
Chemically Degradable Vitrimers Based on Divanillin Imine Diepoxy Monomer and Aliphatic Diamines for Enhanced Carbon Fiber Composite Applications




Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
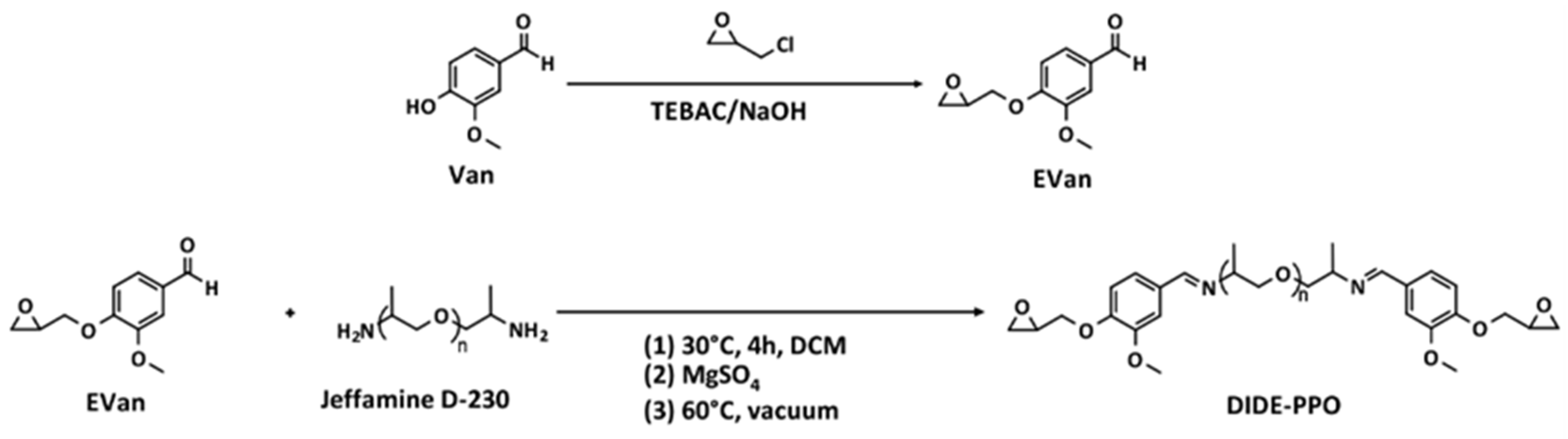
2.2. Synthesis of Vanillin Glycidyl Ether (EVan)
2.3. Synthesis of the Diimine–Diglycidyl Ether Derivative (DIDE-PPO)
2.4. General Procedure for the Preparation of Vitrimeric Samples
2.5. General Procedure for the Preparation of Composite Materials
2.6. Characterization Techniques
3. Results and Discussion
3.1. Synthesis of the Diimine Diepoxy Monomer DIDE-PPO
3.2. Study of the Curing Procedure
3.3. Thermal Stability Characterization of the Materials
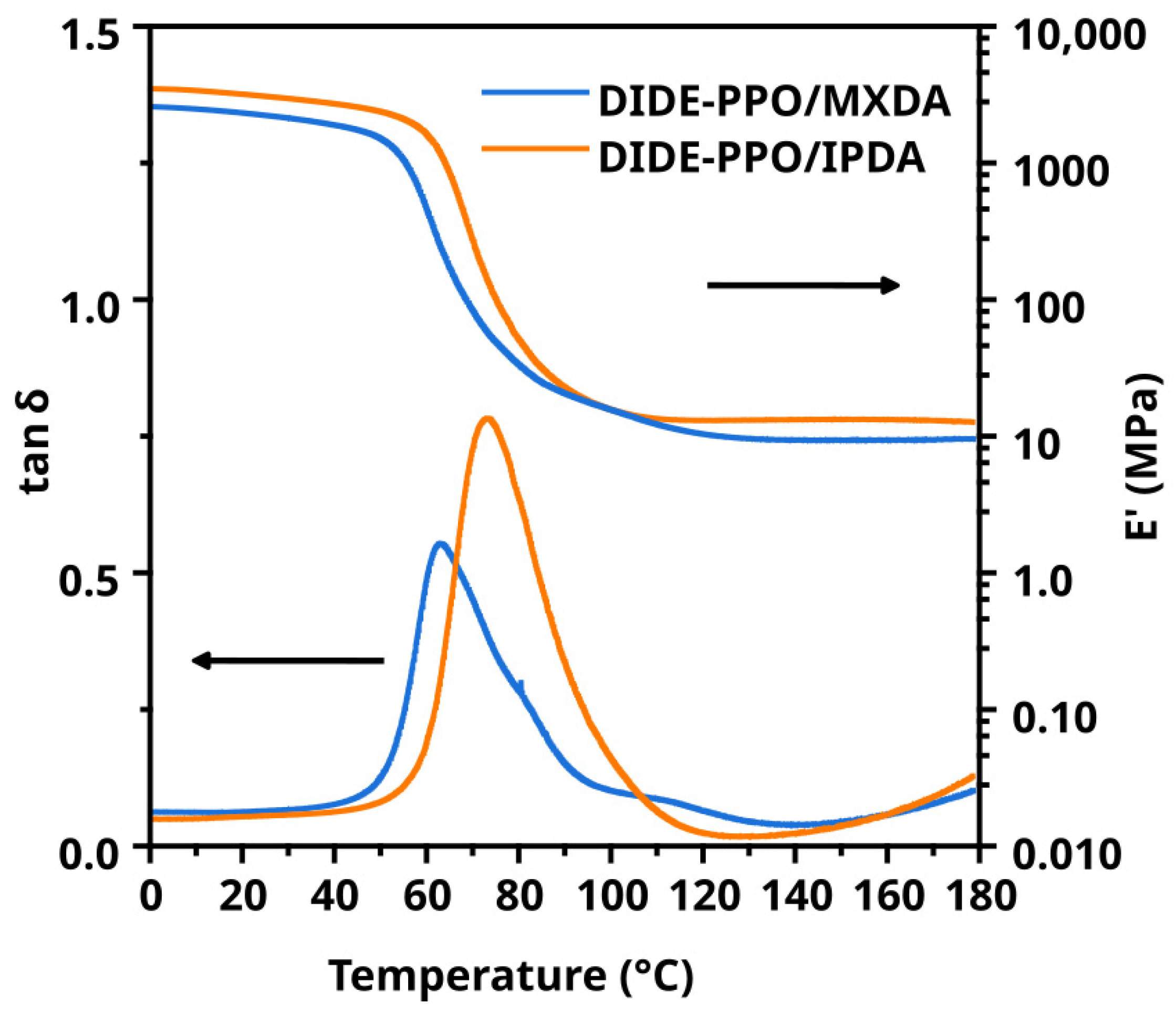
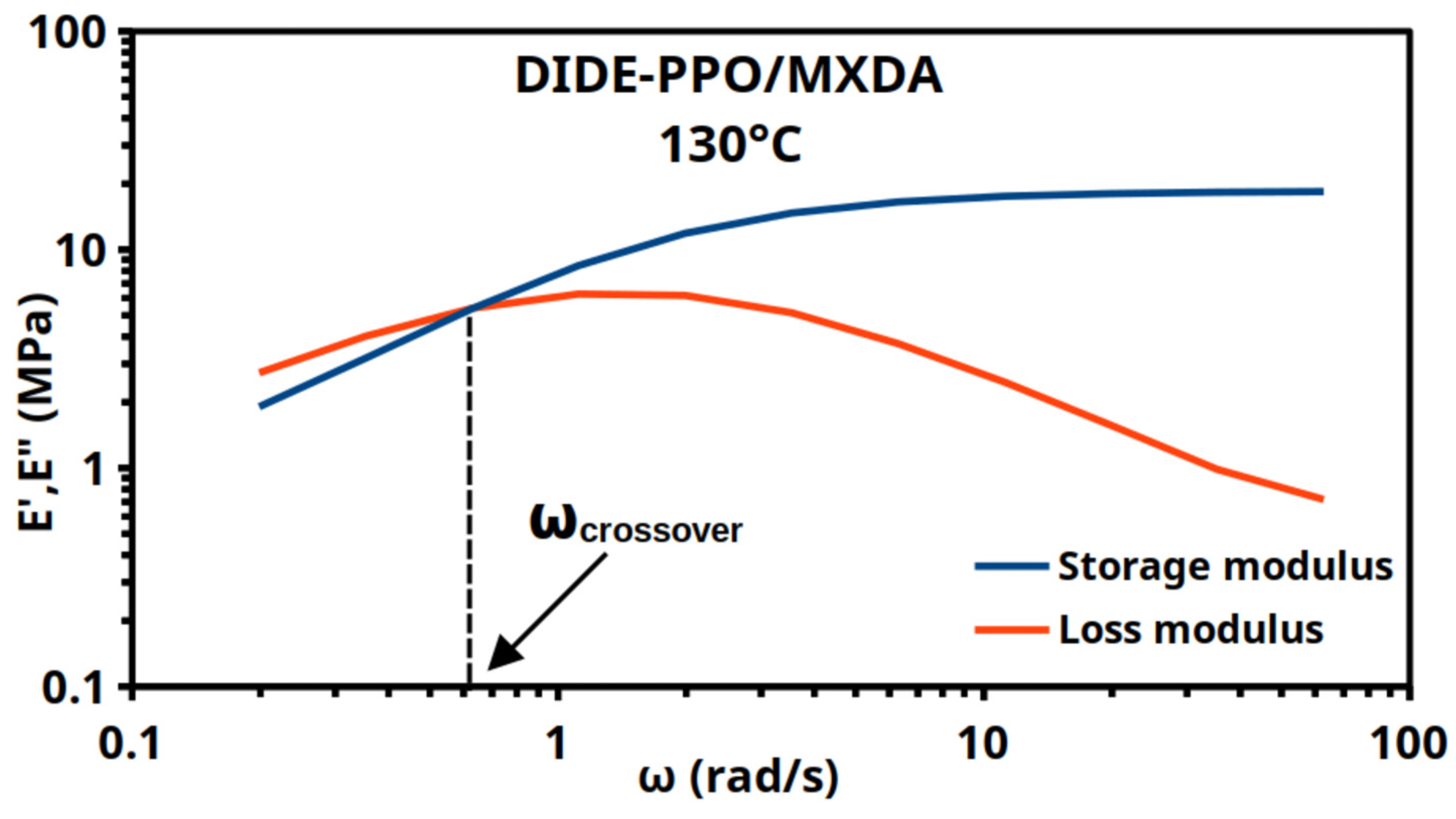
3.4. Thermo-Mechanical Properties of the Materials
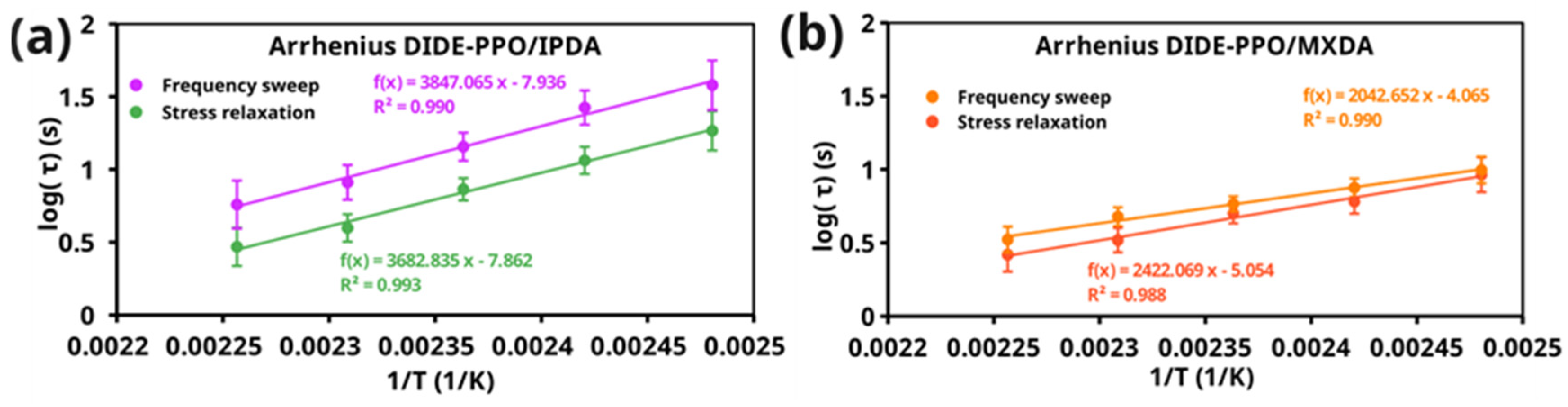
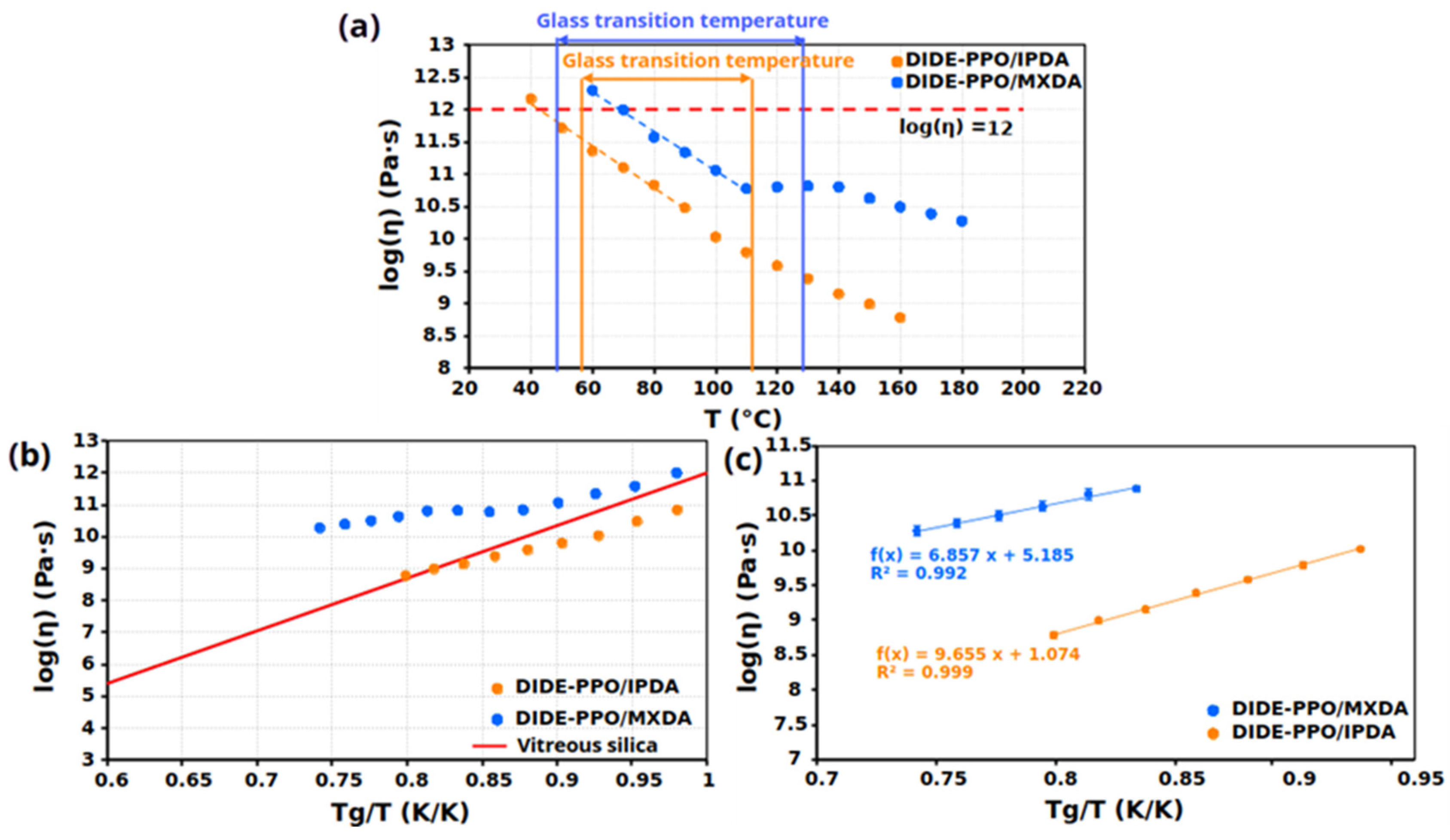
3.5. Vitrimeric Properties
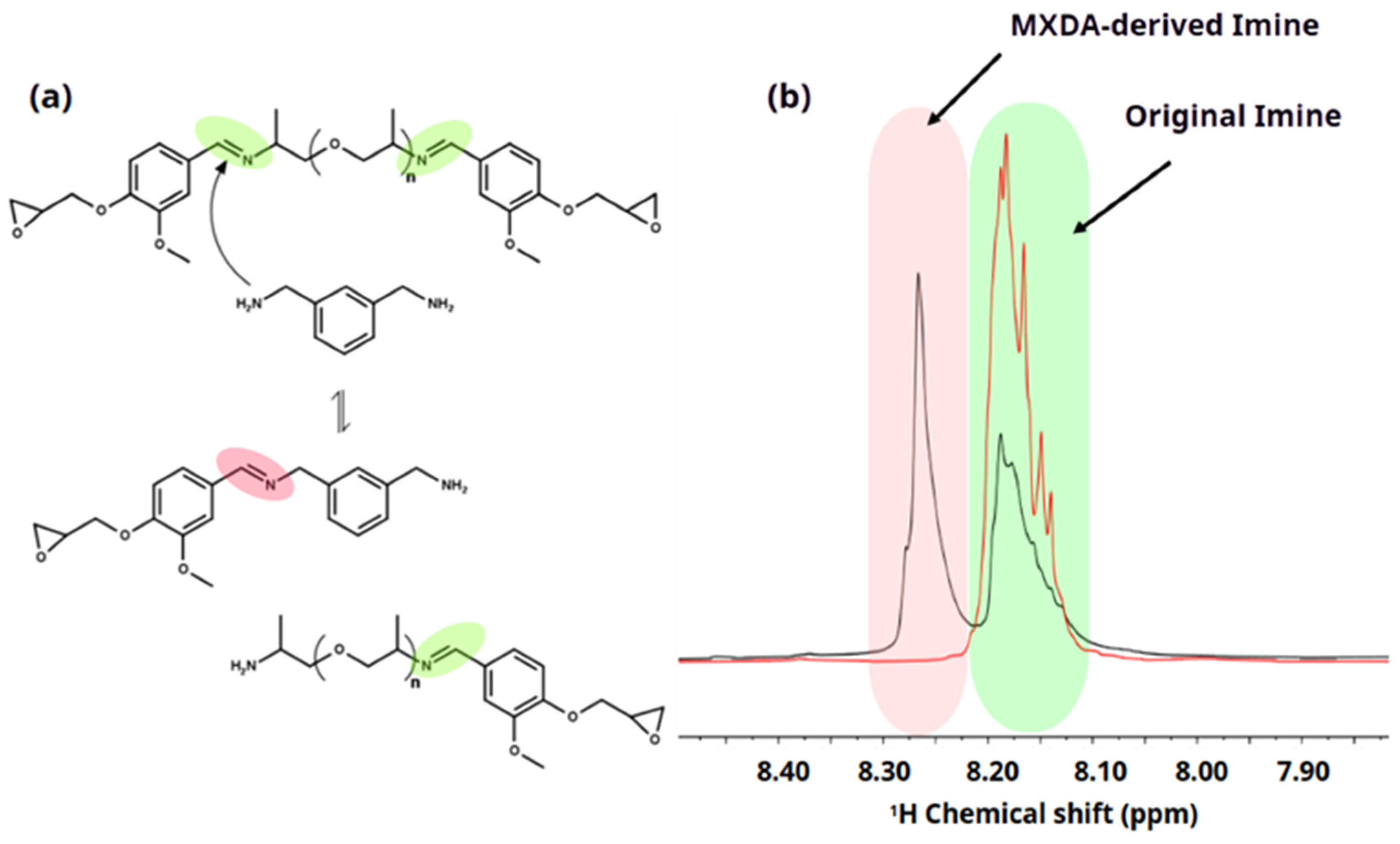
3.6. Chemical Degradation
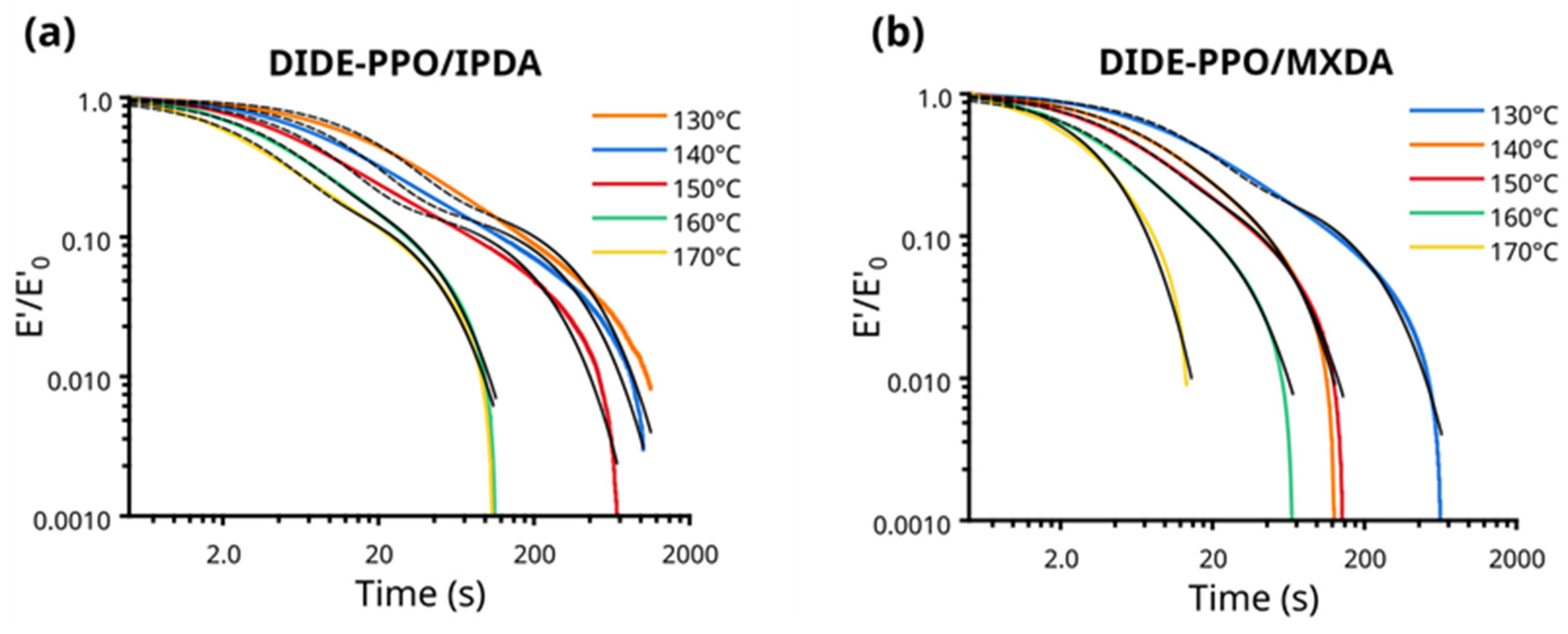
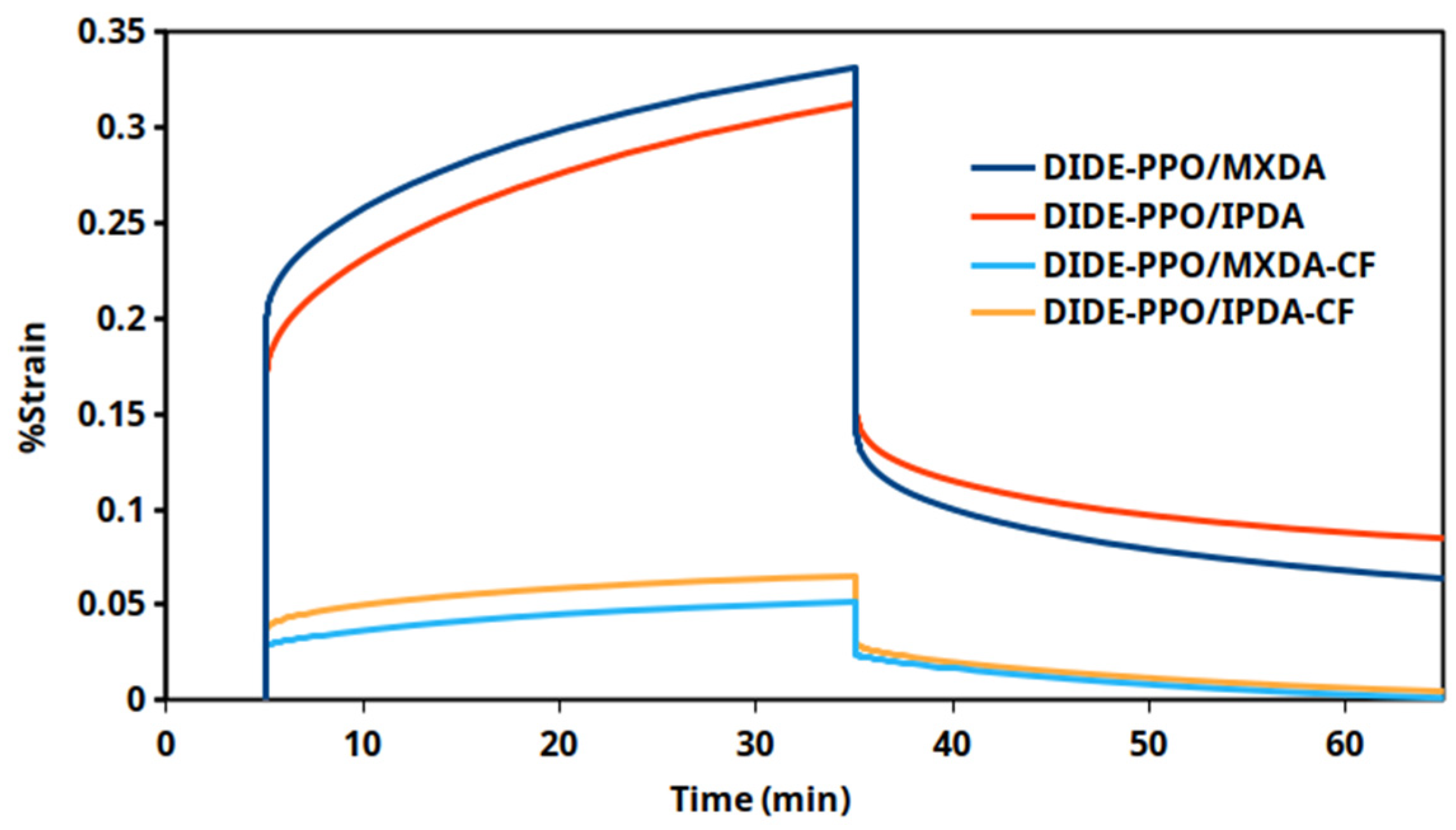
3.7. Creep Experiments
3.8. Carbon-Fiber-Reinforced Composite Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Guerre, M.; Taplan, C.; Winne, J.M.; Du Prez, F.E. Vitrimers: Directing chemical reactivity to control material properties. Chem. Sci. 2020, 11, 4855–4870. [Google Scholar] [CrossRef] [PubMed]
- Lucherelli, M.A.; Duval, A.; Avérous, L. Biobased vitrimers: Towards sustainable and adaptable performing polymer materials. Prog. Polym. Sci. 2022, 127, 101515. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef]
- Zheng, J.; Png, Z.M.; Ng, S.H.; Tham, G.X.; Ye, E.; Goh, S.S.; Loh, X.J.; Li, Z. Vitrimers: Current research trends and their emerging applications. Mater. Today 2021, 51, 568–625. [Google Scholar] [CrossRef]
- Lessard, J.J.; Scheutz, G.M.; Sung, S.H.; Lantz, K.A.; Epps, T.H., III; Sumerlin, B.S. Block copolymer vitrimers. J. Am. Chem. Soc. 2020, 142, 283–289. [Google Scholar] [CrossRef]
- Scheutz, G.M.; Lessard, J.J.; Sims, M.B.; Sumerlin, B.S. Adaptable crosslinks in polymeric materials: Resolving the intersection of thermoplastics and thermosets. J. Am. Chem. Soc. 2019, 141, 16181–16196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-L.; Tian, P.-X.; Li, Y.-D.; Zeng, J.-B. Biobased covalent adaptable networks: Towards better sustainability of thermosets. Green Chem. 2022, 24, 4363–4387. [Google Scholar] [CrossRef]
- Engelen, S.; Van Lijsebetten, F.; Aksakal, R.; Winne, J.M.; Du Prez, F.E. Enhanced Viscosity Control in Thermosets Derived from Epoxy and Acrylate Monomers Based on Thermoreversible Aza-Michael Chemistry. Macromolecules 2023, 56, 7055–7064. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Taynton, P.; Ni, H.; Zhu, C.; Yu, K.; Loob, S.; Jin, Y.; Qi, H.J.; Zhang, W. Repairable Woven Carbon Fiber Composites with Full Recyclability Enabled by Malleable Polyimine Networks. Adv. Mat. 2016, 28, 2904–2909. [Google Scholar] [CrossRef]
- Ciaccia, M.; Di Stefano, S. Mechanisms of imine exchange reactions in organic solvents. Org. Biomol. Chem. 2015, 13, 646–654. [Google Scholar] [CrossRef]
- Taynton, P.; Yu, K.; Shoemaker, R.K.; Jin, Y.; Qi, H.J.; Zhang, W. Heat or water driven malleability in a highly recyclable covalent network polymer. Adv. Mater. 2014, 26, 3938–3942. [Google Scholar] [CrossRef]
- Taynton, P.; Zhu, C.P.; Loob, S.; Shoemaker, R.; Pritchard, J.; Jin, Y.H.; Zhang, W. Re-healable polyimine thermosets polymer composition and moisture sensitivity. Polym. Chem. 2016, 7, 7052–7056. [Google Scholar] [CrossRef]
- Whiteley, J.M.; Taynton, P.; Zhang, W.; Lee, S.H. Ultra-thin solid-state Li-ion electrolyte membrane facilitated by a self- healing polymer matrix. Adv. Mater. 2015, 27, 6922–6927. [Google Scholar] [CrossRef]
- Liang, K.; Zhang, G.; Zhao, J.; Shi, L.; Cheng, V.; Zhang, J. Malleable, recyclable, and robust poly(amide–imine) vitrimers prepared through a green polymerization process. ACS Sustain. Chem. Eng. 2021, 9, 5673–5683. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wang, H.; Huang, X.; Huang, G.; Wu, J. Weldable, malleable and programmable epoxy vitrimers with high mechanical properties and water insensitivity. Chem. Eng. J. 2019, 368, 61–70. [Google Scholar] [CrossRef]
- Telatin, T.; De la Flor, S.; Serra, À.; Montané, X. Poly(epoxy-imine) vitrimers. Effect of the structure on the stress relaxation and creep resistance. Polym. Test. 2024, 135, 108465. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Ramis, X. Latent curing of epoxy-thiol thermosets. Polymer 2017, 116, 191–203. [Google Scholar] [CrossRef]
- Zhao, S.; Abu-Omar, M.M. Recyclable and Malleable Epoxy Thermoset Bearing Aromatic Imine Bonds. Macromolecules 2023, 51, 9816–9824. [Google Scholar] [CrossRef]
- Miao, P.; Leng, X.; Liu, J.; Song, G.; He, M.; Li, Y. Regulating the Dynamic Behaviors of Transcarbamoylation-Based Vitrimers via Mono-Variation in Density of Exchangeable Hydroxyl. Macromolecules 2022, 55, 4956–4966. [Google Scholar] [CrossRef]
- Roig, A.; Hidalgo, P.; Ramis, X.; De la Flor, S.; Serra, À. Vitrimeric epoxy-amine polyimine networks based on a renewable vanillin derivative. ACS Appl. Polym. Mater. 2022, 4, 9341–9350. [Google Scholar] [CrossRef]
- Zhao, X.L.; Liu, Y.Y.; Weng, Y.; Li, Y.D.; Zeng, J.B. Sustainable epoxy vitrimers from epoxidized soybean oil and vanillin. ACS Sustain. Chem. Eng. 2020, 8, 15020–15029. [Google Scholar] [CrossRef]
- Ding, X.-M.; Chen, L.; Xu, Y.-J.; Shi, X.-H.; Luo, X.; Song, X.; Wang, Y.-Z. Robust Epoxy Vitrimer with Simultaneous Ultrahigh Impact Property, Fire Safety, and Multipath Recyclability via Rigid-Flexible Imine Networks. ACS Sustain. Chem. Eng. 2023, 11, 14445–14456. [Google Scholar] [CrossRef]
- Yu, Q.; Peng, X.; Wang, Y.; Geng, H.; Xu, A.; Zhang, X.; Xu, W.; Ye, D. Vanillin-based degradable epoxy vitrimers: Reprocessability and mechanical properties study. Eur. Polym. J. 2019, 117, 55–63. [Google Scholar] [CrossRef]
- Roig, A.; Petrauskaitė, A.; Ramis, X.; De la Flor, S.; Serra, À. Synthesis and characterization of new bio-based poly-(acylhydrazone) vanillin vitrimers. Polym. Chem. 2022, 13, 1510–1519. [Google Scholar] [CrossRef]
- Verdugo, P.; Santiago, D.; De la Flor, S.; Serra, À. A Biobased Epoxy Vitrimer with Dual Relaxation Mechanism: A Promising Material for Renewable, Reusable, and Recyclable Adhesives and Composites. ACS Sustain. Chem. Eng. 2024, 12, 5965. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Wei, T.; Venerus, D.C.; Torkelson, J.M. Reprocessable Polyhydroxyurethane Network Composites: Effect of Filler Surface Functionality on Cross-link Density Recovery and Stress Relaxation. ACS Appl. Mater. Inter. 2019, 11, 2398–2407. [Google Scholar] [CrossRef]
- Meng, F.; Saed, M.O.; Terentjev, E.M. Elasticity and Relaxation in Full and Partial Vitrimer Networks. Macromolecules 2019, 52, 7423–7429. [Google Scholar] [CrossRef]
- Van Lijsebetten, F.; Debsharma, T.; Winne, J.M.; Du Prez, F.E. A Highly Dynamic Covalent Polymer Network without Creep: Mission Impossible? Angew. Chem. Int. 2022, 61, e202210405. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, G.; Wu, S.; Tang, Z.; Guo, B.; Zhang, L. Effects of dynamic covalent bond multiplicity on the performance of vitrimeric elastomers. J. Mater. Chem. A 2020, 8, 20503–20512. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Jin, K.; Torkelson, J.M. Vitrimers designed both to strongly suppress creep and to recover original cross-link density after reprocessing: Quantitative theory and experiments. Macromolecules 2018, 51, 5537–5546. [Google Scholar] [CrossRef]
- Porath, L.E.; Evans, C.M. Importance of Broad Temperature Windows and Multiple Rheological Approaches for Probing Viscoelasticity and Entropic Elasticity in Vitrimers. Macromolecules 2021, 54, 4782–4791. [Google Scholar] [CrossRef]
- Yuan, Y.; Sun, Y.; Yan, S.; Zhao, J.; Liu, S.; Zhang, M.; Zheng, X.; Jia, L. Multiply fully recyclable carbon fibre reinforced heat-resistant covalent thermosetting advanced composites. Nat. Commun. 2017, 8, 14657. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, F.; Xu, N.; Wang, B.; Yang, L.; Huang, Y.; Hu, Z. Mechanically robust, hydrothermal aging resistant, imine-containing epoxy thermoset for recyclable carbon fiber reinforced composites. Mater. Des. 2022, 224, 111357. [Google Scholar] [CrossRef]
- Shaw, M.T.; MacKnight, W.J. Introduction to Polymer Viscoelasticity; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Sperling, L.H. Introduction to Physical Polymer Science; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kelton, K.F. Kinetic and structural fragility—A correlation between structures and dynamics in metallic liquids and glasses. J. Phys. Condens. Matter 2017, 29, 023002. [Google Scholar] [CrossRef]
- Appelt, B.K.; Cook, P.J. Defining B-stage and total cure for a dicy based epoxy resin by DSC. In Analytical Calorimetry Volume 5; Gill, P.S., Johnson Julian, F., Eds.; Springer: New York, NY, USA, 1984; pp. 57–66. [Google Scholar] [CrossRef]
- Ruiz De Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Grunenfelder, L.K.; Nutt, S.R. Prepreg age monitoring via differential scanning calorimetry. J. Reinf. Plast. Compos. 2012, 31, 295–302. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T1% a (°C) | T2% b (°C) | Tmax c (°C) | Char Yield d (%) | Ttan δ e (°C) | FWHM f (°C) | E′Glassy g (MPa) | E′Rubbery h (MPa) |
|---|---|---|---|---|---|---|---|---|
| DIDE-PPO/IPDA | 275 ± 1 | 289 ± 2 | 342 ± 5 | 27.7 ± 0.5 | 73.0 ± 3.0 | 24 ± 4 | 3117 ± 80 | 13 ± 2 |
| DIDE-PPO/MXDA | 273 ± 3 | 286 ± 3 | 343 ± 4 | 37.9 ± 1.0 | 63.0 ± 2.0 | 25 ± 3 | 2437 ± 95 | 9 ± 1 |
| Sample | T (°C) | A a | τ1 b (s) | τ2 c (s) | R2 | τcrossover d (s) |
|---|---|---|---|---|---|---|
| DIDE-PPO/IPDA | 130 | 0.797 | 308.74 | 18.45 | 0.992 | 37.92 |
| 140 | 0.18 | 258.01 | 11.54 | 0.991 | 26.6 | |
| 150 | 0.181 | 160.08 | 7.31 | 0.993 | 14.33 | |
| 160 | 0.271 | 27.93 | 3.96 | 0.996 | 8.16 | |
| 170 | 0.227 | 26.87 | 2.94 | 0.991 | 5.74 | |
| DIDE-PPO/MXDA | 130 | 0.302 | 155.54 | 9.21 | 0.992 | 9.91 |
| 140 | 0.619 | 34.03 | 6.03 | 0.999 | 7.51 | |
| 150 | 0.719 | 39.77 | 5.01 | 0.998 | 5.82 | |
| 160 | 0.73 | 18.99 | 3.3 | 0.993 | 4.77 | |
| 170 | 0.231 | 3.79 | 2.62 | 0.989 | 3.33 |
| Stress Relaxation | Frequency Sweep | |||||
|---|---|---|---|---|---|---|
| Sample | Ea (kJ/mol) | logA (s) | R2 | Ea (kJ/mol) | logA (s) | R2 |
| DIDE-PPO/IPDA | 70 ± 11 | 7.9 ± 1.3 | 0.993 | 74 ± 13 | 7.9 ± 1.6 | 0.99 |
| DIDE-PPO/MXDA | 46 ± 9 | 5.0 ± 1.2 | 0.988 | 39 ± 7 | 4.1 ± 0.9 | 0.99 |
| Sample | Tv a (°C) | Ea (kJ/mol) | logA (s) | R2 |
|---|---|---|---|---|
| DIDE-PPO/IPDA | 43 | 64 ± 2 | 1.1 ± 0.3 | 0.999 |
| DIDE-PPO/MXDA | 71 | 44 ± 5 | 5.2 ± 0.6 | 0.992 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telatin, T.; De la Flor, S.; Montané, X.; Serra, À. Chemically Degradable Vitrimers Based on Divanillin Imine Diepoxy Monomer and Aliphatic Diamines for Enhanced Carbon Fiber Composite Applications. Polymers 2024, 16, 2754. https://doi.org/10.3390/polym16192754
Telatin T, De la Flor S, Montané X, Serra À. Chemically Degradable Vitrimers Based on Divanillin Imine Diepoxy Monomer and Aliphatic Diamines for Enhanced Carbon Fiber Composite Applications. Polymers. 2024; 16(19):2754. https://doi.org/10.3390/polym16192754
Chicago/Turabian StyleTelatin, Tommaso, Silvia De la Flor, Xavier Montané, and Àngels Serra. 2024. "Chemically Degradable Vitrimers Based on Divanillin Imine Diepoxy Monomer and Aliphatic Diamines for Enhanced Carbon Fiber Composite Applications" Polymers 16, no. 19: 2754. https://doi.org/10.3390/polym16192754
APA StyleTelatin, T., De la Flor, S., Montané, X., & Serra, À. (2024). Chemically Degradable Vitrimers Based on Divanillin Imine Diepoxy Monomer and Aliphatic Diamines for Enhanced Carbon Fiber Composite Applications. Polymers, 16(19), 2754. https://doi.org/10.3390/polym16192754