
3D-Printed Hydrogels as Photothermal Actuators

 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Gold Nanorod Synthesis and Functionalization
2.2. Preparation of Hydrogel Macromer
2.3. Preparation of PNIPAAm/GNR Composite Ink for 3D Printing
2.4. Preparation of Acetylated Gelatin Microgel Sacrificial Support Matrix
2.5. Printing Process and Removal of Acetylated Gelatin Microgel Support Matrix
2.6. Characterization of GNRs
2.7. Characterization of 3D-Printed Materials
2.8. Convective Heating
2.9. Photothermal Heating
3. Results and Discussion
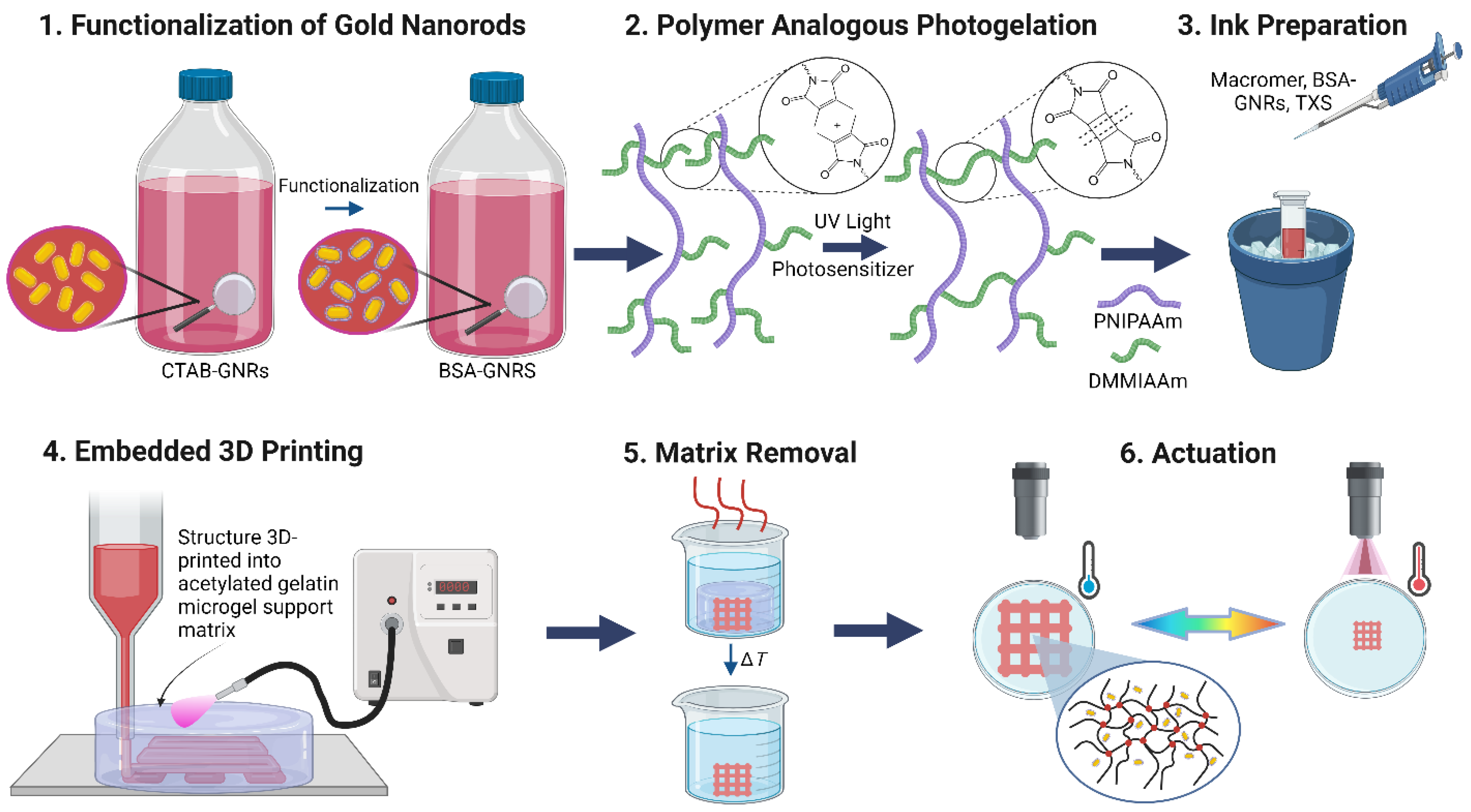
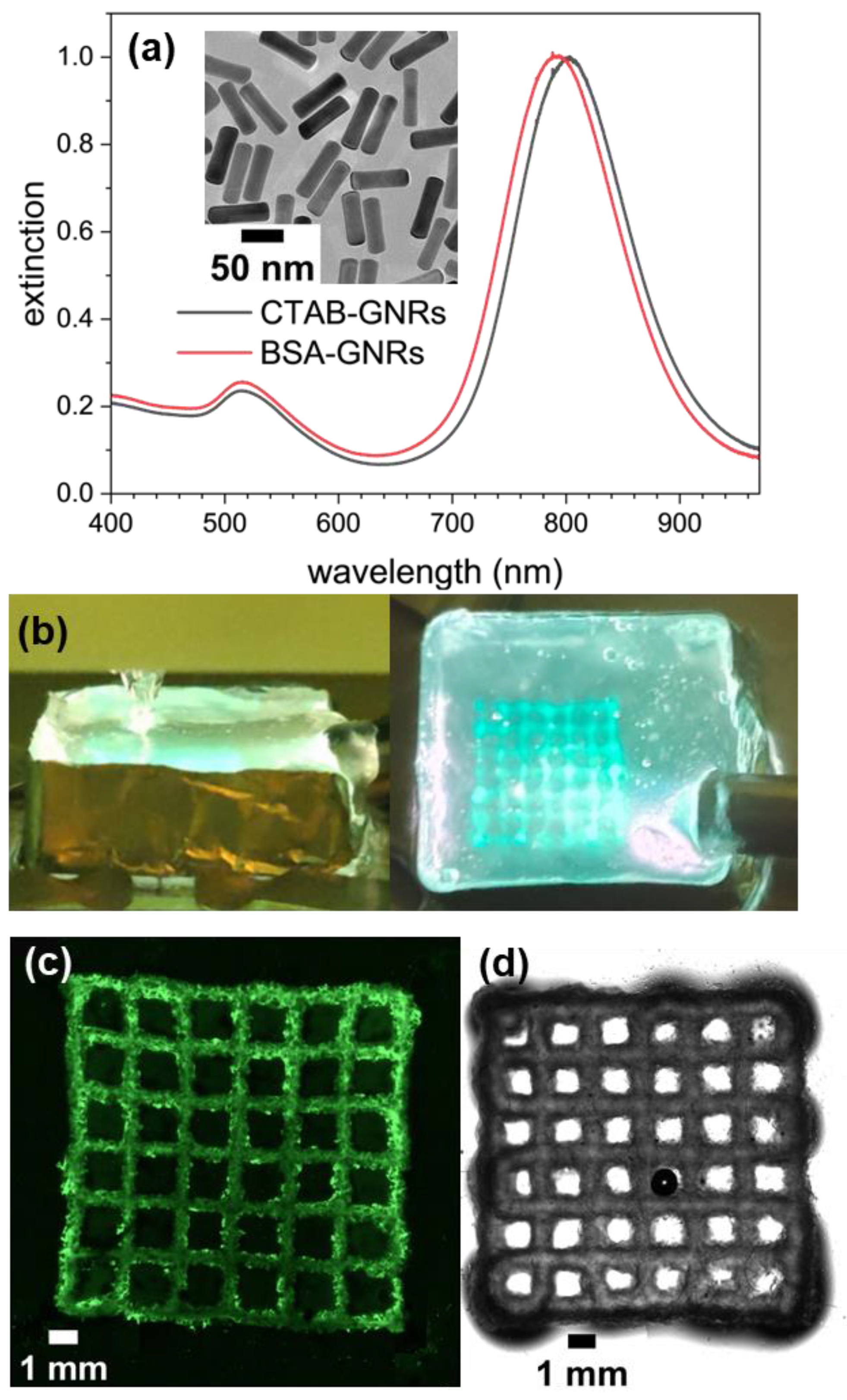
3.1. Preparation of Ink and Sacrificial Support Matrix
3.2. Embedded 3D Printing and Characterization
3.3. Photothermal Heating and Convective Heating
3.4. Effect of Loading of GNRs
3.5. Kinetics of Collapse and Reswelling
3.6. Buckling during Reswelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallin, T.J.; Pikul, J.; Shepherd, R.F. 3D Printing of Soft Robotic Systems. Nat. Rev. Mater. 2018, 3, 84–100. [Google Scholar] [CrossRef]
- Zhao, Y.; Hua, M.; Yan, Y.; Wu, S.; Alsaid, Y.; He, X. Stimuli-Responsive Polymers for Soft Robotics. Annu. Rev. Control Robot. Auton. Syst. 2022, 5, 515–545. [Google Scholar] [CrossRef]
- Soleymani Eil Bakhtiari, S.; Bakhsheshi-Rad, H.R.; Karbasi, S.; Razzaghi, M.; Tavakoli, M.; Ismail, A.F.; Sharif, S.; RamaKrishna, S.; Chen, X.; Berto, F. 3-Dimensional Printing of Hydrogel-Based Nanocomposites: A Comprehensive Review on the Technology Description, Properties, and Applications. Adv. Eng. Mater. 2021, 23, 2100477. [Google Scholar] [CrossRef]
- Budhlall, B.M.; Marquez, M.; Velev, O.D. Microwave, Photo- and Thermally Responsive PNIPAm−Gold Nanoparticle Microgels. Langmuir 2008, 24, 11959–11966. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.W.; Evans, A.A.; Na, J.-H.; Hayward, R.C. Photothermally Reprogrammable Buckling of Nanocomposite Gel Sheets. Angew. Chem. Int. Ed. 2015, 54, 5434–5437. [Google Scholar] [CrossRef] [PubMed]
- Goldhahn, C.; Schubert, J.; Schlaad, H.; Ferri, J.K.; Fery, A.; Chanana, M. Synthesis of Metal@Protein@Polymer Nanoparticles with Distinct Interfacial and Phase Transfer Behavior. Chem. Mater. 2018, 30, 6717–6727. [Google Scholar] [CrossRef]
- Huang, X.; Neretina, S.; El-Sayed, M.A. Gold Nanorods: From Synthesis and Properties to Biological and Biomedical Applications. Adv. Mater. 2009, 21, 4880–4910. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xia, H.; Liu, Y.; Liu, B.; Chen, W.; Zhao, Y. Applications of Gold Nanorods in Biomedical Imaging and Related Fields. Chin. Sci. Bull. 2013, 58, 2530–2536. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, F.; Chen, L.; Wei, X.; Xue, M.; Yang, F.; Jiang, S. 3D Printing Hydrogels for Actuators: A Review. Chin. Chem. Lett. 2021, 32, 2923–2932. [Google Scholar] [CrossRef]
- Shang, J.; Le, X.; Zhang, J.; Chen, T.; Theato, P. Trends in Polymeric Shape Memory Hydrogels and Hydrogel Actuators. Polym. Chem. 2019, 10, 1036–1055. [Google Scholar] [CrossRef]
- Sun, W.; Schaffer, S.; Dai, K.; Yao, L.; Feinberg, A.; Webster-Wood, V. 3D Printing Hydrogel-Based Soft and Biohybrid Actuators: A Mini-Review on Fabrication Techniques, Applications, and Challenges. Front. Robot. AI 2021, 8, 673533. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dong, M.; Zheng, Q.; Wu, Z.L. Digital Light Processing 3D Printing of Hydrogels: A Minireview. Mol. Syst. Des. Eng. 2022, 7, 1017–1029. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-Isopropylacrylamide): Experiment, Theory and Application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Ron, E.S.; Bromberg, L.E. Temperature-Responsive Gels and Thermogelling Polymer Matrices for Protein and Peptide Delivery. Adv. Drug Deliv. Rev. 1998, 31, 197–221. [Google Scholar] [PubMed]
- Qiu, Y.; Park, K. Environment-Sensitive Hydrogels for Drug Delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Sanson, N.; Fava, D.; Kumacheva, E. Microgels Loaded with Gold Nanorods: Photothermally Triggered Volume Transitions under Physiological Conditions. Langmuir 2007, 23, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Mourran, A.; Zhang, H.; Vinokur, R.; Möller, M. Soft Microrobots Employing Nonequilibrium Actuation via Plasmonic Heating. Adv. Mater. 2017, 29, 1604825. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, A.; Zhang, H.; Schweizerhof, S.; Schulte, M.F.; Mourran, A.; Möller, M. 4D Printing of a Light-Driven Soft Actuator with Programmed Printing Density. ACS Appl. Mater. Interfaces 2020, 12, 12176–12185. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Jiang, S.; Zhang, Y.; Dai, Z.; Dai, Y.; Xia, F.; Zhang, X. Gold Nanorods Crosslinking PNIPAM Hydrogels via Dynamic Au-Thiolate Interaction with Stretchable, Adhesive, Self-Healing, and Photothermal Properties. Gold Bull. 2021, 54, 59–67. [Google Scholar] [CrossRef]
- Han, D.; Lu, Z.; Chester, S.A.; Lee, H. Micro 3D Printing of a Temperature-Responsive Hydrogel Using Projection Micro-Stereolithography. Sci. Rep. 2018, 8, 1963. [Google Scholar] [CrossRef]
- Guvendiren, M.; Molde, J.; Soares, R.M.D.; Kohn, J. Designing Biomaterials for 3D Printing. ACS Biomater. Sci. Eng. 2016, 2, 1679–1693. [Google Scholar] [CrossRef] [PubMed]
- Ligon, S.C.; Liska, R.; Stampfl, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef] [PubMed]
- Bakarich, S.E.; Gorkin Iii, R.; Panhuis, M.i.h.; Spinks, G.M. 4D Printing with Mechanically Robust, Thermally Actuating Hydrogels. Macromol. Rapid Commun. 2015, 36, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Gladman, A.S.; Matsumoto, E.A.; Nuzzo, R.G.; Mahadevan, L.; Lewis, J.A. Biomimetic 4D Printing. Nat. Mater. 2016, 15, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Naficy, S.; Gately, R.; Gorkin III, R.; Xin, H.; Spinks, G.M. 4D Printing of Reversible Shape Morphing Hydrogel Structures. Macromol. Mater. Eng. 2017, 302, 1600212. [Google Scholar] [CrossRef]
- Ratri, M.C.; Suh, J.; Ryu, J.; Chung, B.G.; Shin, K. Formulation of three-dimensional, photo-responsive printing ink: Gold nanorod-hydrogel nanocomposites and their four-dimensional structures that respond quickly to stimuli. J. Appl. Polym. Sci. 2023, 140, e53799. [Google Scholar] [CrossRef]
- O’Bryan, C.S.; Bhattacharjee, T.; Niemi, S.R.; Balachandar, S.; Baldwin, N.; Ellison, S.T.; Taylor, C.R.; Sawyer, W.G.; Angelini, T.E. Three-Dimensional Printing with Sacrificial Materials for Soft Matter Manufacturing. MRS Bull. 2017, 42, 571–577. [Google Scholar] [CrossRef]
- Prendergast, M.E.; Solorzano, R.D.; Cabrera, D. Bioinks for Biofabrication: Current State and Future Perspectives. J. 3D Print. Med. 2016, 1, 49–62. [Google Scholar] [CrossRef]
- Miller, J.S.; Stevens, K.R.; Yang, M.T.; Baker, B.M.; Nguyen, D.-H.T.; Cohen, D.M.; Toro, E.; Chen, A.A.; Galie, P.A.; Yu, X.; et al. Rapid Casting of Patterned Vascular Networks for Perfusable Engineered Three-Dimensional Tissues. Nat. Mater. 2012, 11, 768–774. [Google Scholar] [CrossRef]
- Patrício, S.G.; Sousa, L.R.; Correia, T.R.; Gaspar, V.M.; Pires, L.S.; Luís, J.L.; Oliveira, J.M.; Mano, J.F. Freeform 3D Printing Using a Continuous Viscoelastic Supporting Matrix. Biofabrication 2020, 12, 035017. [Google Scholar] [CrossRef]
- Hinton, T.J.; Jallerat, Q.; Palchesko, R.N.; Park, J.H.; Grodzicki, M.S.; Shue, H.-J.; Ramadan, M.H.; Hudson, A.R.; Feinberg, A.W. Three-Dimensional Printing of Complex Biological Structures by Freeform Reversible Embedding of Suspended Hydrogels. Sci. Adv. 2015, 1, e1500758. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Hudson, A.R.; Shiwarski, D.J.; Tashman, J.W.; Hinton, T.J.; Yerneni, S.; Bliley, J.M.; Campbell, P.G.; Feinberg, A.W. 3D Bioprinting of Collagen to Rebuild Components of the Human Heart. Science 2019, 365, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Riffe, M.B.; Davidson, M.D.; Seymour, G.; Dhand, A.P.; Cooke, M.E.; Zlotnick, H.M.; McLeod, R.R.; Burdick, J.A. Multi-Material Volumetric Additive Manufacturing of Hydrogels using Gelatin as a Sacrificial Network and 3D Suspension Bath. Adv. Mater. 2024, 36, 2309026. [Google Scholar] [CrossRef] [PubMed]
- Hua, M.; Wu, D.; Wu, S.; Ma, Y.; Alsaid, Y.; He, X. 4D Printable Tough and Thermoresponsive Hydrogels. ACS Appl. Mater. Interfaces 2021, 13, 12689–12697. [Google Scholar] [CrossRef] [PubMed]
- Podstawczyk, D.; Nizioł, M.; Szymczyk-Ziółkowska, P.; Fiedot-Toboła, M. Development of Thermoinks for 4D Direct Printing of Temperature-Induced Self-Rolling Hydrogel Actuators. Adv. Funct. Mater. 2021, 31, 2009664. [Google Scholar] [CrossRef]
- Arslan, H.; Nojoomi, A.; Jeon, J.; Yum, K. 3D Printing of Anisotropic Hydrogels with Bioinspired Motion. Adv. Sci. 2019, 6, 1800703. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.E.; Murphy, C.J. The Quest for Shape Control: A History of Gold Nanorod Synthesis. Chem. Mater. 2013, 25, 1250–1261. [Google Scholar] [CrossRef]
- Dickerson, E.B.; Dreaden, E.C.; Huang, X.; El-Sayed, I.H.; Chu, H.; Pushpanketh, S.; McDonald, J.F.; El-Sayed, M.A. Gold Nanorod Assisted Near-Infrared Plasmonic Photothermal Therapy (PPTT) of Squamous Cell Carcinoma in Mice. Cancer Lett. 2008, 269, 57–66. [Google Scholar] [CrossRef]
- Yavuz, M.S.; Cheng, Y.; Chen, J.; Cobley, C.M.; Zhang, Q.; Rycenga, M.; Xie, J.; Kim, C.; Song, K.H.; Schwartz, A.G.; et al. Gold Nanocages Covered by Smart Polymers for Controlled Release with Near-Infrared Light. Nat. Mater. 2009, 8, 935–939. [Google Scholar] [CrossRef]
- Conde, J.; Oliva, N.; Zhang, Y.; Artzi, N. Local Triple-Combination Therapy Results in Tumour Regression and Prevents Recurrence in a Colon Cancer Model. Nat. Mater. 2016, 15, 1128–1138. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Ben-Shahar, Y.; Scotognella, F.; Waiskopf, N.; Kriegel, I.; Dal Conte, S.; Cerullo, G.; Banin, U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS–Au Nanorods. Small 2015, 11, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shahar, Y.; Scotognella, F.; Kriegel, I.; Moretti, L.; Cerullo, G.; Rabani, E.; Banin, U. Optimal Metal Domain Size for Photocatalysis with Hybrid Semiconductor-Metal Nanorods. Nat. Commun. 2016, 7, 10413. [Google Scholar] [CrossRef] [PubMed]
- Lapotko, D.; Lukianova-Hleb, E.; Zhdanok, S.; Rostro, B.; Simonette, R.; Hafner, J.; Konopleva, M.; Andreeff, M.; Conjusteau, A.; Oraevsky, A. Photothermolysis by Laser-Induced Microbubbles Generated Around Gold Nanorod Clusters Selectively Formed in Leukemia Cells. In Photons Plus Ultrasound: Imaging and Sensing 2008, Proceedings of the Ninth Conference on Biomedical Thermoacoustics, Optoacoustics, and Acousto-Optics, San Jose, CA, USA, 20–23 January 2008; SPIE: Bellingham, WA, USA, 2008; Volume 6856, p. 68560K. [Google Scholar]
- Neumann, O.; Urban, A.S.; Day, J.; Lal, S.; Nordlander, P.; Halas, N.J. Solar Vapor Generation Enabled by Nanoparticles. ACS Nano 2013, 7, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhen, Y.-R.; Neumann, O.; Polman, A.; García de Abajo, F.J.; Nordlander, P.; Halas, N.J. Evolution of Light-Induced Vapor Generation at a Liquid-Immersed Metallic Nanoparticle. Nano Lett. 2013, 13, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Neumann, O.; Feronti, C.; Neumann, A.D.; Dong, A.; Schell, K.; Lu, B.; Kim, E.; Quinn, M.; Thompson, S.; Grady, N.; et al. Compact Solar Autoclave Based on Steam Generation using Broadband Light-Harvesting Nanoparticles. Proc. Natl. Acad. Sci. USA 2013, 110, 11677. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, H.; Fei, G.; Yu, B.; Tong, X.; Xia, H.; Zhao, Y. Liquid-Crystalline Dynamic Networks Doped with Gold Nanorods Showing Enhanced Photocontrol of Actuation. Adv. Mater. 2018, 30, 1706597. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.R.; Tracy, J.B. Sequential Actuation of Shape-Memory Polymers through Wavelength-Selective Photothermal Heating of Gold Nanospheres and Nanorods. ACS Appl. Nano Mater. 2018, 1, 3063–3067. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Nie, X.; Wen, T.; Ji, Y.; Wu, X.; Zhao, Y.; Chen, C. Near Infrared Laser-Induced Targeted Cancer Therapy Using Thermoresponsive Polymer Encapsulated Gold Nanorods. J. Am. Chem. Soc. 2014, 136, 7317–7326. [Google Scholar] [CrossRef]
- Qian, X.; Zhao, Y.; Alsaid, Y.; Wang, X.; Hua, M.; Galy, T.; Gopalakrishna, H.; Yang, Y.; Cui, J.; Liu, N.; et al. Artificial Phototropism for Omnidirectional Tracking and Harvesting of Light. Nat. Nanotechnol. 2019, 14, 1048–1055. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Li, L.; Liu, Y.; Wang, D.; Xu, L.; Bao, J.; Zhang, A. Fabrication of Photothermally Responsive Nanocomposite Hydrogel through 3D Printing. Macromol. Mater. Eng. 2020, 305, 1900718. [Google Scholar] [CrossRef]
- Kozek, K.A.; Kozek, K.M.; Wu, W.-C.; Mishra, S.R.; Tracy, J.B. Large-Scale Synthesis of Gold Nanorods through Continuous Secondary Growth. Chem. Mater. 2013, 25, 4537–4544. [Google Scholar] [CrossRef] [PubMed]
- Tebbe, M.; Kuttner, C.; Männel, M.; Fery, A.; Chanana, M. Colloidally Stable and Surfactant-Free Protein-Coated Gold Nanorods in Biological Media. ACS Appl. Mater. Interfaces 2015, 7, 5984–5991. [Google Scholar] [CrossRef] [PubMed]
- Neuendorf, T.A.; Weigel, N.; Vigogne, M.; Thiele, J. Additive Soft Matter Design by UV-Induced Polymer Hydrogel Inter-Crosslinking. Gels 2022, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Hauck, N.; Beck, T.; Cojoc, G.; Schlüßler, R.; Ahmed, S.; Raguzin, I.; Mayer, M.; Schubert, J.; Müller, P.; Guck, J.; et al. PNIPAAm Microgels with Defined Network Architecture as Temperature Sensors in Optical Stretchers. Mater. Adv. 2022, 3, 6179–6190. [Google Scholar] [CrossRef] [PubMed]
- Bello, J.; Vinograd, J.R. Selective Acetylation of the Hydroxyl Groups in Gelatin. J. Am. Chem. Soc. 1956, 78, 1369–1372. [Google Scholar] [CrossRef]
- Claaßen, C.; Claaßen, M.H.; Truffault, V.; Sewald, L.; Tovar, G.E.M.; Borchers, K.; Southan, A. Quantification of Substitution of Gelatin Methacryloyl: Best Practice and Current Pitfalls. Biomacromolecules 2018, 19, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Kuckling, D.; Adler, H.-J.P.; Ling, L.; Habicher, W.D.; Arndt, K.-F. Temperature Sensitive Polymers Based on 2-(Dimethyl Maleinimido)-N-Ethyl-Acrylamide: Copolymers with N-Isopropylacrylamide. Polym. Bull. 2000, 44, 269–276. [Google Scholar] [CrossRef]
- Seiffert, S.; Weitz, D.A. Controlled Fabrication of Polymer Microgels by Polymer-Analogous Gelation in Droplet Microfluidics. Soft Matter 2010, 6, 3184–3190. [Google Scholar] [CrossRef]
- Erbil, C.; Yıldız, Y.; Uyanık, N. Effects of Synthesis-Solvent Composition and Initiator Concentration on the Swelling Behaviour of Poly(N-isopropylacrylamide) P(NIPAAM), Poly(NIPAAM-co-Dimethyl Itaconate), and Poly(NIPAAM-co Itaconic Acid) Gels. Polym. Int. 2000, 49, 795–800. [Google Scholar] [CrossRef]
- Kuckling, D.; Harmon, M.E.; Frank, C.W. Photo-Cross-Linkable PNIPAAm Copolymers. 1. Synthesis and Characterization of Constrained Temperature-Responsive Hydrogel Layers. Macromolecules 2002, 35, 6377–6383. [Google Scholar] [CrossRef]
- Kuckling, D.; Hoffmann, J.; Plötner, M.; Ferse, D.; Kretschmer, K.; Adler, H.-J.P.; Arndt, K.-F.; Reichelt, R. Photo Cross-Linkable Poly(N-Isopropylacrylamide) Copolymers III: Micro-Fabricated Temperature Responsive Hydrogels. Polymer 2003, 44, 4455–4462. [Google Scholar] [CrossRef]
- Rizvi, M.H.; Wang, R.; Schubert, J.; Crumpler, W.D.; Rossner, C.; Oldenburg, A.L.; Fery, A.; Tracy, J.B. Magnetic Alignment for Plasmonic Control of Gold Nanorods Coated with Iron Oxide Nanoparticles. Adv. Mater. 2022, 34, 2203366. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, S.; Oppermann, W.; Saalwaechter, K. Hydrogel Formation by Photocrosslinking of Dimethylmaleimide Functionalized Polyacrylamide. Polymer 2007, 48, 5599–5611. [Google Scholar] [CrossRef]
- Bello, J.; Riese, H.C.A.; Vinograd, J.R. Mechanism of Gelation of Gelatin. Influence of Certain Electrolytes on the Melting Points of Gels of Gelatin and Chemically Modified Gelatins. J. Phys. Chem. 1956, 60, 1299–1306. [Google Scholar] [CrossRef]
- Strek, T.; Jopek, H.; Wojciechowski, K.W. The Influence of Large Deformations on Mechanical Properties of Sinusoidal Ligament Structures. Smart Mater. Struct. 2016, 25, 054002. [Google Scholar] [CrossRef]
- Lakes, R. Foam Structures with a Negative Poisson’s Ratio. Science 1987, 235, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Das, R.; Tran, P.; Ngo, T.D.; Xie, Y.M. Auxetic Metamaterials and Structures: A Review. Smart Mater. Struct. 2018, 27, 023001. [Google Scholar] [CrossRef]
- Chansoria, P.; Blackwell, J.; Etter, E.L.; Bonacquisti, E.E.; Jasiewicz, N.; Neal, T.; Kamal, S.A.; Hoque, J.; Varghese, S.; Egan, T.; et al. Rationally Designed Anisotropic and Auxetic Hydrogel Patches for Adaptation to Dynamic Organs. Adv. Funct. Mater. 2022, 32, 2207590. [Google Scholar] [CrossRef]
- Ma, Y.; Sikdar, D.; Fedosyuk, A.; Velleman, L.; Zhao, M.; Tang, L.; Kornyshev, A.A.; Edel, J.B. Auxetic Thermoresponsive Nanoplasmonic Optical Switch. ACS Appl. Mater. Interfaces 2019, 11, 22754–22760. [Google Scholar] [CrossRef]
- Cheng, Q.; Liu, Y.; Lyu, J.; Lu, Q.; Zhang, X.; Song, W. 3D Printing-Directed Auxetic Kevlar Aerogel Architectures with Multiple Functionalization Options. J. Mater. Chem. A 2020, 8, 14243–14253. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Wang, K.; Ho, C.-C.; Kao, C.-T.; Ng, H.Y.; Shie, M.-Y. Cyclic Tensile Stimulation Enrichment of Schwann Cell-Laden Auxetic Hydrogel Scaffolds Towards Peripheral Nerve Tissue Engineering. Mater. Des. 2020, 195, 108982. [Google Scholar] [CrossRef]
- Wei, Y.-L.; Yang, Q.-S.; Ma, L.-H.; Tao, R.; Shang, J.-J. Design and Analysis of 2D/3D Negative Hydration Expansion Metamaterial Driven by Hydrogel. Mater. Des. 2020, 196, 109084. [Google Scholar] [CrossRef]
- Pruksawan, S.; Chee, H.L.; Wang, Z.; Luo, P.; Chong, Y.T.; Thitsartarn, W.; Wang, F. Toughened Hydrogels for 3D Printing of Soft Auxetic Structures. Chem. Asian J. 2022, 17, e202200677. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, J.-M. Microfluidic Technology for Single-Cell Capture and Isolation. In Microfluidics for Single-Cell Analysis; Lin, J.-M., Ed.; Springer: Singapore, 2019; pp. 27–51. [Google Scholar]
- Niazi, M.; Alizadeh, E.; Zarebkohan, A.; Seidi, K.; Ayoubi-Joshaghani, M.H.; Azizi, M.; Dadashi, H.; Mahmudi, H.; Javaheri, T.; Jaymand, M.; et al. Advanced Bioresponsive Multitasking Hydrogels in the New Era of Biomedicine. Adv. Funct. Mater. 2021, 31, 2104123. [Google Scholar] [CrossRef]
- Wu, Y.; Yakov, S.; Fu, A.; Yossifon, G. A Magnetically and Electrically Powered Hybrid Micromotor in Conductive Solutions: Synergistic Propulsion Effects and Label-Free Cargo Transport and Sensing. Adv. Sci. 2023, 10, 2204931. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghelardini, M.M.; Geisler, M.; Weigel, N.; Hankwitz, J.P.; Hauck, N.; Schubert, J.; Fery, A.; Tracy, J.B.; Thiele, J. 3D-Printed Hydrogels as Photothermal Actuators. Polymers 2024, 16, 2032. https://doi.org/10.3390/polym16142032
Ghelardini MM, Geisler M, Weigel N, Hankwitz JP, Hauck N, Schubert J, Fery A, Tracy JB, Thiele J. 3D-Printed Hydrogels as Photothermal Actuators. Polymers. 2024; 16(14):2032. https://doi.org/10.3390/polym16142032
Chicago/Turabian StyleGhelardini, Melanie M., Martin Geisler, Niclas Weigel, Jameson P. Hankwitz, Nicolas Hauck, Jonas Schubert, Andreas Fery, Joseph B. Tracy, and Julian Thiele. 2024. "3D-Printed Hydrogels as Photothermal Actuators" Polymers 16, no. 14: 2032. https://doi.org/10.3390/polym16142032
APA StyleGhelardini, M. M., Geisler, M., Weigel, N., Hankwitz, J. P., Hauck, N., Schubert, J., Fery, A., Tracy, J. B., & Thiele, J. (2024). 3D-Printed Hydrogels as Photothermal Actuators. Polymers, 16(14), 2032. https://doi.org/10.3390/polym16142032