
The Thermo-Oxidative Degradation of Polyurethane Open-Cell Soft Foam Investigated Through Gas Chromatography and Mass Spectrometry of Volatile Organic Compounds




Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
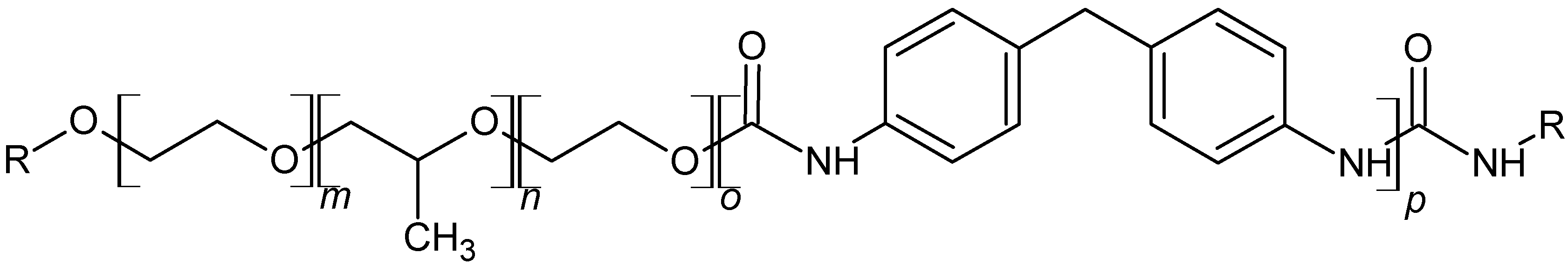
2.1.1. Chemical Compounds
2.1.2. Polyethers
2.1.3. Polyisocyanate
2.1.4. Synthesis of Qualitative and Quantitative References
Dioxolanes
Glycol Esters
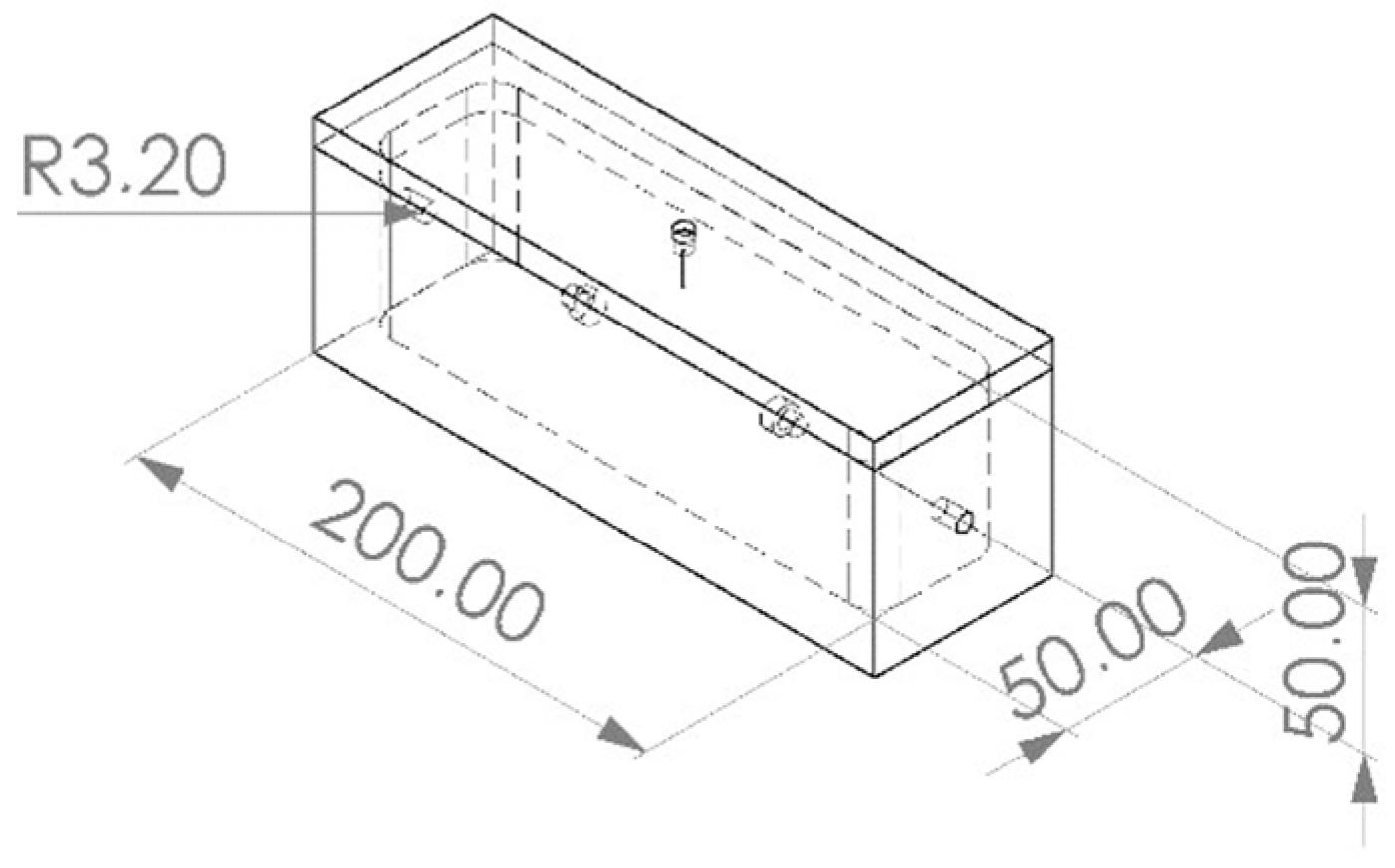
2.1.5. Sample Chamber Creation
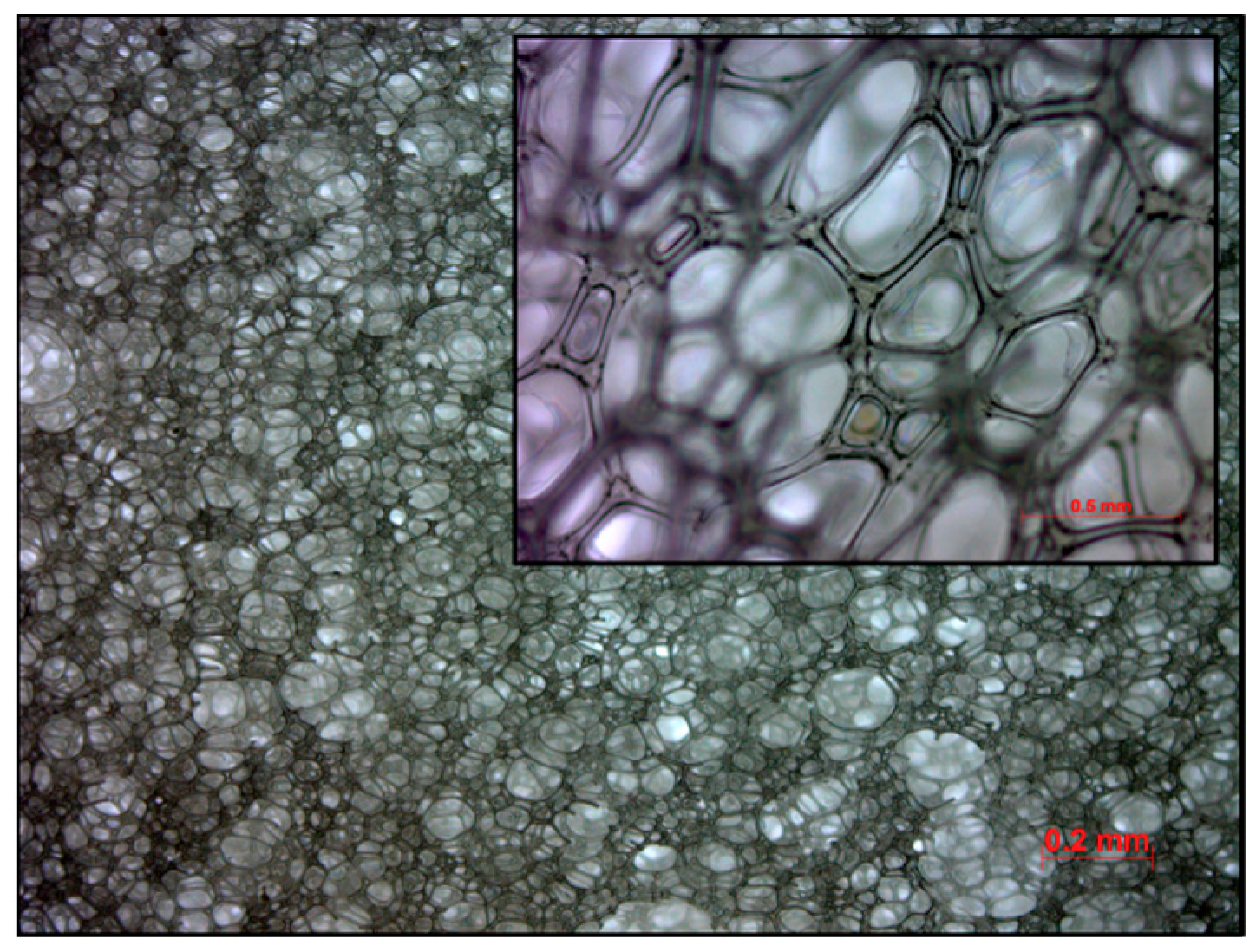
2.1.6. Polyurethane Foam Slab Synthesis
2.2. Sample Preparation
2.3. Methods
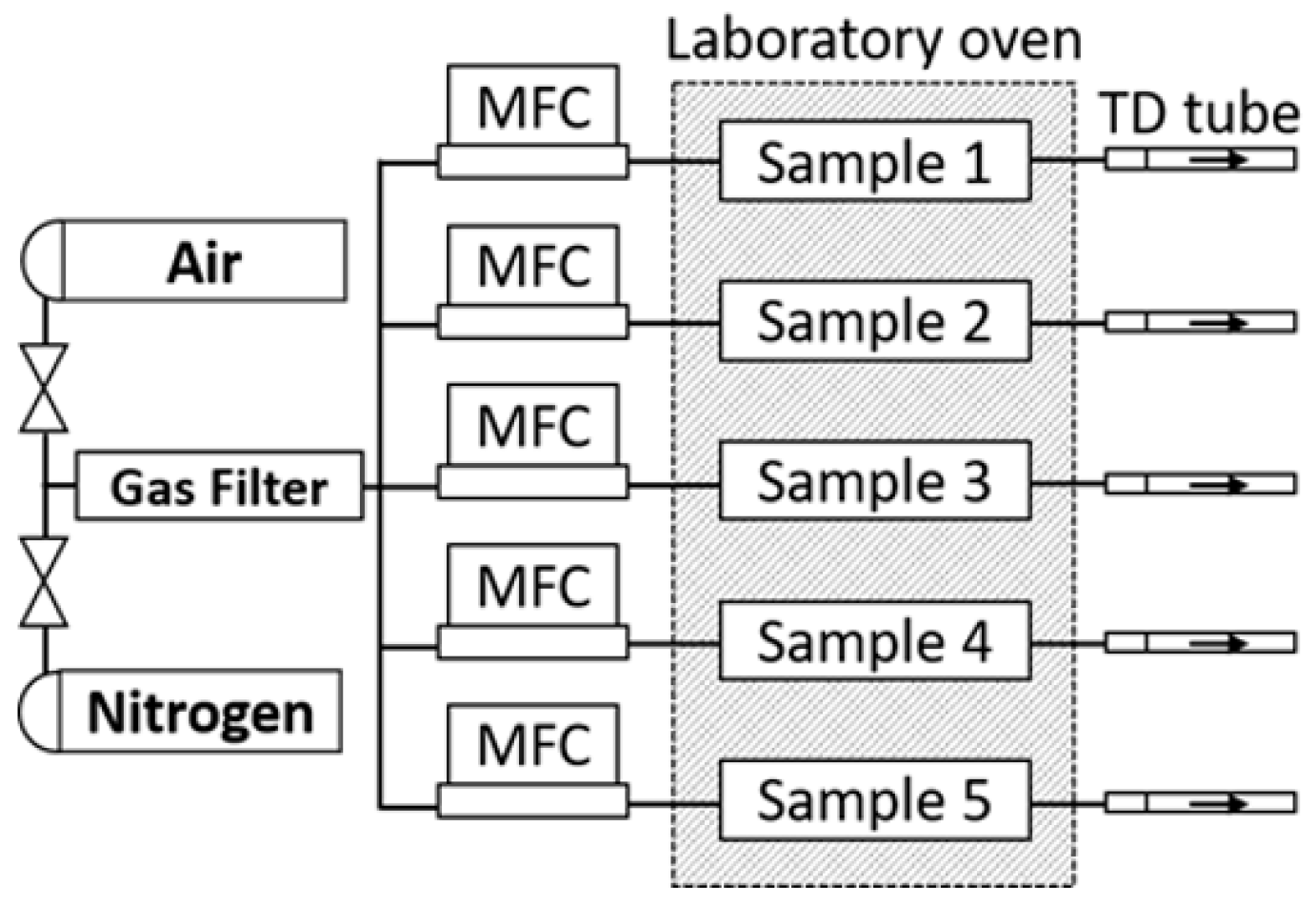
2.3.1. Aging Methodology
2.3.2. Thermo-Oxidation at 120 °C
2.3.3. Reproducibility Investigations
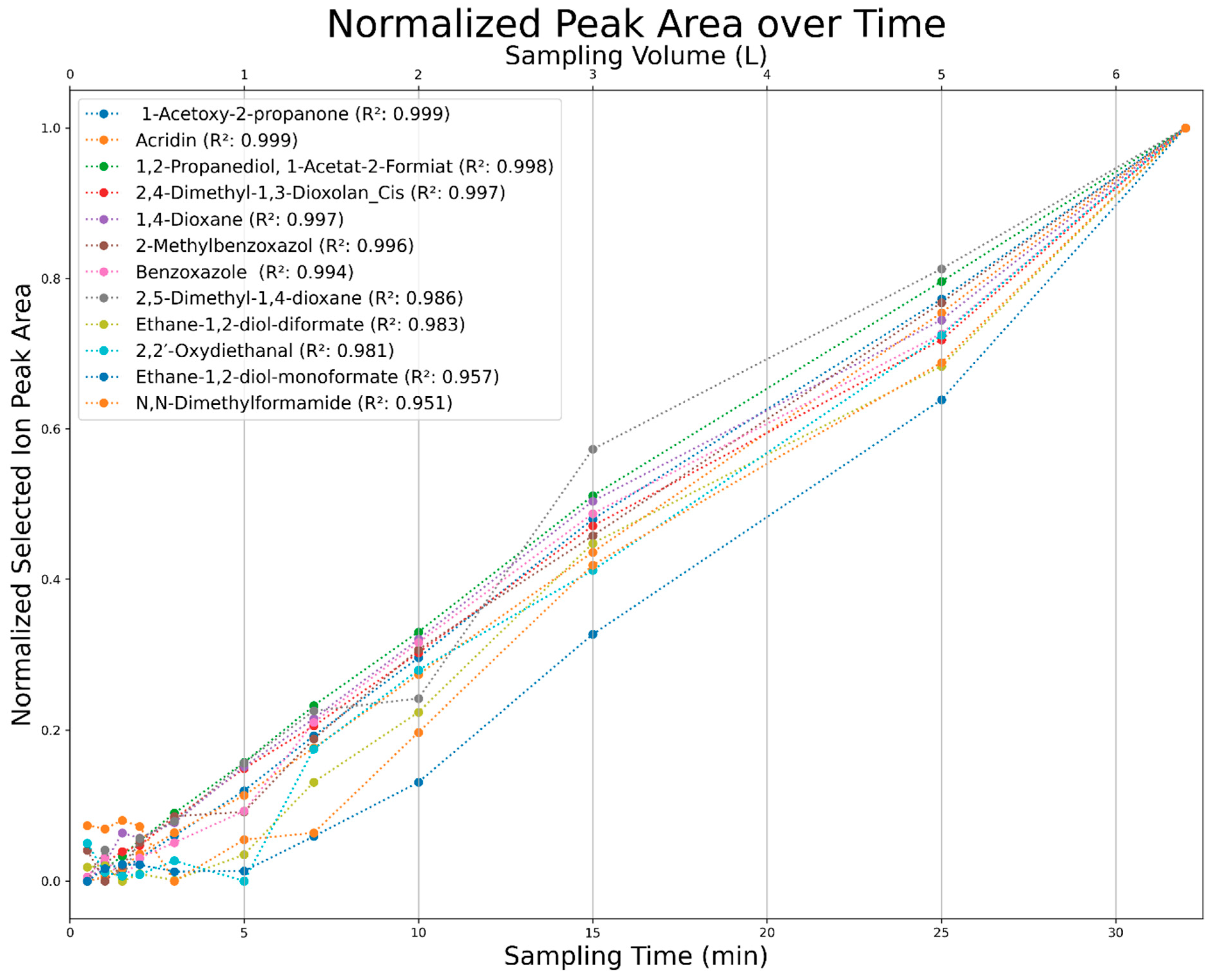
2.3.4. Sampling Time Variation
2.3.5. Identification of Oxidation Products
2.3.6. Thermal Desorption Unit (TD)
2.3.7. Gas Chromatography and Mass Spectrometry (GC-MS) Parameters
2.3.8. External Calibration Protocol for Quantification
3. Results
3.1. Qualitative Results
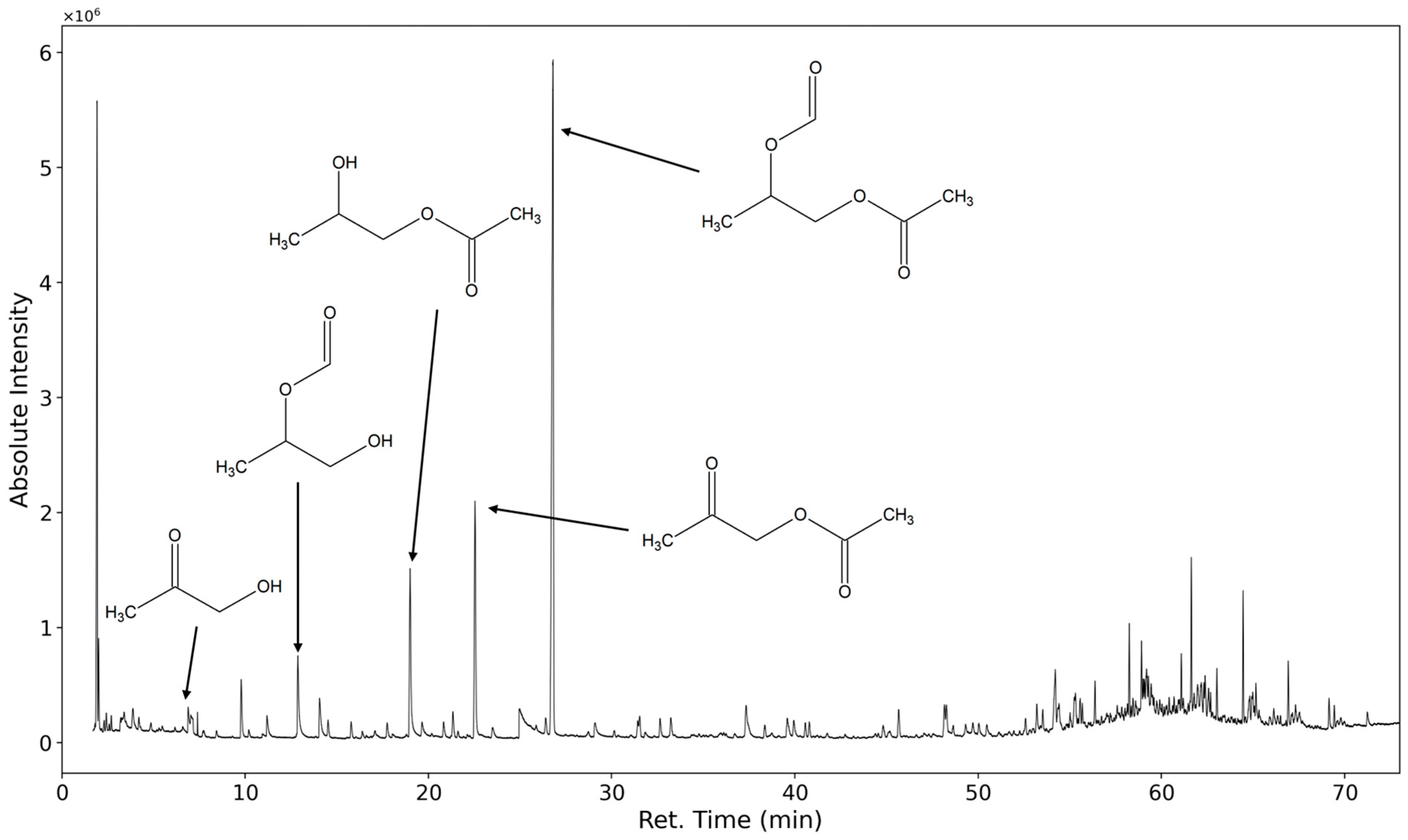
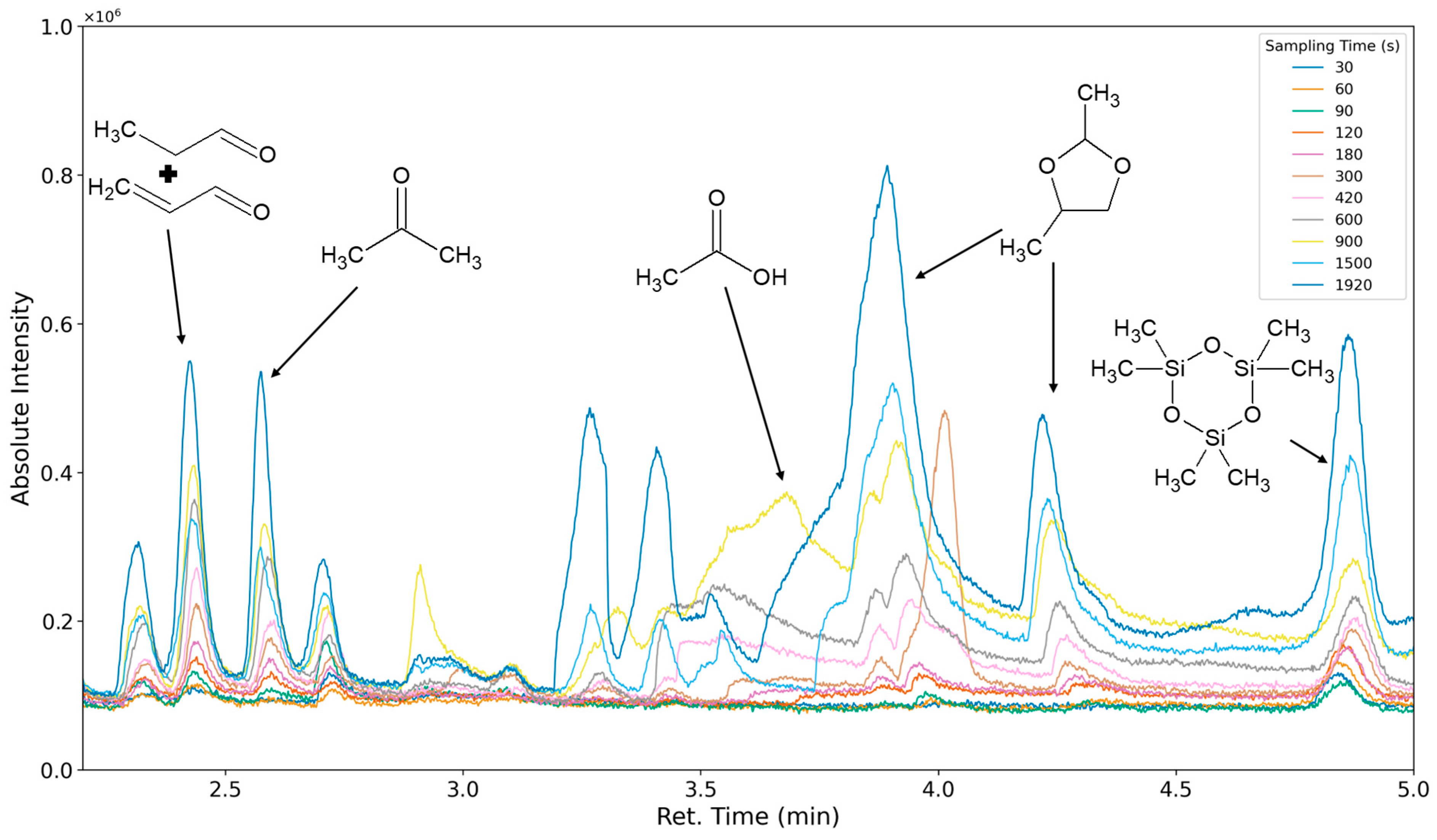
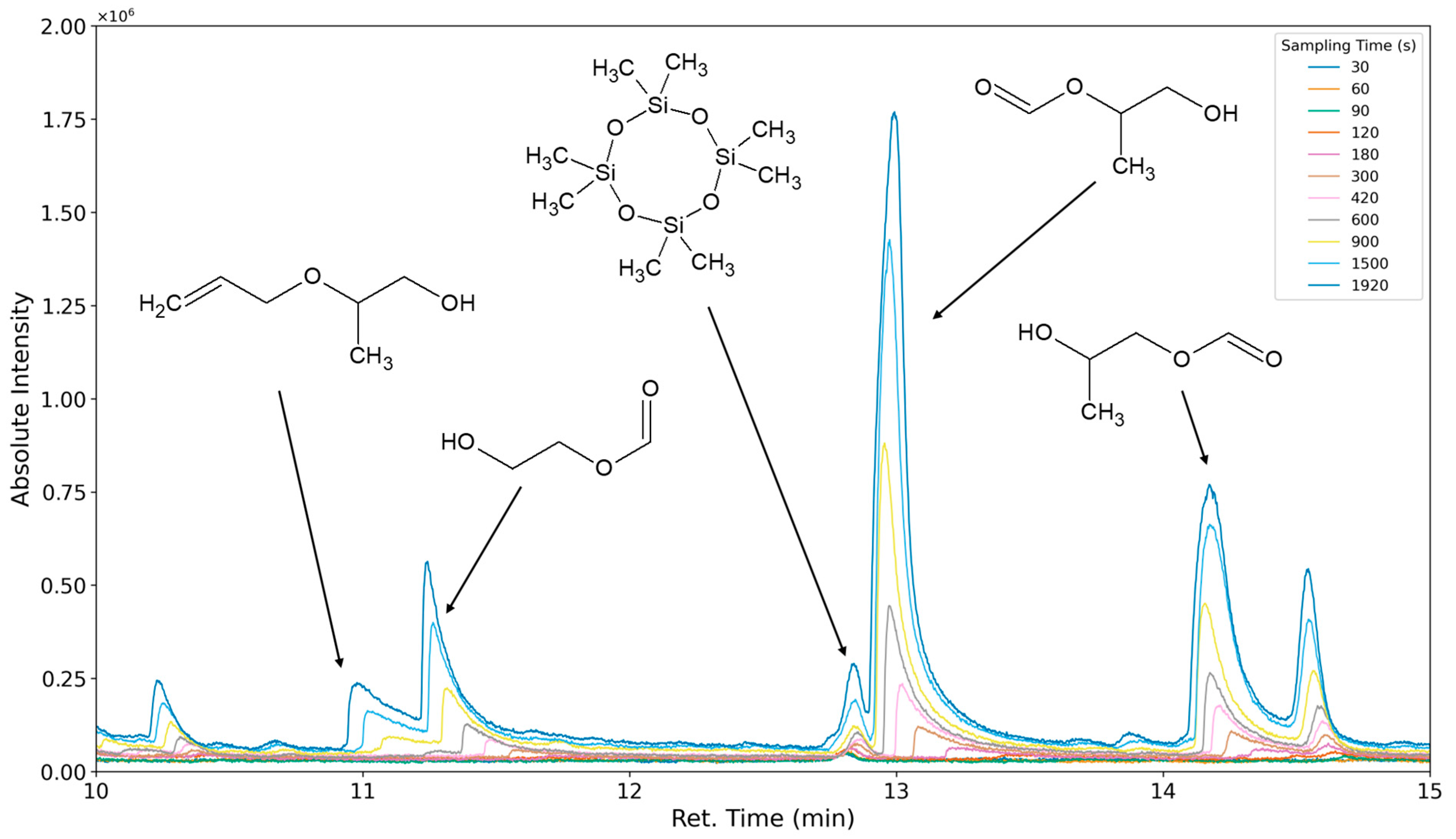
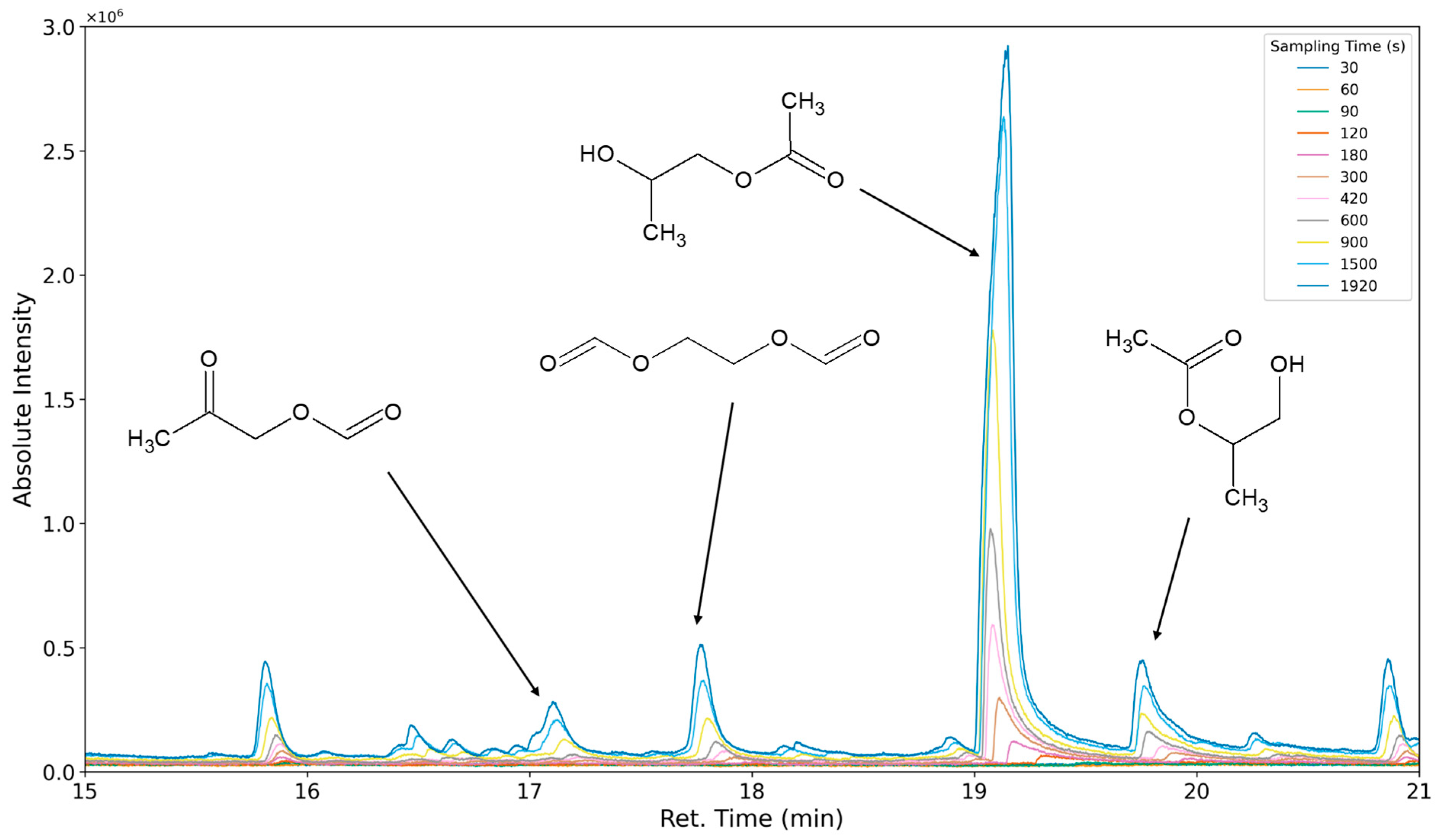
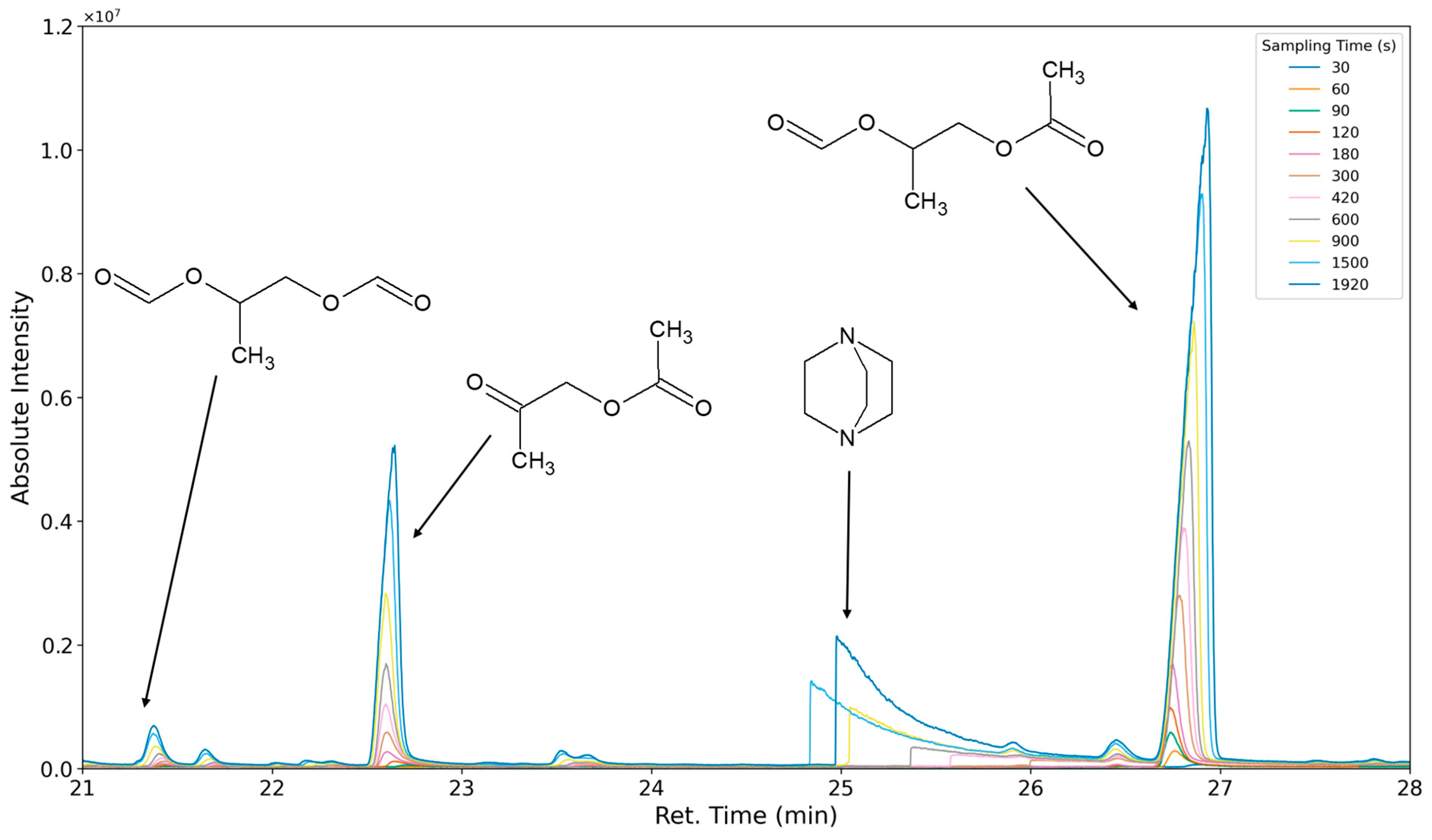
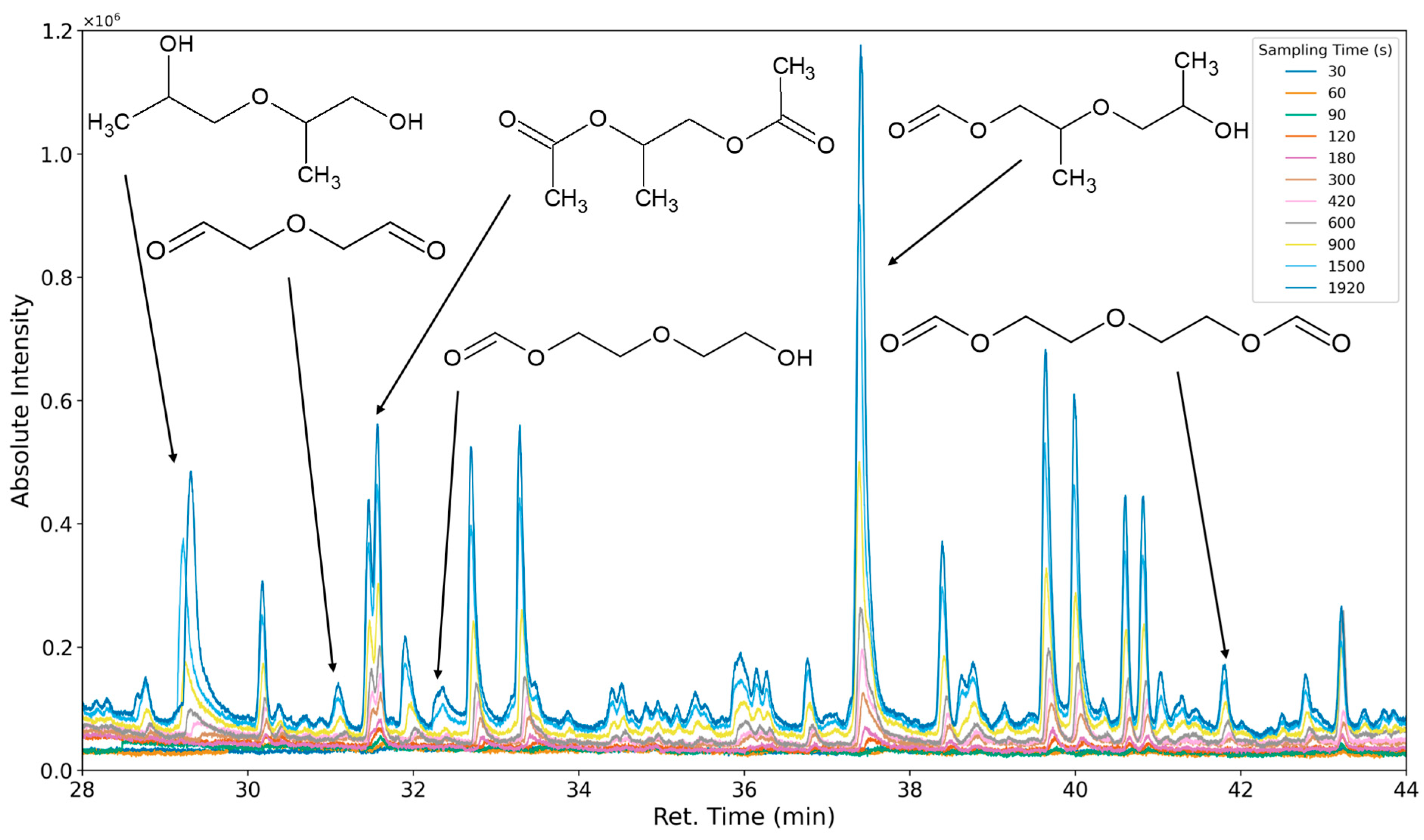
3.1.1. Most Prominent Peaks
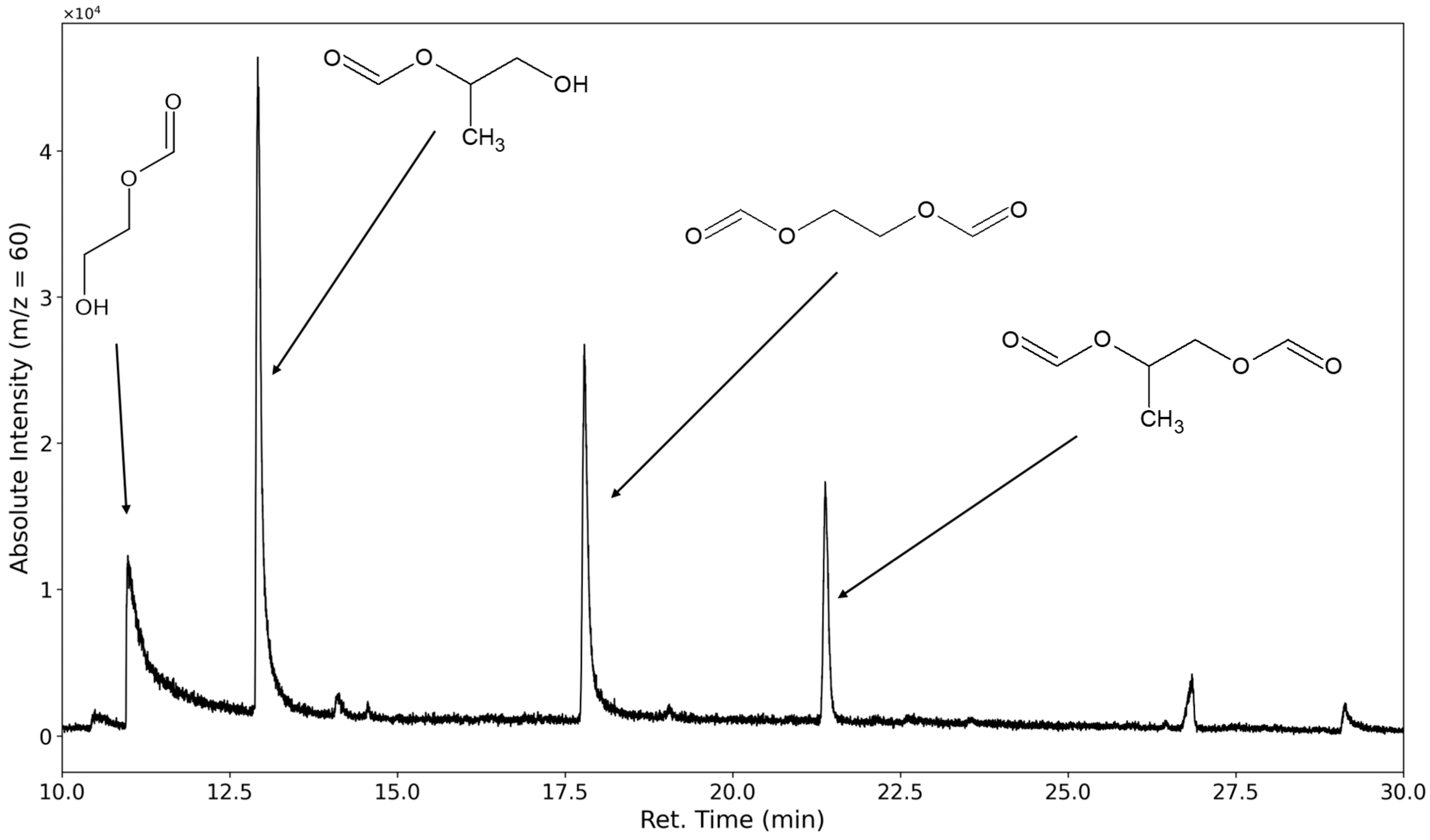
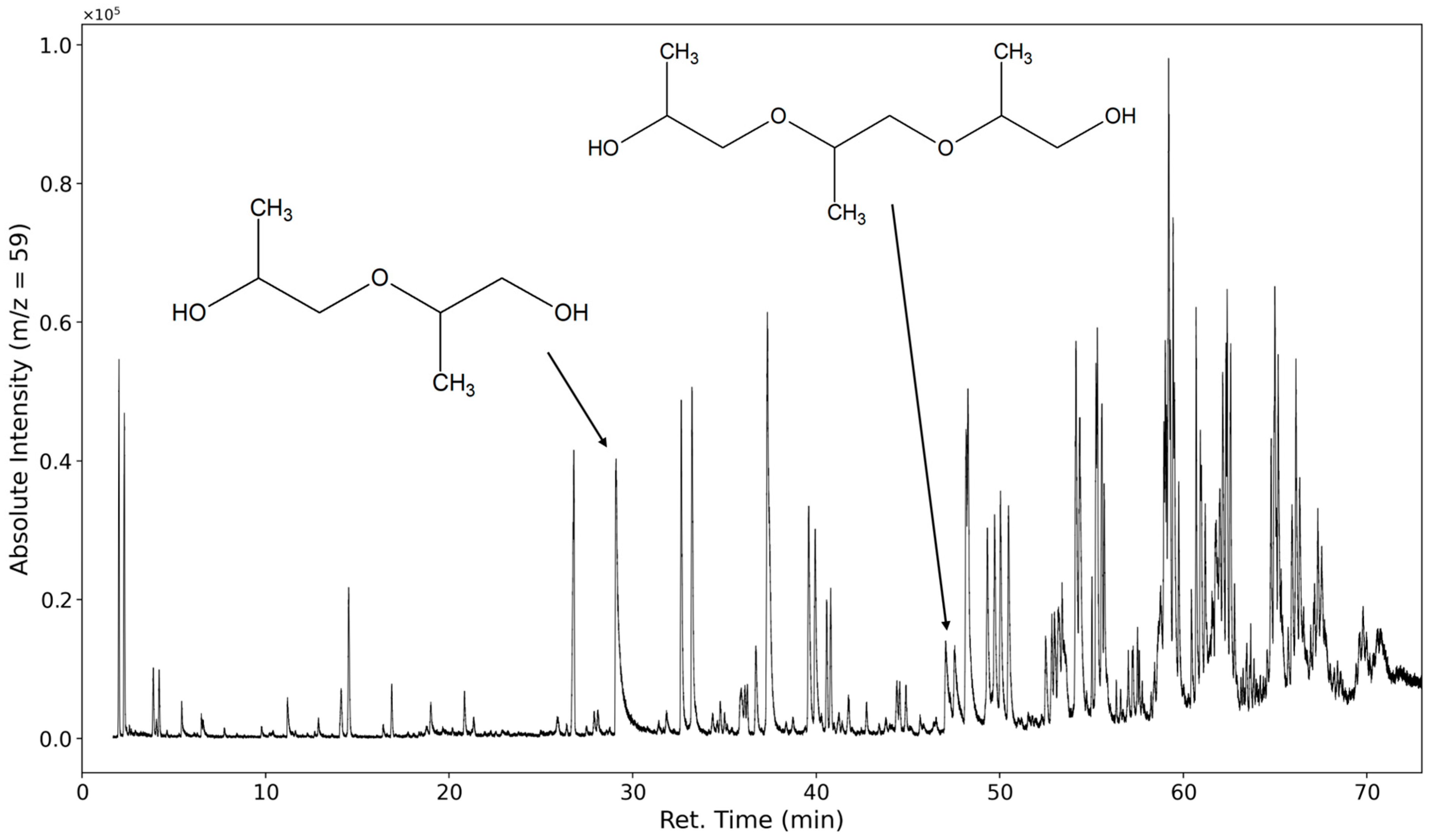
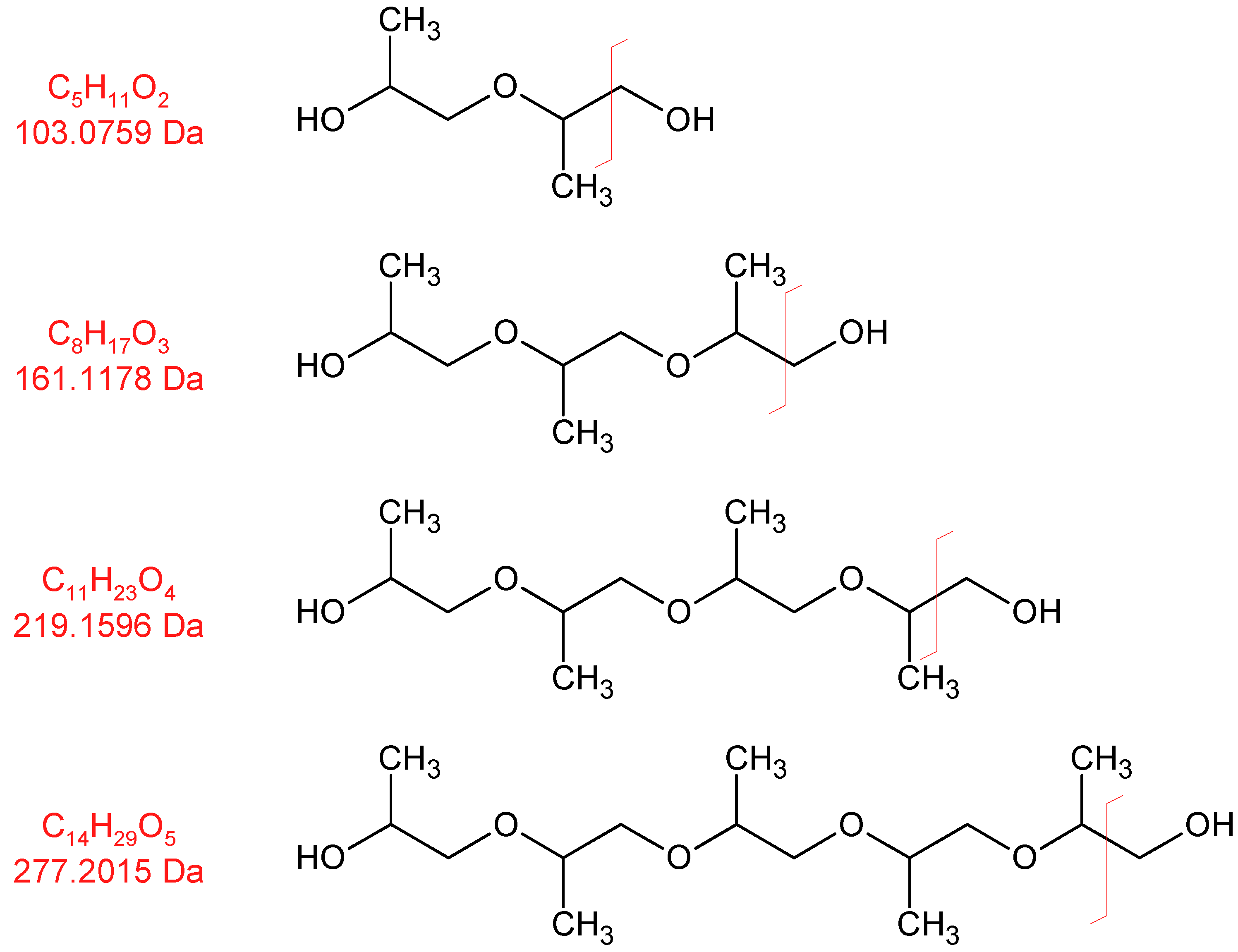
3.1.2. Degradation Products of the Soft Segment (EO Phase)
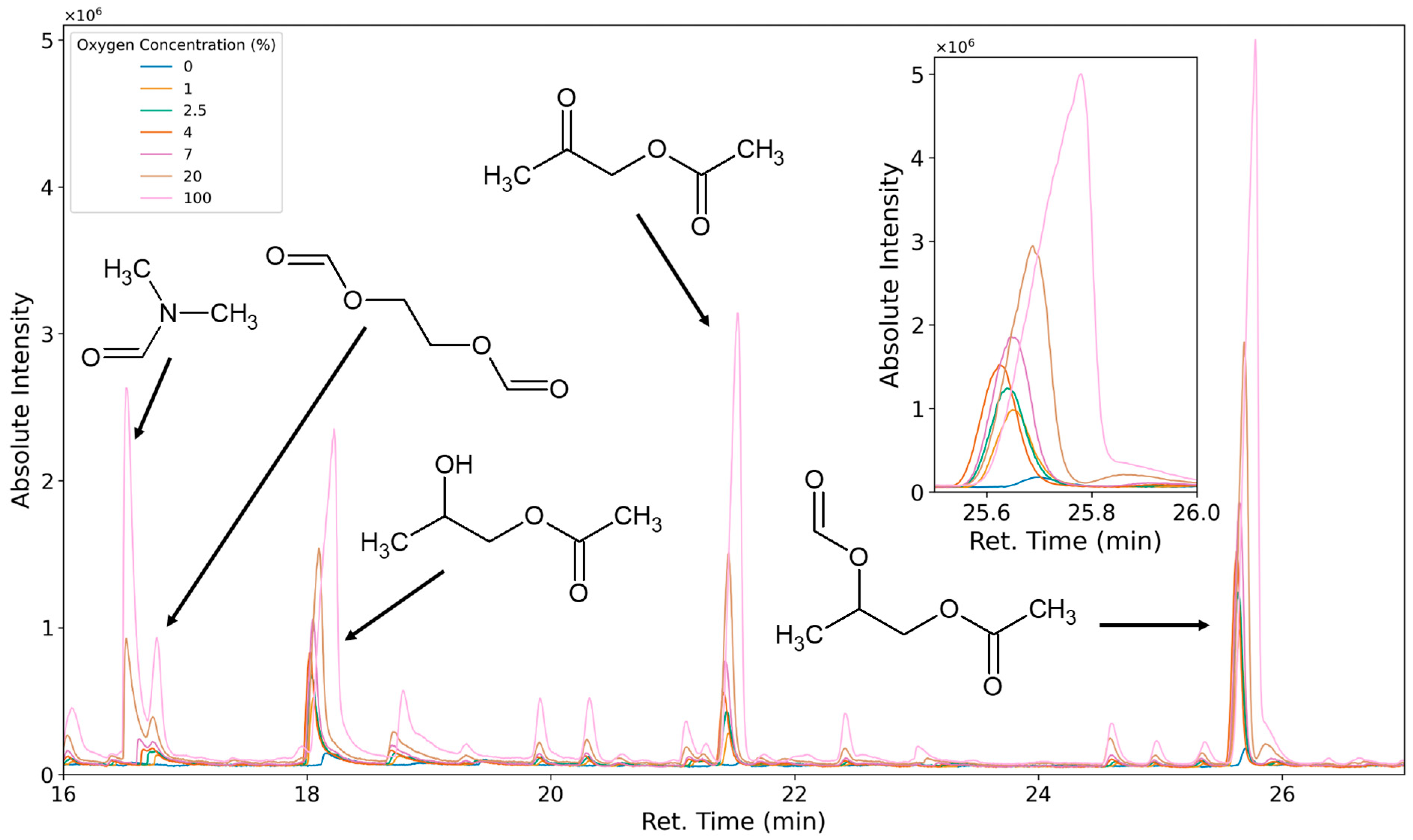
3.1.3. Degradation Products of the Soft Segment (PO Phase)
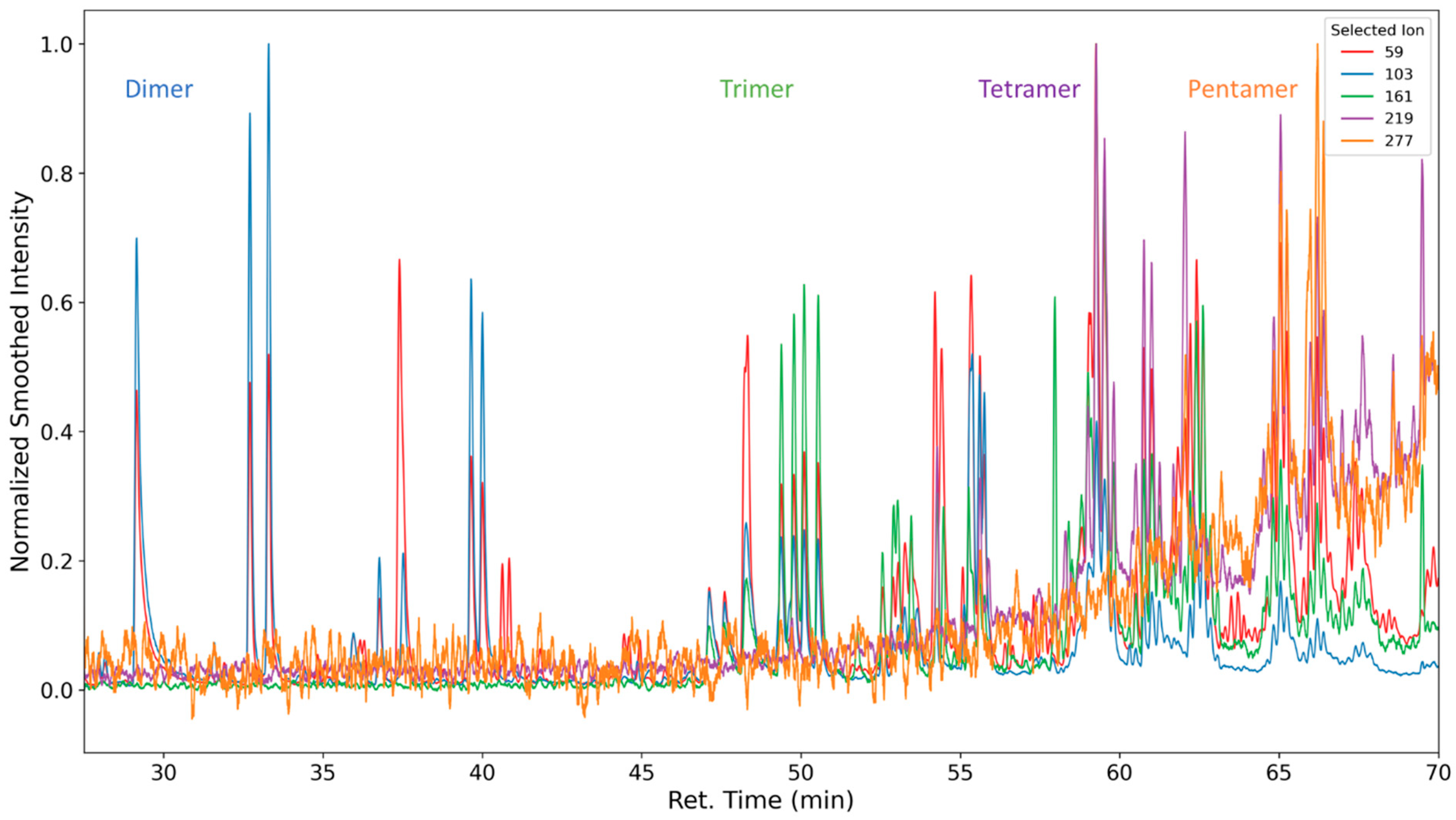
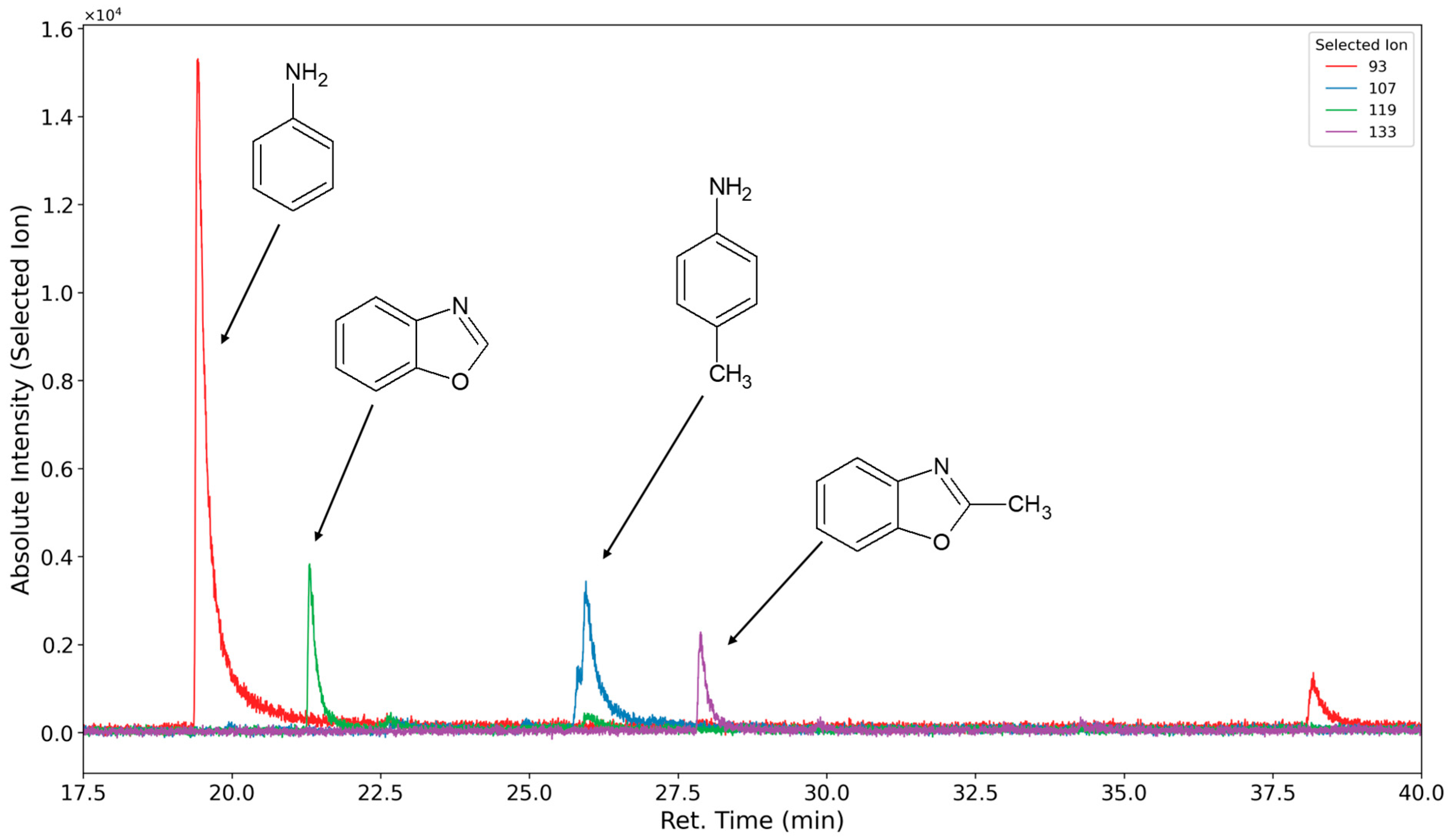
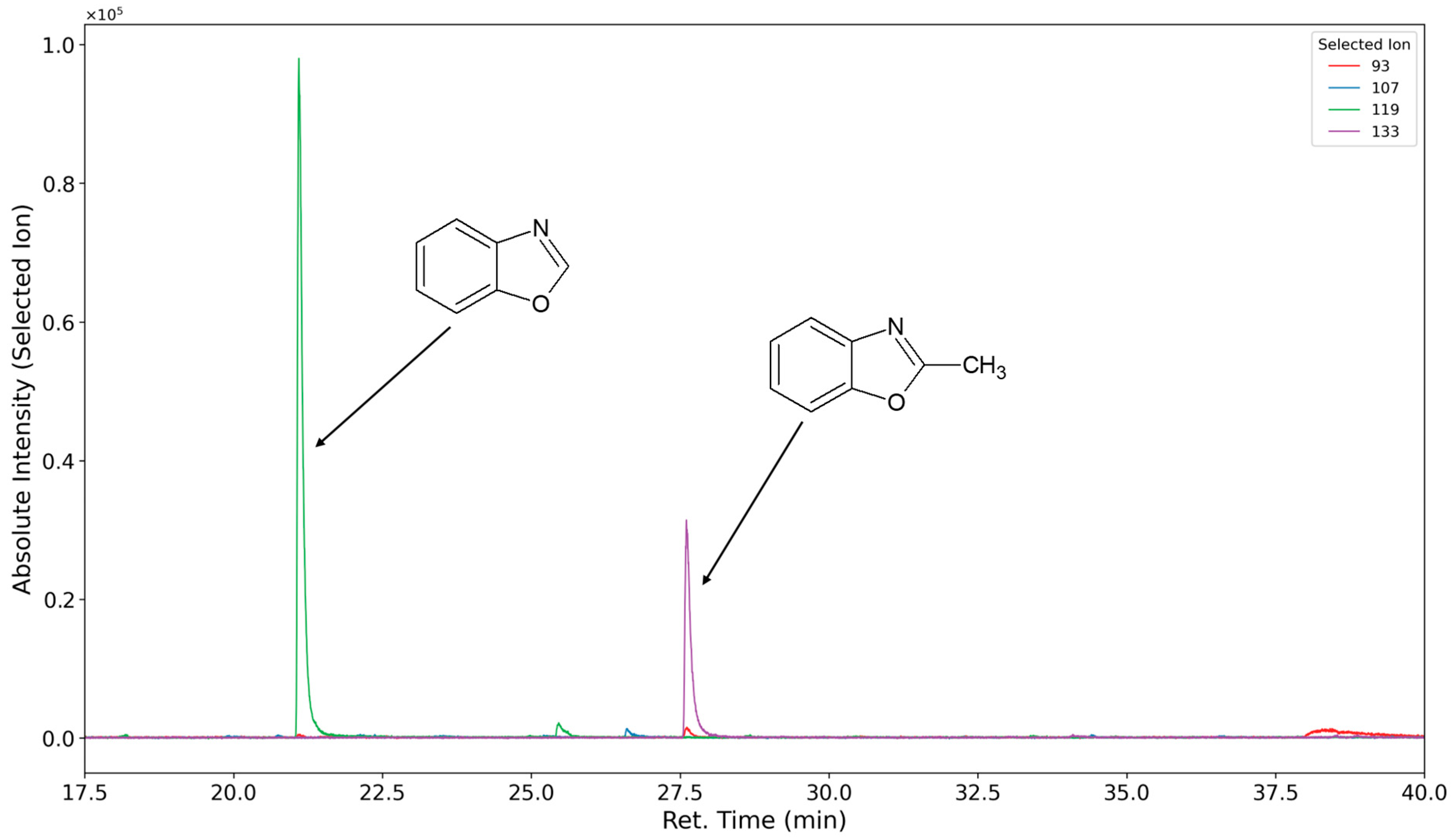
3.1.4. Degradation of the Hard Phase Segment
3.1.5. Classification of Emittents as Oxidation Products
3.2. Reproducibility
3.3. Results of Sampling Time Variation
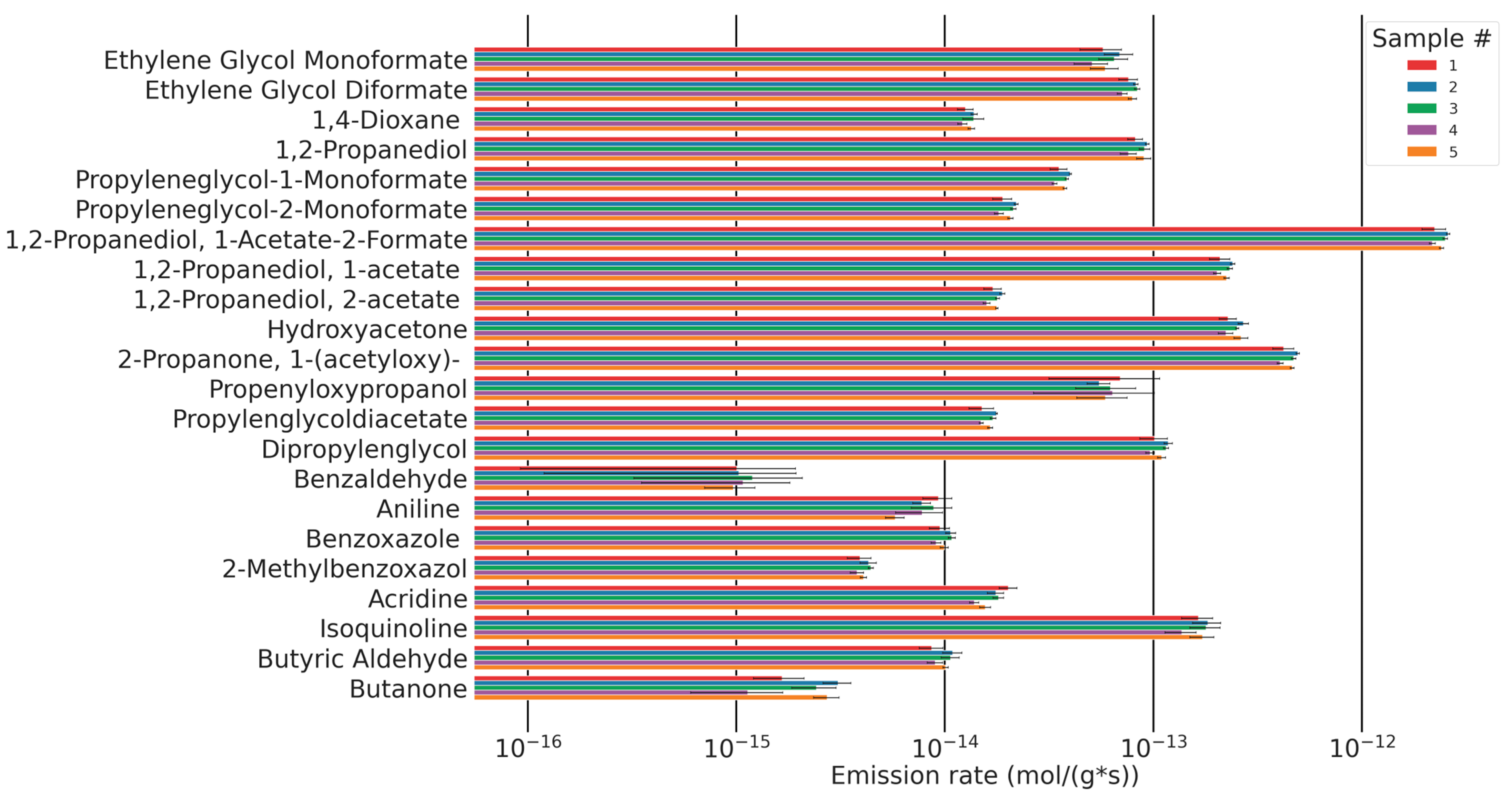
3.4. Analyte Quantification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oertel, G. (Ed.) Polyurethane: Kunststoff Handbuch 7, 3rd ed.; Hanser: München, Germany, 1993. [Google Scholar]
- Ionescu, M. Chemistry and Technology of Polyols for Polyurethanes, 2nd ed.; A Smithers Group Company: Shropshire, UK, 2016. [Google Scholar]
- Ashida, K. Polyurethane and Related Foams: Chemistry and Technology; CRC/Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Defonseka, C. Flexible Polyurethane Foams: A Practical Guide; De Gruyter: Berlin, Germany; Boston, MA, USA, 2019. [Google Scholar]
- Engels, H.W.; Pirkl, H.G.; Albers, R.; Albach, R.W.; Krause, J.; Hoffmann, A.; Casselmann, H.; Dormish, J. Polyurethanes: Versatile materials and sustainable problem solvers for today’s challenges. Angew. Chem. 2013, 52, 9422–9441. [Google Scholar] [CrossRef]
- Gama, N.V.; Ferreira, A.; Barros-Timmons, A. Polyurethane Foams: Past, Present, and Future. Materials 2018, 11, 1841. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, E.; Zafar, F. Polyurethane: An Introduction. In Polyurethane; Zafar, F., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Szycher, M. Szycher’s Handbook of Polyurethanes, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Sandten, C.S.; Kreyenschmidt, M.; Albach, R. The autoxidation of polyether-polyurethane open cell soft foam: An analytical aging method to reproducibly determine VOC emissions caused by thermo-oxidative degradation. J. Hazard. Mater. 2024, 474, 134747. [Google Scholar] [CrossRef] [PubMed]
- Grosborne, P.; Sérée de Roch, I.; Sajus, L. No 282.-Étude cinétique de l’oxydation des polyéthers en phase liquide. Bull. Soc. Chim. Fr. 1968, 1968, 2020–2029. [Google Scholar]
- Erlandsson, B. Stability-indicating changes in poloxamers: The degradation of ethylene oxide-propylene oxide block copolymers at 25 and 40 °C. Polym. Degrad. Stab. 2002, 78, 571–575. [Google Scholar] [CrossRef]
- Goglev, R.S.; Neiman, M.B. Thermal-oxidative degradation of the simpler polyalkyleneoxides. Polym. Sci. USSR 1968, 9, 2351–2364. [Google Scholar] [CrossRef]
- Gallet, G.; Carroccio, S.; Rizzarelli, P.; Karlsson, S. Thermal degradation of poly(ethylene oxide–propylene oxide–ethylene oxide) triblock copolymer: Comparative study by SEC/NMR, SEC/MALDI-TOF-MS and SPME/GC-MS. Polymer 2002, 43, 1081–1094. [Google Scholar] [CrossRef]
- Gallet, G.; Erlandsson, B.; Albertsson, A.-C.; Karlsson, S. Thermal oxidation of poly(ethylene oxide–propylene oxide–ethylene oxide) triblock copolymer: Focus on low molecular weight degradation products. Polym. Degrad. Stab. 2002, 77, 55–66. [Google Scholar] [CrossRef]
- Decker, C. Radiation-induced oxidation of solid poly(ethylene oxide). II. Mechanism. J. Polym. Sci. Polym. Chem. Ed. 1977, 15, 799–813. [Google Scholar] [CrossRef]
- Decker, C.; Marchal, J. Caracterisation de reactions primaries de degradation oxydante au cours de l’autoxidation des polyoxyethylenes a 25 °C: Etude en solution aqueuse avec amorcage par radiolyse du solvant: VI. Polyoxyethylene: Produits d’oxydation et schema cinetique. Die Makromol. Chem. 1973, 166, 155–178. [Google Scholar] [CrossRef]
- Griffiths, P.J.; Hughes, J.G.; Park, G.S. The autoxidation of poly(propylene oxide)s. Eur. Polym. J. 1993, 29, 437–442. [Google Scholar] [CrossRef]
- Santacesaria, E.; Gelosa, D.; Di Serio, M.; Tesser, R. Thermal stability of nonionic polyoxyalkylene surfactants. J. Appl. Polym. Sci. 1991, 42, 2053–2061. [Google Scholar] [CrossRef]
- Hähner, U.; Habicher, W.D.; Schwetlick, K. Studies on the thermooxidation of ethers and polyethers: Part I—The uninhibited thermooxidation of a polyether. Polym. Degrad. Stab. 1991, 34, 111–118. [Google Scholar] [CrossRef]
- Toivonen, H.; Lajunen, L.H.J.; Holster, T.; Hietaniemi, L.; Nupponen, H.; Theve, N.O. Autoxidation of Allyl Ether Compounds. Part 1. Reactivity of Allyl Ether Alcohols. Acta Chem. Scand. 1984, 38, 37–42. [Google Scholar] [CrossRef]
- Stahl, S.S.; Alsters, P.L. Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016. [Google Scholar]
- Yang, L.; Heatley, F.; Blease, T.G.; Thompson, R.I. A study of the mechanism of the oxidative thermal degradation of poly(ethylene oxide) and poly(propylene oxide) using 1H- and 13C-NMR. Eur. Polym. J. 1996, 32, 535–547. [Google Scholar] [CrossRef]
- Gauvin, P.; Lemaire, J.; Sallet, D. Photo-oxydation de polyéther-bloc-polyamides, 3†. Propriétés des hydroperoxydes dans les homopolym`res de polyéthers correspondants. Die Makromol. Chem. 1987, 188, 1815–1824. [Google Scholar] [CrossRef]
- Toivonen, H.; Ahlberg, P.; Rømming, C.; Sköldefors, H.; Wilking, N.; Theve, N.O. Autoxidation of Allyl Ether Compounds. Part II. Reactivity of Alkyl Allyl Ethers. Acta Chem. Scand. 1984, 38b, 63–66. [Google Scholar] [CrossRef]
- Aleksandrov, Y.A.; Sadikov, G.B.; Tomadze, T.V.; Golov, V.G. Nature of the primary products of the autoxidation and thermal polymerization of 4,4′-diisocyanatediphenylmethane. Khimiya Elementoorg. Soedin. 1976, 4, 78–80. [Google Scholar]
- Aleksandrov, Y.A.; Sadikov, G.B.; Golov, V.G. Peroxide product of the autoxidation of 4,4′-diisocyanatodiphenylmethane. Zhurnal Org. Khimii 1978, 14, 1873–1877. [Google Scholar]
- Schultze, H. Über den photochemischen Abbau von Polyurethanen. Die Makromol. Chem. 1973, 172, 57–75. [Google Scholar] [CrossRef]
- Hillier, K.; Schupp, T.; Carney, I. An Investigation into VOC Emissions from Polyurethane Flexible Foam Mattresses. Cell. Polym. 2003, 22, 237–259. [Google Scholar] [CrossRef]
- Thiébaut, B.; Lattuati-Derieux, A.; Hocevar, M.; Vilmont, L.-B. Application of headspace SPME-GC-MS in characterisation of odorous volatile organic compounds emitted from magnetic tape coatings based on poly(urethane-ester) after natural and artificial ageing. Polym. Test. 2007, 26, 243–256. [Google Scholar] [CrossRef]
- Choi, Y.J.; Alagi, P.; Jang, J.H.; Lee, S.J.; Yoon Hy Hong, S.C. Compositional elements of thermoplastic polyurethanes for reducing the generation of acetaldehyde during thermo-oxidative degradation. Polym. Test. 2018, 68, 279–286. [Google Scholar] [CrossRef]
- Yarahmadi, N.; Vega, A.; Jakubowicz, I. Accelerated ageing and degradation characteristics of rigid polyurethane foam. Polym. Degrad. Stab. 2017, 138, 192–200. [Google Scholar] [CrossRef]
- Poppendieck, D.; Gong, M.; Lawson, L. Lessons learned from spray polyurethane foam emission testing using micro-chambers. In Proceedings of the 59th Annual Polyurethanes Technical Conference, Baltimore, Maryland, 26–28 September 2016. [Google Scholar]
- Poppendieck, D.; Gong, M.; Emmerich, S. Characterization of Emissions from Spray Polyurethane Foam—Final Report to U.S. Consumer Product Safety Commission; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar]
- Even, M.; Hutzler, C.; Wilke, O.; Luch, A. Emissions of volatile organic compounds from polymer-based consumer products: Comparison of three emission chamber sizes. Indoor Air 2020, 30, 40–48. [Google Scholar] [CrossRef]
- Afshari, A.; Lundgren, B.; Ekberg, L.E. Comparison of three small chamber test methods for the measurement of VOC emission rates from paint. Indoor Air 2003, 13, 156–165. [Google Scholar] [CrossRef]
- Salem, M.Z.M.; Böhm, M.; Barcík, Š.; Srba, J. Inter-laboratory comparison of formaldehyde emission from particleboard using ASTM D 6007-02 method. Holz Als Roh- Und Werkst. 2012, 70, 621–628. [Google Scholar] [CrossRef]
- Even, M.; Wilke, O.; Kalus, S.; Schultes, P.; Hutzler, C.; Luch, A. Formaldehyde Emissions from Wooden Toys: Comparison of Different Measurement Methods and Assessment of Exposure. Materials 2021, 14, 262. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.-J. Comparison of standard methods and gas chromatography method in determination of formaldehyde emission from MDF bonded with formaldehyde-based resins. Bioresour. Technol. 2005, 96, 1457–1464. [Google Scholar] [CrossRef]
- Risholm-Sundman, M.; Larsen, A.; Vestin, E.; Weibull, A. Formaldehyde emission—Comparison of different standard methods. Atmos. Environ. 2007, 41, 3193–3202. [Google Scholar] [CrossRef]
- Risholm-Sundman, M.; Wallin, N. Comparison of different laboratory methods for determining the formaldehyde emission from three-layer parquet floors. Holz Als Roh- Und Werkst. 1999, 57, 319–324. [Google Scholar] [CrossRef]
- Haug, H.; Klein, L.; Sauerwald, T.; Poelke, B.; Beauchamp, J.; Roloff, A. Sampling Volatile Organic Compound Emissions from Consumer Products: A Review. Crit. Rev. Anal. Chem. 2022, 54, 1895–1916. [Google Scholar] [CrossRef]
- Fitzer, E.; Fritz, W.; Emig, G. Technische Chemie: Einführung in die Chemische Reaktionstechnik; Springer: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- VDA 278; Thermodesorptionsanalyse Organischer Emissionen zur Charakterisierung von Nichtmetallischen KFZ-Werkstoffen. Verband der Automobilindustrie: Eisenach, Germany, 2002.
- DIN ISO 12219-1; Innenraumluft von Straßenfahrzeugen: Teil 1 Gesamtfahrzeugprüfkammer Spezifikation und Verfahren zur Bestimmung von Flüchtigen Organischen Verbindungen in Fahrzeugkabinen; 13.040.20; 43.020 (ISO 12219-1:2021). Beuth Verlag GmbH: Berlin, Germany, 2022.
- Knauf, T.; Wershofen, S.; Gruner, K.-G.; Hartjes, V. Title (EN): Method of Producing Diamines and Polyamines of the Diphenylmethane Series at Different Production Capacities. C07C 209/78, C07C 209/60, C07C 211/50, C07C 209/54, C07C 211/48 (WP 2018/114767 A1). 2017. Available online: https://data.epo.org/publication-server/rest/v1.2/patents/EP3558929NWA1/document.html (accessed on 21 October 2024).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyether Polyol Mixture | g/mol | Mass [g] | mmol | mmol OH/NH | mmol PO | mmol EO |
|---|---|---|---|---|---|---|
| Propylene glycol ← PO58 ← EO13 | 4000 | 241.0 | 60 | 121 | 3073 | 663 |
| Glycerol ← PO87 ← EO19 | 6000 | 120.5 | 20 | 60 | 1908 | 362 |
| Glycerol ← PO70 ← EO15 | 4800 | 11.5 | 2 | 7 | 182 | 34 |
| Water | 18 | 18.3 | 1017 | 2033 | ||
| Triethanolamine | 149 | 1.0 | 7 | 20 | ||
| Triethylenediamine | 112 | 1 | 0.009 | |||
| Dabco NE300 (Evonik) | 203 | 0.4 | 2 | 2 | ||
| Polyether-modified polydimethylsiloxane Tegostab 8734 LF 2 (Evonik) | 500 | 3.7 | 7 | 7 | unknown | unknown |
| TOTAL | 396.4 | 2251 | 5163 | 1058 | ||
| Isocyanate | mmol NCO | |||||
| A blend of 4,4′-MDI, 2,4′-MDI, 2,2′-MDI, and polymeric MDI (2,2 mol NCO/mol) | 253.5 | 2028 |
| Target Analyte | Limiting Compounds | Excess Compound/Solvent |
|---|---|---|
| Monomer esters | ||
| 1,2-Ethanediol monoformate | Formic acid | 1,2-Ethanediol |
| 1,2-Ethanediol diformate | 1,2-Ethanediol | Formic acid |
| 1,2-Propanediol-1-formate | Formic acid | 1,2-Propanediol |
| 1,2-Propanediol-2-formate | Formic acid | 1,2-Propanediol |
| 1,2-Propanediol diformate | 1,2-Propanediol | Formic acid |
| Hydroxyacetoneformate | Hydroxyacetone | Formic acid |
| Dimer esters | ||
| Diethylene glycol monoformate | Formic acid | Diethylene glycol |
| Diethylene glycol diformate | Diethylene glycol | Formic acid |
| Dipropylene glycol monoformate | Formic acid | Dipropylene glycol |
| Dipropylene glycol diformate | Dipropylene glycol | Formic acid |
| Trimer esters | ||
| Triethylene glycol monoformate | Formic acid | Triethylene glycol |
| Triethylene glycol diformate | Triethylene glycol | Formic acid |
| Tripropylene glycol monoformate | Formic acid | Tripropylene glycol |
| Tripropylene glycol diformate | Tripropylene glycol | Formic acid |
| Peak # | Identified Compound | Retention Time (min) | M (g/mol) | Selected Ion | Yield (fmol/g·s) ± 1 σ |
|---|---|---|---|---|---|
| Polyether related products | |||||
| 1 (a) | Acetaldehyde | 2 | 44 | 44 | |
| 2 (a) | Acetic acid | 6 | 60 | 60 | |
| 3 (a) | Propionic aldehyde | 2.4 | 58 | 29 | |
| 4 (a) | Acrolein | 2.4 | 56 | 56 | |
| 5(a) | Acetone | 2.58 | 58 | 43 | |
| 6 (c) | cis-2,4-Dimethyl-1,3-dioxolane | 3.6 | 102 | 87 | |
| 7 (c) | trans-2,4-Dimethyl-1,3-dioxolane | 3.9 | 102 | 87 | |
| 8 (c) | 1,4-Dioxane (no oxidation product) | 5.33 | 88 | 88 | 13.1 ± 0.8 |
| 9 (c) | 2,5-Dimethyl-1,4-dioxane (no oxidation product) | 7.53 | 116 | 116 | |
| 10 (c) | Ethyleneglycolmonoformate | 10.4 | 60 | 60 ± 10 | |
| 11 (c) | Ethyleneglycol diformate | 17.4 | 72 | 79 ± 4 | |
| 12 (c) | Diethyleneglycol dialdehyde | 30 | 102 | ||
| 13 (a) | 1,2-Propyleneglycol | 7.5 | 76 | 45 | 87 ± 6 |
| 14 (a) | Hydroxyacetone | 6.73 | 74 | 74 | 248 ± 16 |
| 15 (c) | Propenyloxypropanol | 11.1 | 116 | 45 | 62 ± 24 |
| 16 (c) | 1,2-Propyleneglycol-1-formate | 13 | 45 | 37 ± 1 | |
| 17 (c) | 1,2-Propyleneglycol-2-formate | 14.2 | 45 | 20 ± 1 | |
| 18 (c) | Hydroxyacetone formate | 16.85 | 102 | ||
| 19 (c) | Hydroxyacetone acetate | 22.19 | 43 | 451 ± 18 | |
| 20 (c) | 1,2-Propyleneglycol-1-acetate | 18.86 | 118 | 43 | 222 ± 10 |
| 21 (c) | 1,2-Propyleneglycol-2-acetate | 19.5 | 118 | 43 | 17 ± 1 |
| 22 (c) | 1,2-Propyleneglycol diformate | 21 | 132 | 60 | |
| 23 (c) | 1,2-Propyleneglycol-1-formate-2-Acetate | 26 | 146 | 87 | |
| 24 (c) | 1,2-Propyleneglycol-1-acetate-2-formate | 26.4 | 146 | 43 | 2386 ± 106 |
| 25 (a) | 1,2-Propyleneglycol diacetate | 31.35 | 160 | 43 | 16 ± 1 |
| 26 (a) | Dipropyleneglycol | 29.5 | 59 | 108 ± 7 | |
| 27(b) | Propenyloxydipropyleneglycol | 32.3 | 41 | ||
| Isocyanate related compounds | |||||
| 28 (a) | Aniline (no oxidation product) | 20 | 93 | 8 ± 1 | |
| 29 (a) | Benzaldehyde | 21.55 | 106 | 1 ± 0.7 | |
| 30 (a) | Benzoxazole | 21.83 | 119 | 10 ± 1 | |
| 31 (a) | 2-Methylbenzoxazole | 28.39 | 133 | 4 ± 0.3 | |
| 32 (a) | Isoquinoline (no oxidation product) | 37.8 | 129 | 167 ± 26 | |
| 33 (a) | Acridine (no oxidation product) | 64 | 179 | 17 ± 1.25 | |
| Additive-related compounds | |||||
| 34 (a) | Diazabicyclooctane (no oxidation product) | 26.1 | 112 | ||
| 35 (a) | BHT (oxidized) | 53.43 | 220 | ||
| 36 (a) | Dimethylformamide | 18 | 73 | 73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandten, C.S.; Kreyenschmidt, M.; Albach, R.; Fittschen, U.E.A. The Thermo-Oxidative Degradation of Polyurethane Open-Cell Soft Foam Investigated Through Gas Chromatography and Mass Spectrometry of Volatile Organic Compounds. Polymers 2024, 16, 3342. https://doi.org/10.3390/polym16233342
Sandten CS, Kreyenschmidt M, Albach R, Fittschen UEA. The Thermo-Oxidative Degradation of Polyurethane Open-Cell Soft Foam Investigated Through Gas Chromatography and Mass Spectrometry of Volatile Organic Compounds. Polymers. 2024; 16(23):3342. https://doi.org/10.3390/polym16233342
Chicago/Turabian StyleSandten, Christian Stefan, Martin Kreyenschmidt, Rolf Albach, and Ursula E. A. Fittschen. 2024. "The Thermo-Oxidative Degradation of Polyurethane Open-Cell Soft Foam Investigated Through Gas Chromatography and Mass Spectrometry of Volatile Organic Compounds" Polymers 16, no. 23: 3342. https://doi.org/10.3390/polym16233342
APA StyleSandten, C. S., Kreyenschmidt, M., Albach, R., & Fittschen, U. E. A. (2024). The Thermo-Oxidative Degradation of Polyurethane Open-Cell Soft Foam Investigated Through Gas Chromatography and Mass Spectrometry of Volatile Organic Compounds. Polymers, 16(23), 3342. https://doi.org/10.3390/polym16233342