuindersma, M.E. Monte Carlo simulation of pulsed laser polymerization. Macromol. Theory Simul. 1994, 3, 469–478. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.-D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization, 1. Propagation rate coefficient for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Olaj, O.F.; Russell, G.T.; Schweer, J.; van Herk, A.M. Critically evaluated rate coefficients for free-radical polymerization, 2. Propagation rate coefficients for methyl methacrylate. Macromol. Chem. Phys. 1997, 198, 1545–1560. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Kajiwara, A.; Klumperman, B.; Russell, G.T. Critically evaluated rate coefficients for free-radical polymerization, 3. Propagation rate coefficients for alkyl methacrylates. Macromol. Chem. Phys. 2000, 201, 1355–1364. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; García, N.; Gilbert, R.G.; Hutchinson, R.A.; Kajiwara, A.; Kamachi, M.; Lacík, I.; Russell, G.T. Critically Evaluated Rate Coefficients for Free-Radical Polymerization, 4. Macromol. Chem. Phys. 2003, 204, 1338–1350. [Google Scholar] [CrossRef]
- Asua, J.M.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Gilbert, R.G.; Hutchinson, R.A.; Leiza, J.R.; Nikitin, A.N.; Vairon, J.-P.; et al. Critically Evaluated Rate Coefficients for Free-Radical Polymerization, 5. Macromol. Chem. Phys. 2004, 205, 2151–2160. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Hesse, P.; Kuchta, F.-D.; Lacík, I.; Herk, A.M.v. Critically evaluated rate coefficients for free-radical polymerization Part 6: Propagation rate coefficient of methacrylic acid in aqueous solution (IUPAC Technical Report). Pure Appl. Chem. 2007, 79, 1463–1469. [Google Scholar] [CrossRef]
- Junkers, T.; Barner-Kowollik, C. Optimum Reaction Conditions for the Synthesis of Macromonomers Via the High-Temperature Polymerization of Acrylates. Macromol. Theory Simul. 2009, 18, 421–433. [Google Scholar] [CrossRef]
- Ahmad, N.M.; Heatley, F.; Lovell, P.A. Chain Transfer to Polymer in Free-Radical Solution Polymerization of n-Butyl Acrylate Studied by NMR Spectroscopy. Macromolecules 1998, 31, 2822–2827. [Google Scholar] [CrossRef]
- Peck, A.N.F.; Hutchinson, R.A. Secondary Reactions in the High-Temperature Free Radical Polymerization of Butyl Acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Alberdi, J.M.; van Herk, A.M.; Leiza, J.R.; Asua, J.M. Evidence of Branching in Poly(butyl acrylate) Produced in Pulsed-Laser Polymerization Experiments. Macromol. Rapid Commun. 2003, 24, 173–177. [Google Scholar] [CrossRef]
- Boschmann, D.; Vana, P. Z-RAFT Star Polymerizations of Acrylates: Star Coupling via Intermolecular Chain Transfer to Polymer. Macromolecules 2007, 40, 2683–2693. [Google Scholar] [CrossRef]
- Wang, W.; Nikitin, A.N.; Hutchinson, R.A. Consideration of Macromonomer Reactions in n-Butyl Acrylate Free Radical Polymerization. Macromol. Rapid Commun. 2009, 30, 2022–2027. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Wang, W.; Kalfas, G.A.; Richards, J.R.; Bruni, C. Effect of Intramolecular Transfer to Polymer on Stationary Free-Radical Polymerization of Alkyl Acrylates, 5—Consideration of Solution Polymerization up to High Temperatures. Macromol. React. Eng. 2010, 4, 691–706. [Google Scholar] [CrossRef]
- Hamzehlou, S.; Ballard, N.; Reyes, Y.; Aguirre, A.; Asua, J.M.; Leiza, J.R. Analyzing the discrepancies in the activation energies of the backbiting and β-scission reactions in the radical polymerization of n-butyl acrylate. Polym. Chem. 2016, 7, 2069–2077. [Google Scholar] [CrossRef]
- Lena, J.-B.; Deschamps, M.; Sciortino, N.F.; Masters, S.L.; Squire, M.A.; Russell, G.T. Effects of Chain Transfer Agent and Temperature on Branching and β-Scission in Radical Polymerization of 2-Ethylhexyl Acrylate. Macromol. Chem. Phys. 2018, 219, 1700579. [Google Scholar] [CrossRef]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Backbiting and β-scission reactions in free-radical polymerization of methyl acrylate. Int. J. Quantum Chem. 2014, 114, 345–360. [Google Scholar] [CrossRef]
- Cuccato, D.; Mavroudakis, E.; Moscatelli, D. Quantum Chemistry Investigation of Secondary Reaction Kinetics in Acrylate-Based Copolymers. J. Phys. Chem. A 2013, 117, 4358–4366. [Google Scholar] [CrossRef]
- Moghadam, N.; Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational Study of Chain Transfer to Monomer Reactions in High-Temperature Polymerization of Alkyl Acrylates. J. Phys. Chem. A 2013, 117, 2605–2618. [Google Scholar] [CrossRef]
- Moghadam, N.; Liu, S.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical Study of Intermolecular Chain Transfer to Polymer Reactions of Alkyl Acrylates. Ind. Eng. Chem. Res. 2015, 54, 4148–4165. [Google Scholar] [CrossRef]
- Mavroudakis, E.; Cuccato, D.; Moscatelli, D. Quantum Mechanical Investigation on Bimolecular Hydrogen Abstractions in Butyl Acrylate-Based Free Radical Polymerization Processes. J. Phys. Chem. A 2014, 118, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Van Cauter, K.; Van Den Bossche, B.J.; Van Speybroeck, V.; Waroquier, M. Ab Initio Study of Free-Radical Polymerization: Defect Structures in Poly(vinyl chloride). Macromolecules 2007, 40, 1321–1331. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Reyniers, M.-F.; Marin, G.B. The Crucial Role of Diffusional Limitations in Controlled Radical Polymerization. Macromol. React. Eng. 2013, 7, 362–379. [Google Scholar] [CrossRef]
- Edeleva, M.; Van Steenberge, P.H.M.; Sabbe, M.K.; D’hooge, D.R. Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art. Polymers 2021, 13, 3027. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- McQuarrie, D.A. Physical Chemistry: A Molecular Approach; University Science Books: Sausalito, CA, USA, 1997. [Google Scholar]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Eckert, F.; Klamt, A. Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE J. 2002, 48, 369–385. [Google Scholar] [CrossRef]
- COSMOlogic GmbH & Co. KG. COSMOthermX, version 19.0.4; COSMOlogic: Leverkusen, Germany, 2019. [Google Scholar]
- Eckart, C. The Penetration of a Potential Barrier by Electrons. Phys. Rev. 1930, 35, 1303–1309. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. Kinetic Monte Carlo Modeling Extracts Information on Chain Initiation and Termination from Complete PLP-SEC Traces. Macromolecules 2017, 50, 1371–1385. [Google Scholar] [CrossRef]
- Vir, A.B.; Marien, Y.W.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’Hooge, D.R. From n-butyl acrylate Arrhenius parameters for backbiting and tertiary propagation to β-scission via stepwise pulsed laser polymerization. Polym. Chem. 2019, 10, 4116–4125. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. Improved kinetic Monte Carlo simulation of chemical composition-chain length distributions in polymerization processes. Chem. Eng. Sci. 2014, 110, 185–199. [Google Scholar] [CrossRef]
- De Smit, K.; Marien, Y.W.; Edeleva, M.; Van Steenberge, P.H.M.; D’hooge, D.R. Roadmap for Monomer Conversion and Chain Length-Dependent Termination Reactivity Algorithms in Kinetic Monte Carlo Modeling of Bulk Radical Polymerization. Ind. Eng. Chem. Res. 2020, 59, 22422–22439. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; Vandenbergh, J.; Reyniers, M.-F.; Junkers, T.; D’hooge, D.R.; Marin, G.B. Kinetic Monte Carlo Generation of Complete Electron Spray Ionization Mass Spectra for Acrylate Macromonomer Synthesis. Macromolecules 2017, 50, 2625–2636. [Google Scholar] [CrossRef]
- Satoh, K.; Kamigaito, M. Stereospecific Living Radical Polymerization: Dual Control of Chain Length and Tacticity for Precision Polymer Synthesis. Chem. Rev. 2009, 109, 5120–5156. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Broadbelt, L.J. Kinetic Study of 1,5-Hydrogen Transfer Reactions of Methyl Acrylate and Butyl Acrylate Using Quantum Chemistry. Macromol. Theory Simul. 2012, 21, 461–469. [Google Scholar] [CrossRef]
- Arzamendi, G.; Plessis, C.; Leiza, J.R.; Asua, J.M. Effect of the Intramolecular Chain Transfer to Polymer on PLP/SEC Experiments of Alkyl Acrylates. Macromol. Theory Simul. 2003, 12, 315–324. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Buback, M.; Hesse, P. Determination of Intramolecular Chain Transfer and Midchain Radical Propagation Rate Coefficients for Butyl Acrylate by Pulsed Laser Polymerization. Macromolecules 2007, 40, 8631–8641. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M. SP–PLP–EPR Investigations into the Chain-Length-Dependent Termination of Methyl Methacrylate Bulk Polymerization. Macromol. Rapid Commun. 2009, 30, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Kattner, H.; Buback, M. Termination, Propagation, and Transfer Kinetics of Midchain Radicals in Methyl Acrylate and Dodecyl Acrylate Homopolymerization. Macromolecules 2018, 51, 25–33. [Google Scholar] [CrossRef]
- Laki, S.; AShamsabadi, A.; Riazi, H.; Grady, M.C.; Rappe, A.M.; Soroush, M. Experimental and Mechanistic Modeling Study of Self-Initiated High-Temperature Polymerization of Ethyl Acrylate. Ind. Eng. Chem. Res. 2020, 59, 2621–2630. [Google Scholar] [CrossRef]
- Cuccato, D.; Mavroudakis, E.; Dossi, M.; Moscatelli, D. A Density Functional Theory Study of Secondary Reactions in n-Butyl Acrylate Free Radical Polymerization. Macromol. Theory Simul. 2013, 22, 127–135. [Google Scholar] [CrossRef]
- Paraskevas, P.D.; Sabbe, M.K.; Reyniers, M.-F.; Papayannakos, N.G.; Marin, G.B. Group Additive Kinetics for Hydrogen Transfer between Oxygenates. J. Phys. Chem. A 2015, 119, 6961–6980. [Google Scholar] [CrossRef]
- Zorn, A.-M.; Junkers, T.; Barner-Kowollik, C. Synthesis of a Macromonomer Library from High-Temperature Acrylate Polymerization. Macromol. Rapid Commun. 2009, 30, 2028–2035. [Google Scholar] [CrossRef]
- Heidarzadeh, N.; Hutchinson, R.A. Maximizing macromonomer content produced by starved-feed high temperature acrylate/methacrylate semi-batch polymerization. Polym. Chem. 2020, 11, 2137–2146. [Google Scholar] [CrossRef]
- Hirano, T.; Yamada, B. Macromonomer formation by sterically hindered radical polymerization of methyl acrylate trimer at high temperature. Polymer 2003, 44, 347–354. [Google Scholar] [CrossRef]
- Ballard, N.; Veloso, A.; Asua, J.M. Mid-Chain Radical Migration in the Radical Polymerization of n-Butyl Acrylate. Polymers 2018, 10, 765. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M. SP-PLP-EPR—A Novel Method for Detailed Studies into the Termination Kinetics of Radical Polymerization. Macromol. React. Eng. 2010, 4, 288–301. [Google Scholar] [CrossRef]
- Wenn, B.; Junkers, T. Kilohertz Pulsed-Laser-Polymerization: Simultaneous Determination of Backbiting, Secondary, and Tertiary Radical Propagation Rate Coefficients for tert-Butyl Acrylate. Macromol. Rapid Commun. 2016, 37, 781–787. [Google Scholar] [CrossRef]
- Quintens, G.; Junkers, T. Pulsed laser polymerization–size exclusion chromatography investigations into backbiting in ethylhexyl acrylate polymerization. Polym. Chem. 2022, 13, 2019–2025. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M.; Hesse, P.; Sergeeva, T. Termination and Transfer Kinetics of Butyl Acrylate Radical Polymerization Studied via SP-PLP-EPR. Macromolecules 2010, 43, 4023–4031. [Google Scholar] [CrossRef]
- Maeder, S.; Gilbert, R.G. Measurement of transfer constant for butyl acrylate free-radical polymerization. Macromolecules 1998, 31, 4410–4418. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Leiza, J.R.; Schoonbrood, H.A.S.; Charmot, D.; Asua, J.M. Modeling of Seeded Semibatch Emulsion Polymerization of n-BA. Ind. Eng. Chem. Res. 2001, 40, 3883–3894. [Google Scholar] [CrossRef]
- Ballard, N.; Hamzehlou, S.; Asua, J.M. Intermolecular Transfer to Polymer in the Radical Polymerization of n-Butyl Acrylate. Macromolecules 2016, 49, 5418–5426. [Google Scholar] [CrossRef]
- Vir, A.B.; Marien, Y.W.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’Hooge, D.R. Access to the β-scission rate coefficient in acrylate radical polymerization by careful scanning of pulse laser frequencies at elevated temperature. React. Chem. Eng. 2018, 3, 807–815. [Google Scholar] [CrossRef]

 this work’s prediction;
this work’s prediction; 
 experimental median;
experimental median;  ab initio median;
ab initio median;  25–75% ab initio percentile area [34,35,56,62];
25–75% ab initio percentile area [34,35,56,62];  25–75% experimental percentile area [28,32,50,57,58,59,60,61].
this work’s prediction; experimental median; ab initio median; 25–75% ab initio percentile area [34,35,56,62]; 25–75% experimental percentile area [28,32,50,57,58,59,60,61].
25–75% experimental percentile area [28,32,50,57,58,59,60,61].
this work’s prediction; experimental median; ab initio median; 25–75% ab initio percentile area [34,35,56,62]; 25–75% experimental percentile area [28,32,50,57,58,59,60,61].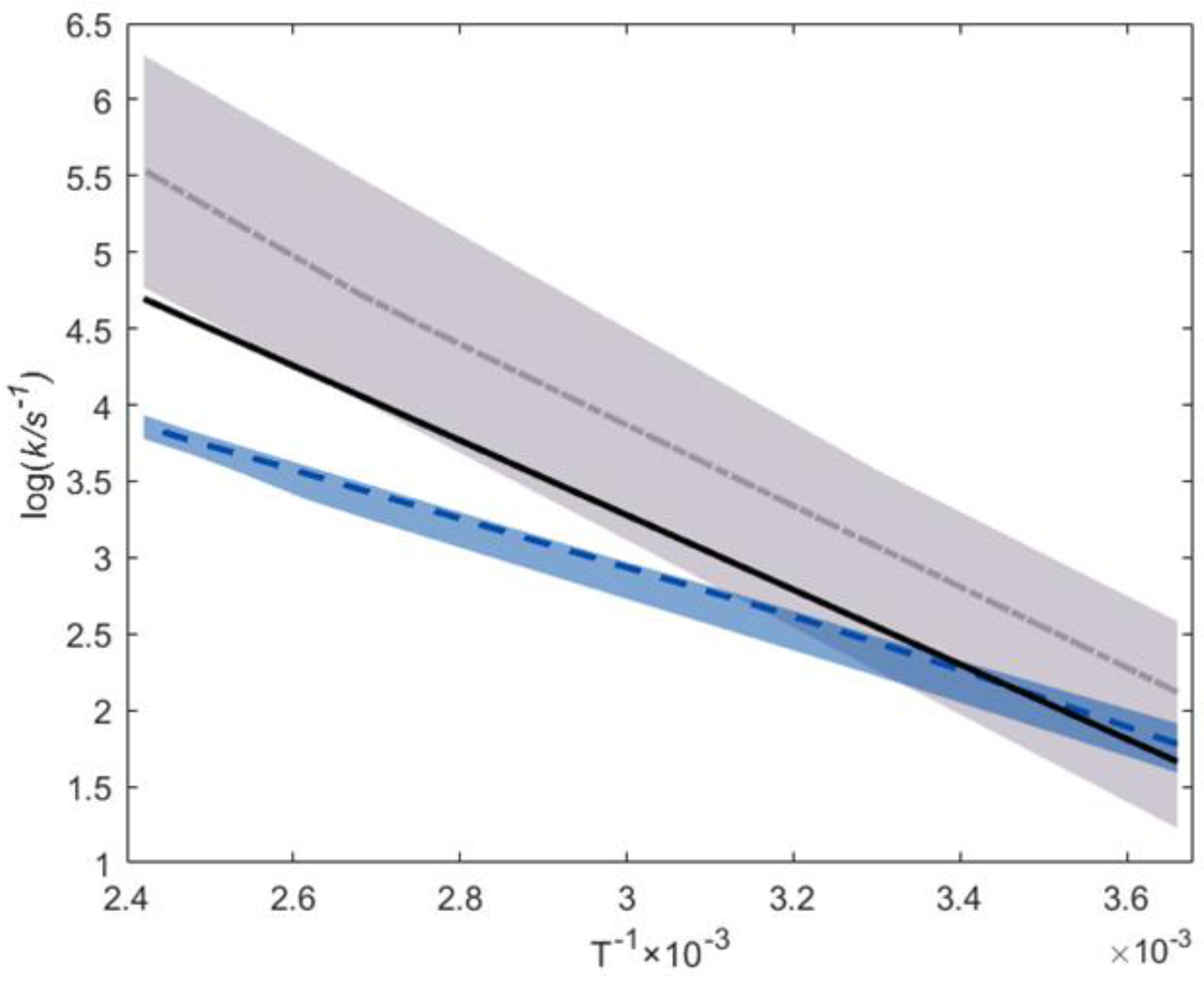
 ) represent the Gibbs energy of a reactant structure with a certain elongation, relative to the non-elongated reactant molecular model. The red circle (
) represent the Gibbs energy of a reactant structure with a certain elongation, relative to the non-elongated reactant molecular model. The red circle ( ) represents the Gibbs energy of the transition-state structure with a certain elongation, relative to the non-elongated transition-state molecular model. The blue circle (
) represents the Gibbs energy of the transition-state structure with a certain elongation, relative to the non-elongated transition-state molecular model. The blue circle ( ) represents the Gibbs reaction barrier at a certain elongation.
) represent the Gibbs energy of a reactant structure with a certain elongation, relative to the non-elongated reactant molecular model. The red circle () represents the Gibbs energy of the transition-state structure with a certain elongation, relative to the non-elongated transition-state molecular model. The blue circle () represents the Gibbs reaction barrier at a certain elongation.
) represents the Gibbs reaction barrier at a certain elongation.
) represent the Gibbs energy of a reactant structure with a certain elongation, relative to the non-elongated reactant molecular model. The red circle () represents the Gibbs energy of the transition-state structure with a certain elongation, relative to the non-elongated transition-state molecular model. The blue circle () represents the Gibbs reaction barrier at a certain elongation.
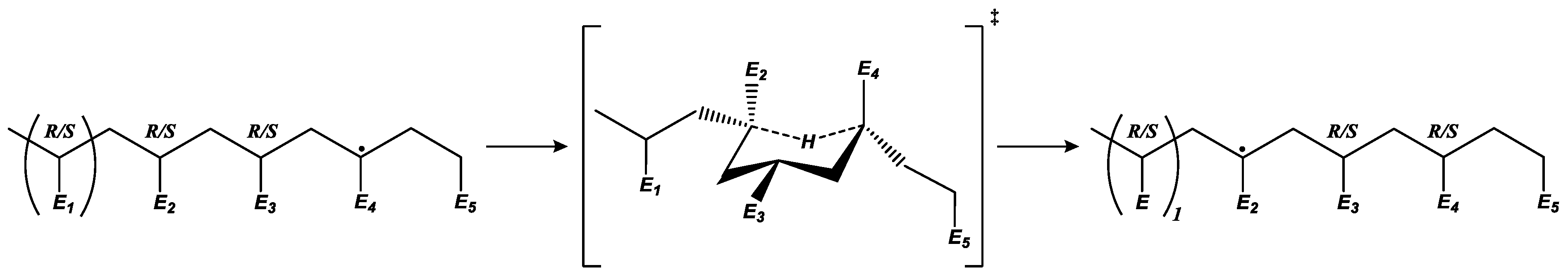
 ) vs. elongation of the radical migration reaction barrier applying the reduced-flexibility approach.
) vs. elongation of the radical migration reaction barrier applying the reduced-flexibility approach.
) vs. elongation of the radical migration reaction barrier applying the reduced-flexibility approach.
) vs. elongation of the radical migration reaction barrier applying the reduced-flexibility approach.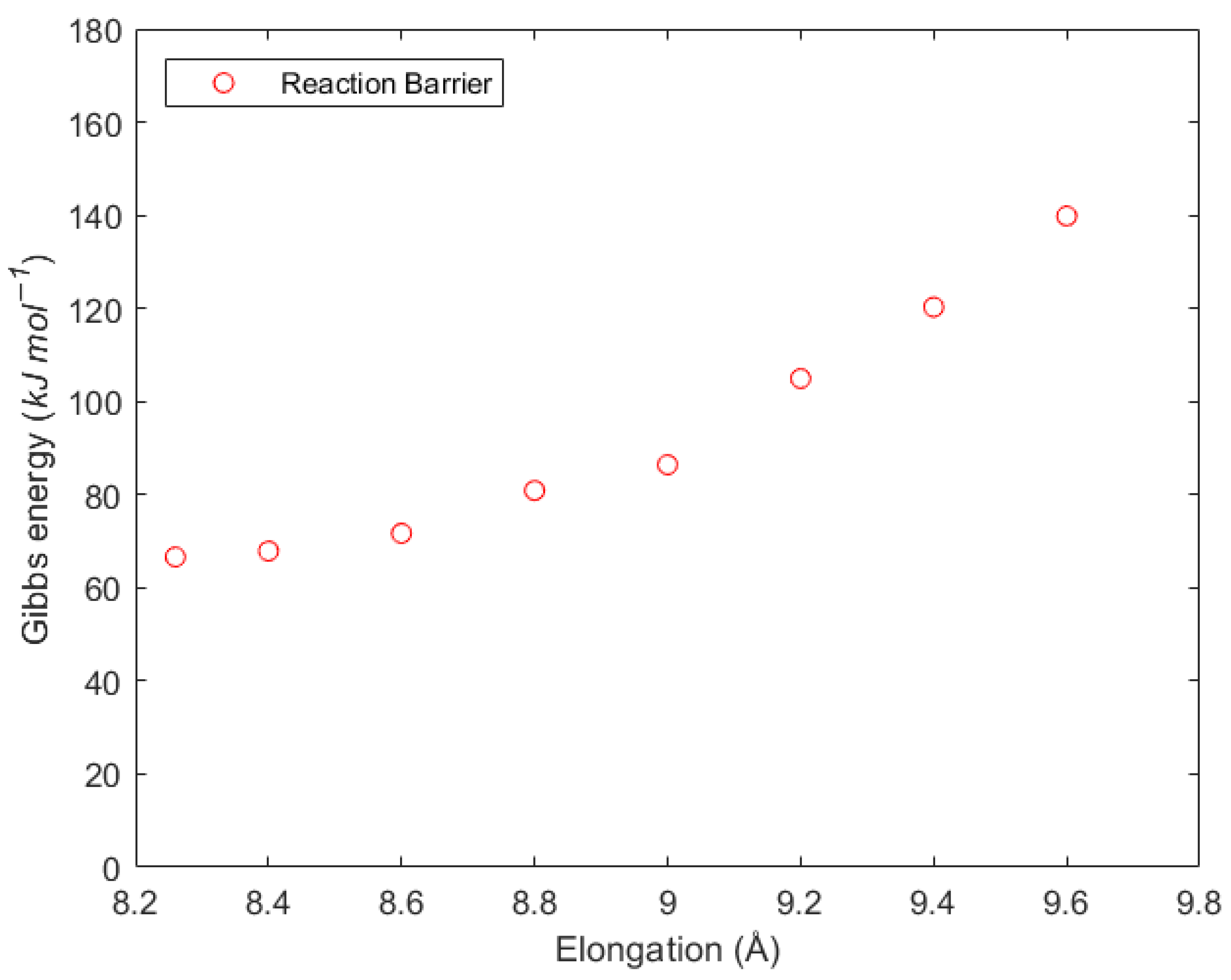

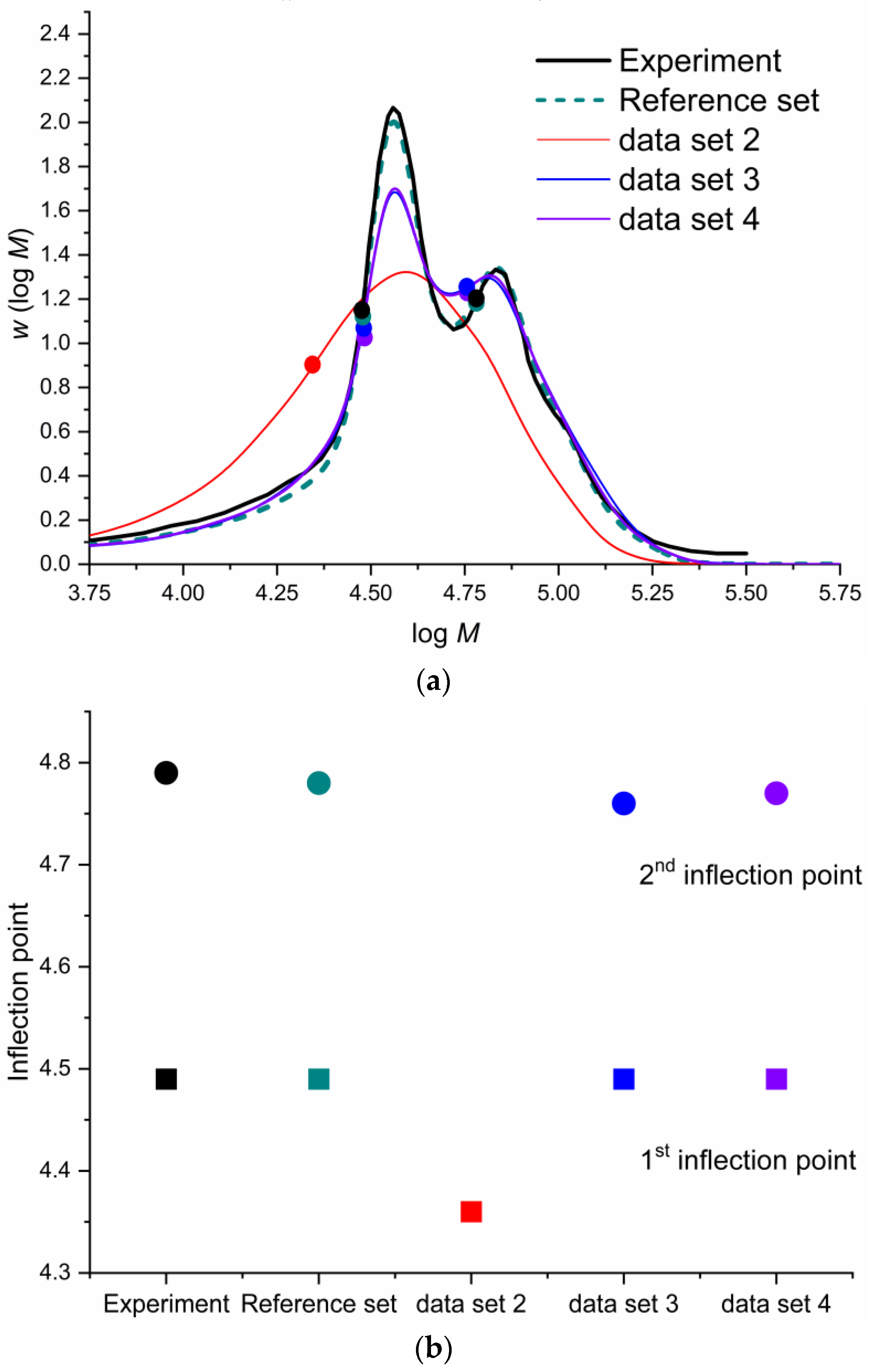
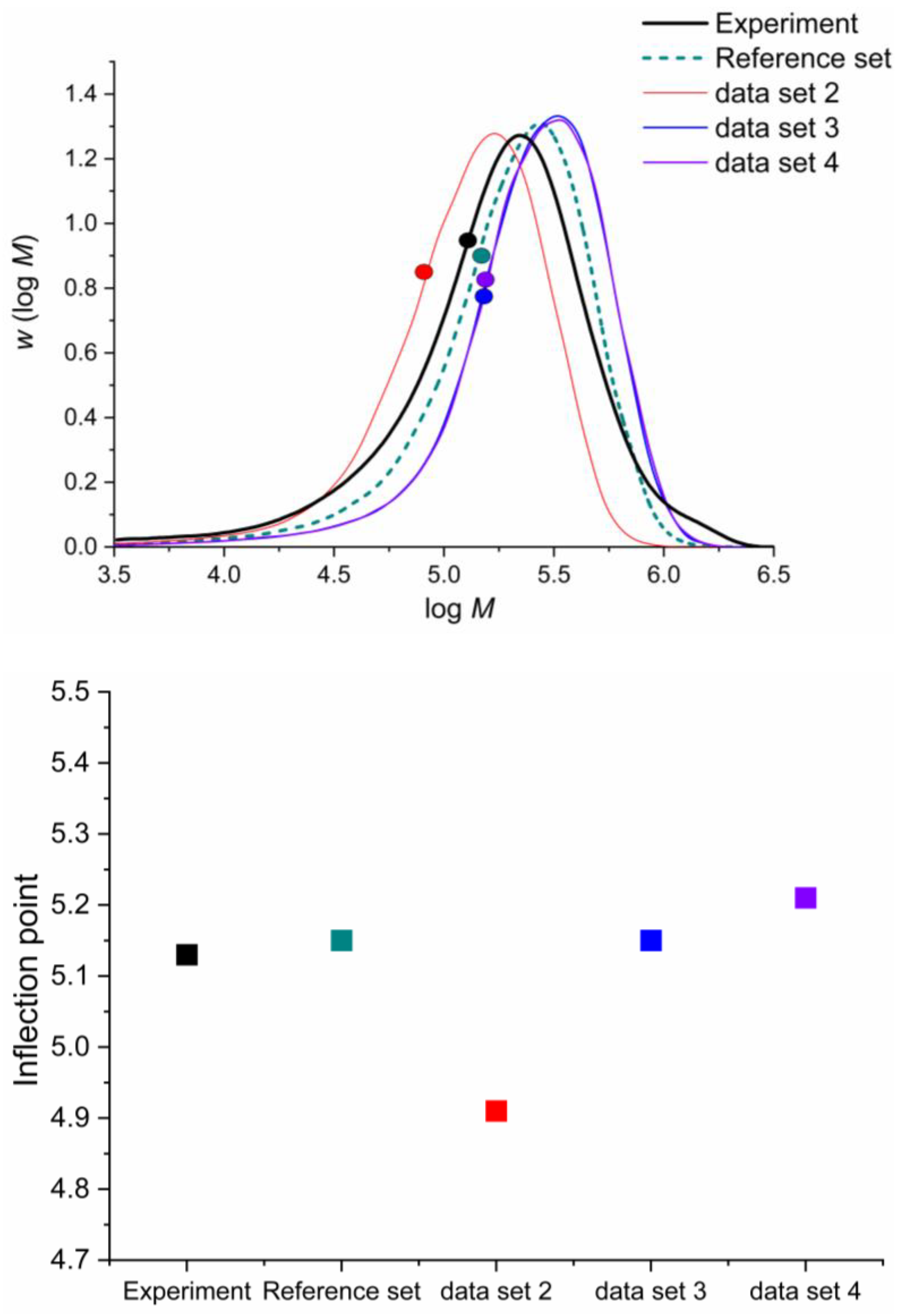

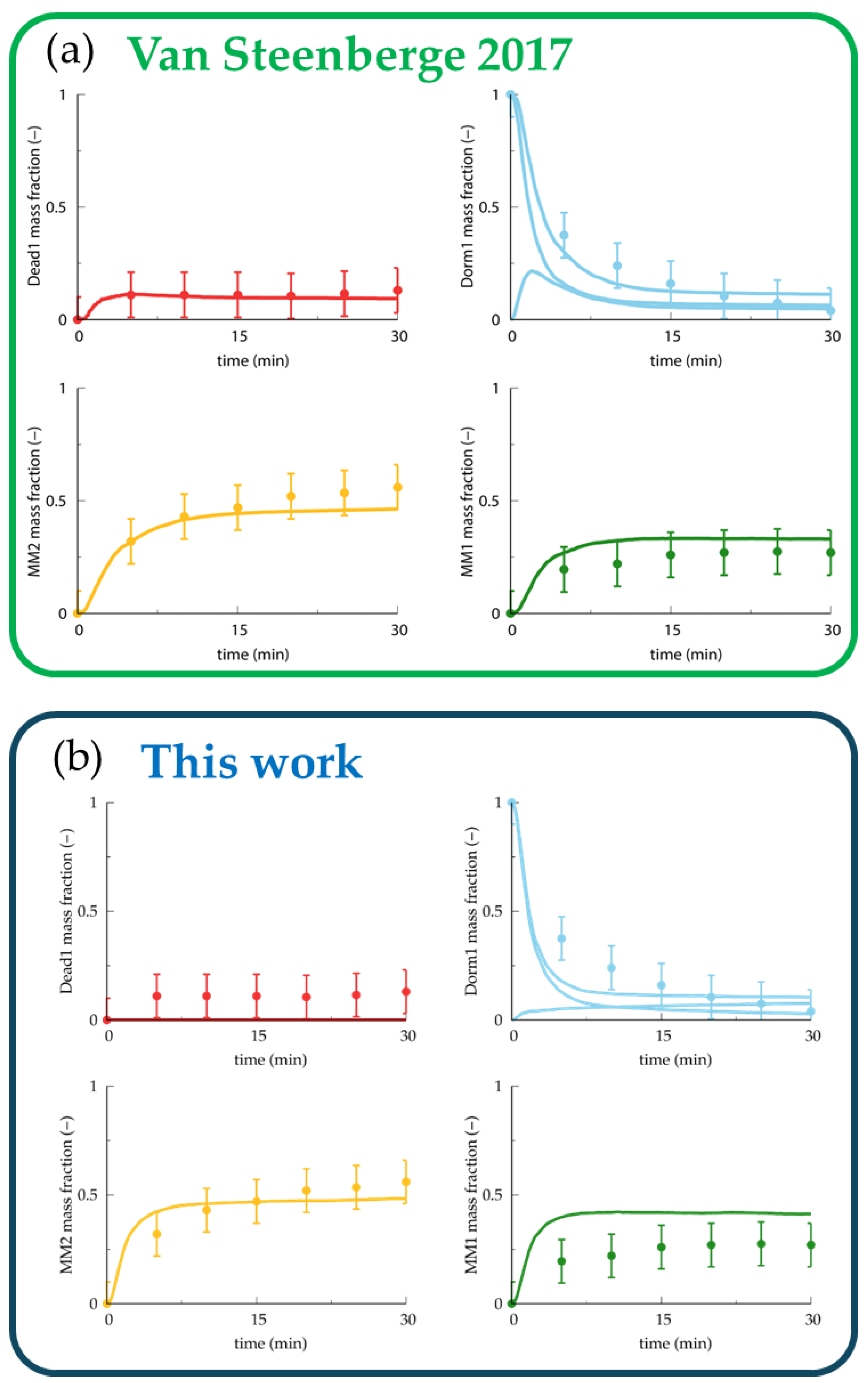
| Addition Probabilities | Experimental Work [55] | This Work |
|---|---|---|
| Identical chirality | 0.52 | 0.34 |
| Alternating chirality | 0.48 | 0.66 |
| Enantiomer | Experiment (%) | This Work (%) |
|---|---|---|
| SSSS/RRRR | 14.1 | 3.9 |
| SSSR/RRRS | 13.0 | 7.6 |
| SSRS/RRSR | 12.0 | 14.8 |
| SRSS/RSRR | 12.0 | 14.8 |
| RSSS/SRRR | 13.0 | 7.6 |
| SSRR/RRSS | 13.0 | 7.6 |
| SRSR/RSRS | 11.1 | 28.7 |
| RSSR/SRRS | 12.0 | 14.8 |
| Enantiomer | k [s−1] 298.15K | k [s−1] 413.15K | A [s−1] | Ea [kJ/mol] | |
|---|---|---|---|---|---|
| SSSS/RRRR | 0.039 | 2.40 × 102 | 3.96 × 104 | 2.29 × 1010 | 45.8 |
| SSSR/RRRS | 0.076 | 5.64 × 102 | 7.87 × 104 | 2.96 × 1010 | 44.3 |
| SSRS/RRSR | 0.148 | 2.35 × 102 | 2.16 × 104 | 2.75 × 109 | 40.5 |
| SRSS/RSRR | 0.148 | 3.16 × 101 | 1.04 × 103 | 1.23 × 107 | 33.8 |
| RSSS/SRRR | 0.076 | 1.08 × 103 | 2.94 × 105 | 5.63 × 1011 | 49.4 |
| SSRR/RRSS | 0.076 | 3.38 × 102 | 5.90 × 104 | 3.92 × 1010 | 46.2 |
| SRSR/RSRS | 0.287 | 4.92 × 101 | 1.55 × 104 | 4.88 × 1010 | 51.6 |
| RSSR/SRRS | 0.148 | 2.89 × 102 | 4.82 × 104 | 2.85 × 1010 | 45.8 |
| 2.57 × 102 | 4.95 × 104 | 4.09 × 1010 * | 46.8 * |
| Elongation between First and Last Carbon Atoms | Ea [kJ mol−1 s−1] | k @ 383.15 K [s−1] |
|---|---|---|
| Non-strained | 116.0 | 5.14 × 10−3 |
| 10.6 | 115.1 | 1.38 × 10−2 |
| 10.8 | 112.6 | 2.89 × 10−2 |
| 11.0 | 109.4 | 7.26 × 10−2 |
| 11.2 | 108.0 | 1.26 × 10−1 |
| 11.4 | 104.5 | 3.84 × 10−1 |
| 11.6 | 101.2 | 8.83 × 10−1 |
| 11.8 | 95.8 | 5.01 × 100 |
| 12.0 | 86.3 | 9.85 × 101 |
| 12.2 | 75.6 | 2.77 × 103 |
| 12.4 | 64.5 | 8.92 × 104 |
| Enantiomer | k [s−1] 298.15K | k [s−1] 413.15K | A [s−1] | Ea [kJ mol−1] | |
|---|---|---|---|---|---|
| SSS/RRR | 0.116 | 5.24 × 104 | 2.46 × 106 | 6.40 × 109 | 29.5 |
| SSR/RRS | 0.224 | 2.52 × 101 | 2.72 × 104 | 2.13 × 1010 | 51.9 |
| SRS/RSR | 0.436 | 6.43 × 10−1 | 1.07 × 103 | 2.13 × 109 | 55.4 |
| RSS/SRR | 0.224 | 1.58 × 100 | 2.31 × 102 | 9.05 × 106 | 39.1 |
| kmig (weighted average) | 6.08 × 103 | 2.92 × 105 | 7.85 × 108 | 29.6 | |
| Van Steenberge et al. [54] | 1.6 × 102 | ||||
| Ballard et al. [67] | 3 × 103 | ||||
| Cuccato et al. [62] | 6.24 × 100 | 6.62 × 10−4 | 2.86 × 1010 | 63.3 |
| Elongation between First and Last Carbon Atoms | G‡ 413.15 K [kJ mol−1 s−1] | 413.15 K [s−1] |
|---|---|---|
| Non-strained | 63.4 | 3.84 × 105 |
| 8.4 | 64.8 | 2.55 × 105 |
| 8.6 | 70.0 | 5.64 × 104 |
| 8.8 | 82.2 | 1.60 × 103 |
| 9.0 | 93.7 | 5.76 × 101 |
| 9.2 | 113.5 | 1.80 × 10−1 |
| 9.4 | 139.1 | 1.03 × 10−4 |
| 9.6 | 171.3 | 8.87 × 10−9 |
| Reaction | Source | k @ 413.15K [L mol−1 s−1] | A [L mol−1 s−1] | Ea [kJ mol−1] |
|---|---|---|---|---|
| MM Propagation | This work | 3.02 × 105 | 3.97 × 107 | 16.8 |
| MM Propagation | Van Steenberge et al. [54] | 2.5 × 105 | ||
| MM Propagation | Wang et al. [30] | 6.63 × 104 | ||
| ECR Propagation | This work | 2.18 × 105 | 1.77 × 108 | 23.0 |
| CTM | k @ 333.15K [L mol−1 s−1] | A [L mol−1 s−1] | Ea [kJ mol−1] |
|---|---|---|---|
| V1 | 2.43 × 10−8 | 2.44 × 106 | 89.1 |
| V2 | 8.11 × 10−8 | 5.72 × 107 | 94.7 |
| A1 | 3.82 × 10−2 | 1.97 × 106 | 49.0 |
| A2 | 6.17 × 10−2 | 3.69 × 106 | 49.5 |
| A3 | 2.24 × 10−2 | 1.87 × 106 | 50.3 |
| A4 | 3.59 × 10−4 | 1.68 × 106 | 61.5 |
| kCTM | 2.45 × 10−1 | 1.82 × 107 | 49.6 |
| Maeder and Gilbert [72] | 2.24 × 100 | 2.9 ± 0.9 × 105 | 32.6 ± 0.8 |
| Laki et al. [61] | 1.48 × 101 | 4.88 × 106 | 35.2 ± 0.61 |
| Reaction | k [s−1] 298.15K | k [s−1] 413.15K | A [s−1] | Ea [kJ mol−1] | |
|---|---|---|---|---|---|
| RRR/SSS | 0.1156 | 1.72 × 10−1 | 1.92 × 101 | 3.91 × 106 | 42.0 |
| RRS/SSR | 0.4488 | 6.97 × 10−4 | 3.25 × 10−1 | 2.70 × 106 | 54.7 |
| RSR/SRS | 0.4356 | 4.67 × 10−4 | 1.77 × 10−1 | 8.48 × 105 | 52.9 |
| Chain transfer to backbone | 2.03 × 10−2 | 2.43 × 100 | 5.97 × 105 | 42.6 | |
| Chain transfer to alkyl branch | 3.32 × 10−2 | 8.47 × 100 | 1.50 × 107 | 49.5 | |
| 5.20 × 10−2 | 1.07 × 101 | 1.05 × 107 | 47.4 |
| A | Ea | A | Ea | A | Ea | A | Ea | |
|---|---|---|---|---|---|---|---|---|
| Data Set 1: reference set (Vir et al. [50]) | 2.2 × 107 | 17.9 | 5.4 × 107 | 30.6 | 7.9 × 1012 | 81.1 | 1.9 × 106 | 30.1 |
| Data Set 2: no special approaches | 1.8 × 108 | 23.0 | 3.1 × 1010 | 49.4 | 1.2 × 1014 | 112.9 | 3.7 × 106 | 28.8 |
| Data Set 3: all enantiomers considered for backbiting–weighted-average (WA) approach | 1.8 × 108 | 23.0 | 4.1 × 1010 | 46.8 | 1.2 × 1014 | 112.9 | 3.7 × 106 | 28.8 |
| Data Set 4: reduced flexibility applied to β-scission | 1.8 × 108 | 23.0 | 4.1 × 1010 | 46.8 | 2.8 × 1013 | 86.3 | 3.7 × 106 | 28.8 |
| Reaction | Rate Used in Van Steenberge et al. [54] k [L mol−1 s−1] or [s−1] @ 413.15K | This Work’s Data Set k [L mol−1 s−1] or [s−1] @ 413.15K |
|---|---|---|
| Activation | 4.0 × 103 | 4.0 × 103 |
| Deactivation | 1.0 × 106 | 1.0 × 106 |
| Reduction | 3.0 × 10−1 | 3.0 × 10−1 |
| Backbiting | 6.5 × 103 | 9.4 × 104 |
| Migration | 1.6 × 102 | 1.6 × 103 |
| Chain transfer to polymer | 6.0 × 102 | 1.1 × 101 |
| β-scission | 1.2 × 100 | 3.4 × 102 |
| Macromonomer addition | 2.5 × 105 | 3.0 × 105 |
| Termination | 1.0 × 108 | 1.0 × 108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugo, F.A.; Edeleva, M.; Van Steenberge, P.H.M.; Sabbe, M.K. Improved Approach for ab Initio Calculations of Rate Coefficients for Secondary Reactions in Acrylate Free-Radical Polymerization. Polymers 2024, 16, 872. https://doi.org/10.3390/polym16070872
Lugo FA, Edeleva M, Van Steenberge PHM, Sabbe MK. Improved Approach for ab Initio Calculations of Rate Coefficients for Secondary Reactions in Acrylate Free-Radical Polymerization. Polymers. 2024; 16(7):872. https://doi.org/10.3390/polym16070872
Chicago/Turabian StyleLugo, Fernando A., Mariya Edeleva, Paul H. M. Van Steenberge, and Maarten K. Sabbe. 2024. "Improved Approach for ab Initio Calculations of Rate Coefficients for Secondary Reactions in Acrylate Free-Radical Polymerization" Polymers 16, no. 7: 872. https://doi.org/10.3390/polym16070872
APA StyleLugo, F. A., Edeleva, M., Van Steenberge, P. H. M., & Sabbe, M. K. (2024). Improved Approach for ab Initio Calculations of Rate Coefficients for Secondary Reactions in Acrylate Free-Radical Polymerization. Polymers, 16(7), 872. https://doi.org/10.3390/polym16070872