


Optimization of Biologically Inspired Electrospun Scaffold for Effective Use in Bone Regenerative Applications
 and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of Hydroxyapatite
2.2. Electrospinning of Polymer Scaffolds
2.3. Characterization Techniques
2.4. Bioactivity Study Using SBF Immersion
2.5. Hemocompatibility Analysis
2.6. Alkaline Phosphatase Assay
2.7. Biocompatibility Assessment
3. Results
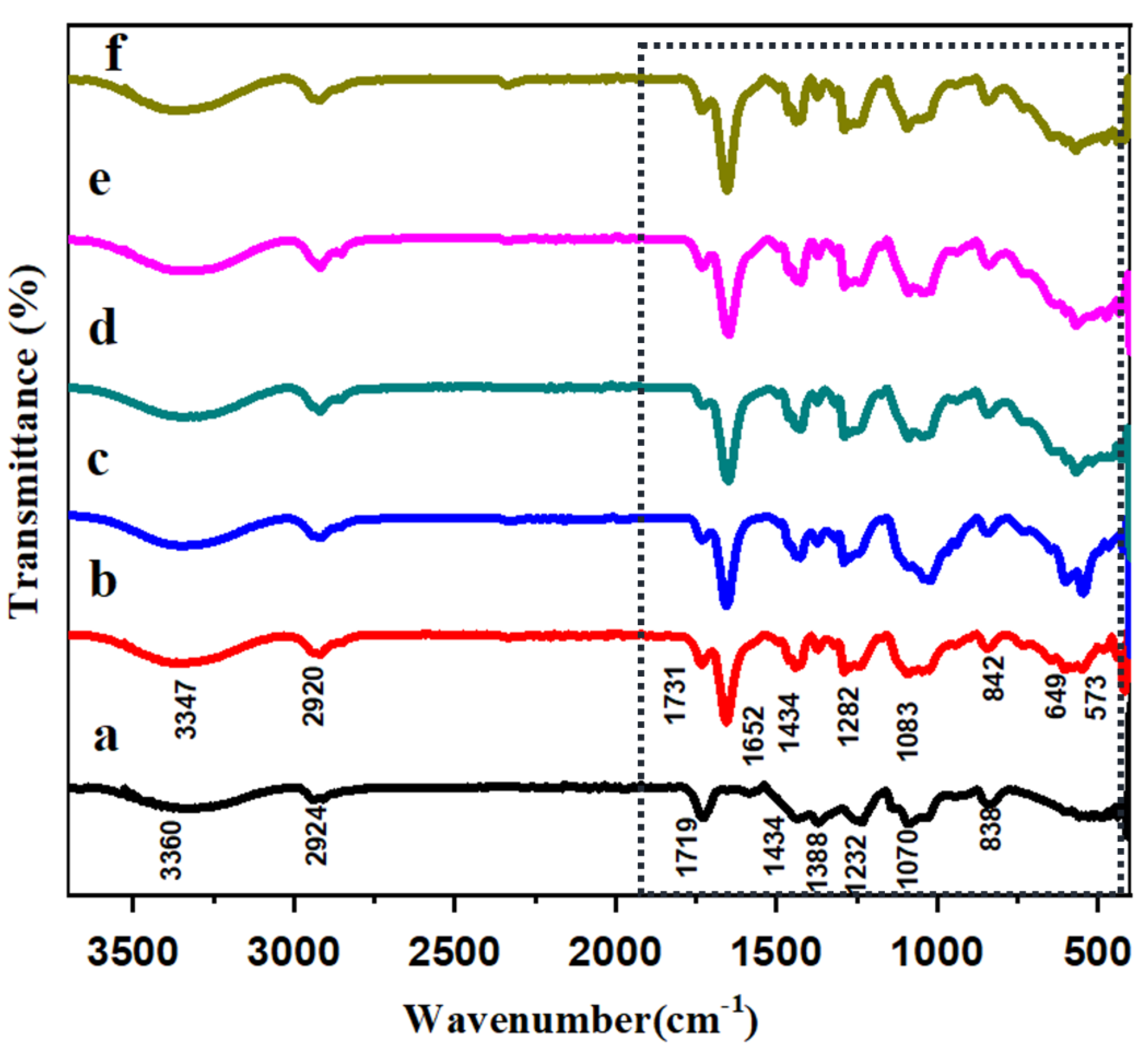
3.1. FT-IR Analysis of HAP-PVA-PVP Nanofibers
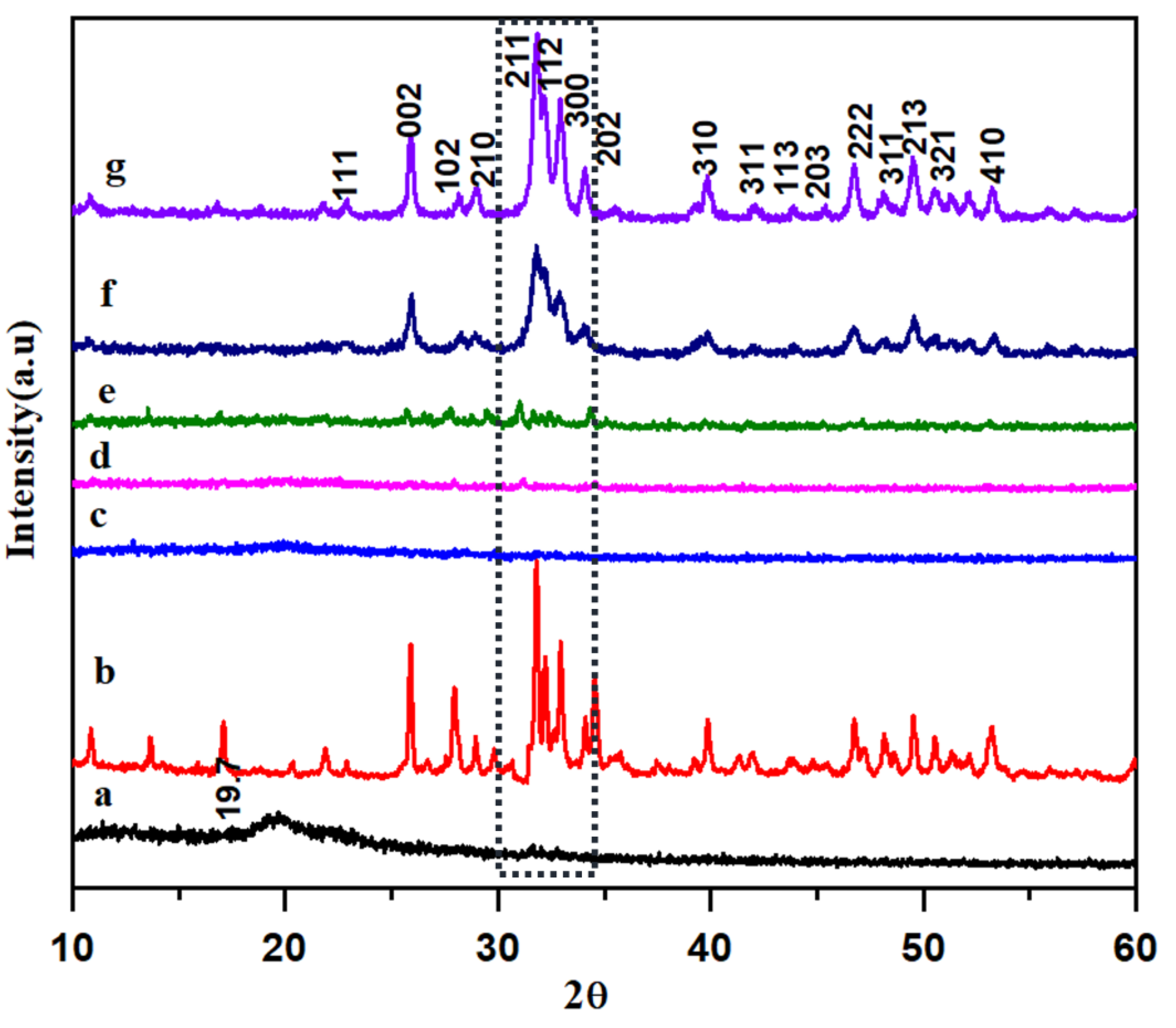
3.2. XRD Analysis of HAP-PVA-PVP Nanofibers
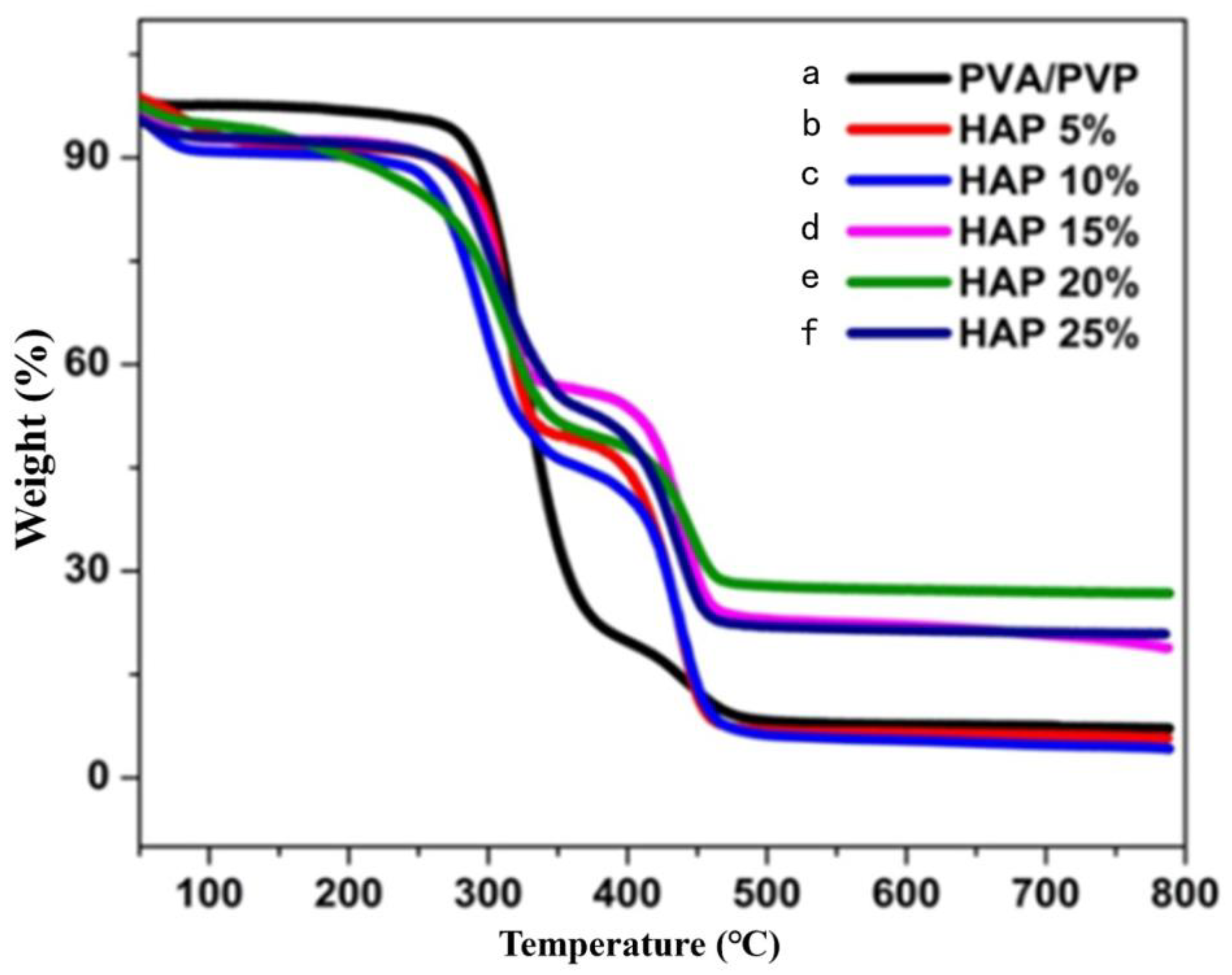
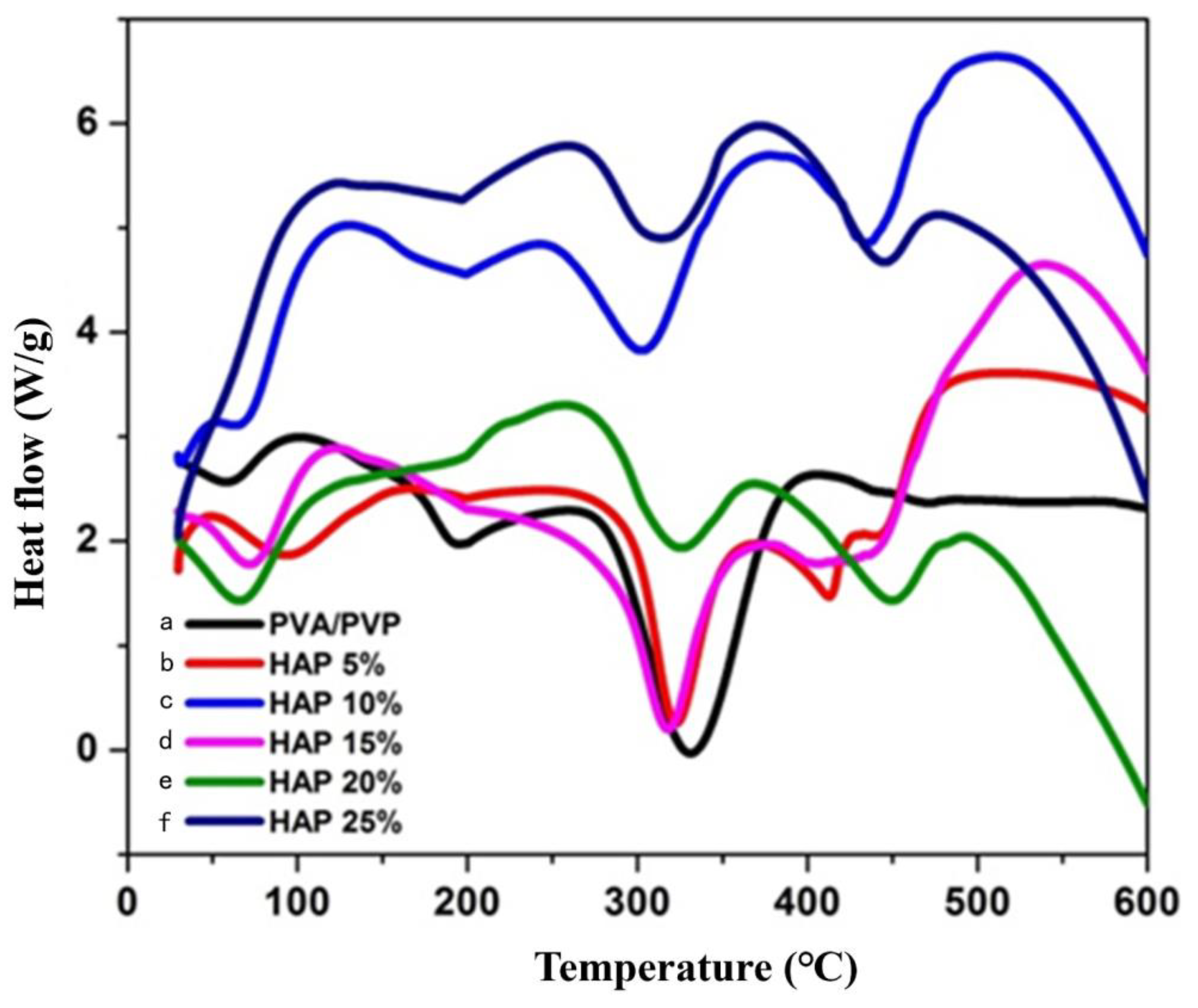
3.3. Thermal Stability of Composite Nanofibers
3.4. Morphology of Nanofibers Using SEM Analysis
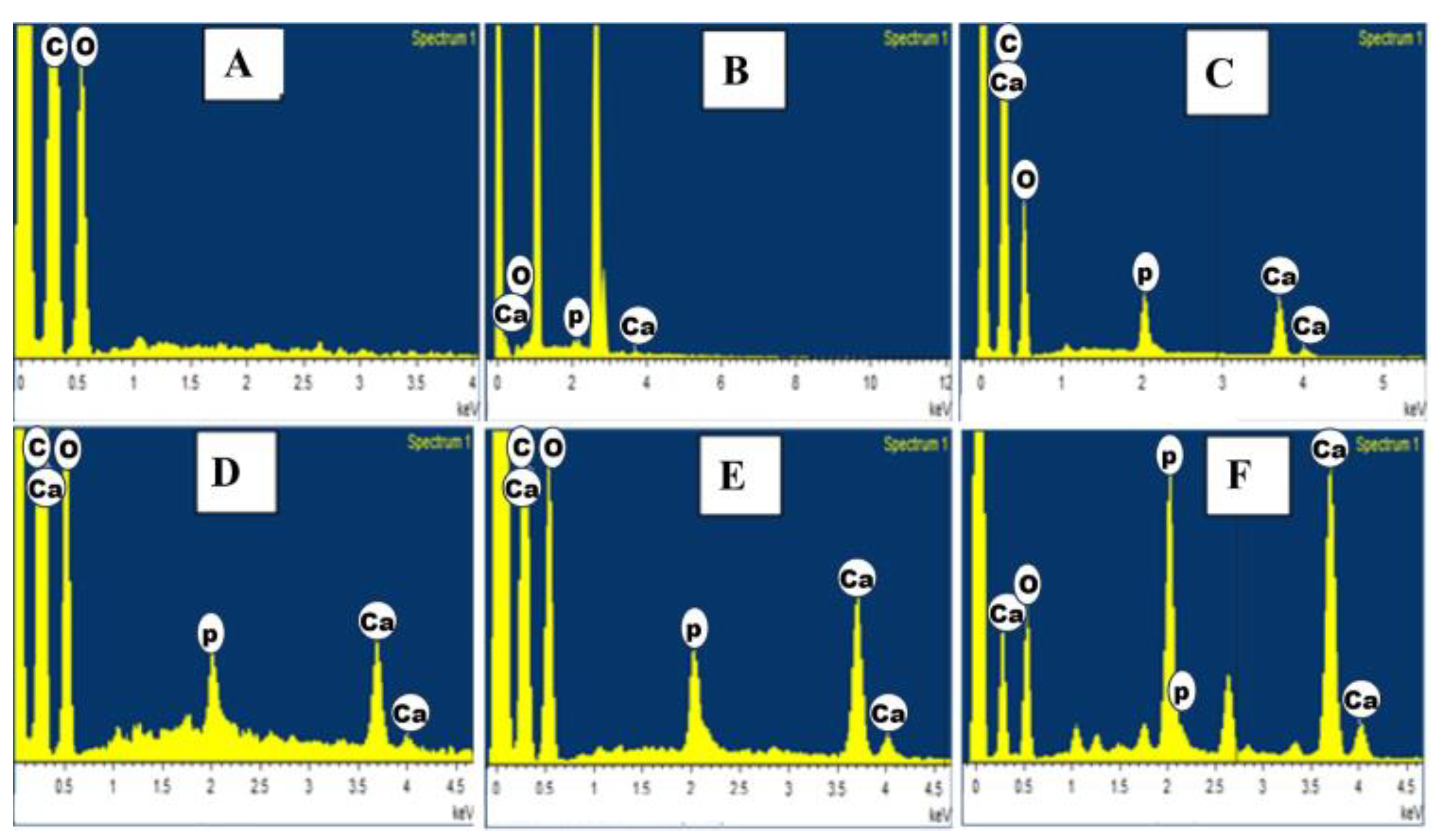
3.5. EDAX Analysis of HAP-PVA-PVP Nanofibers
3.6. Morphology of Nanofibers Using TEM Analysis
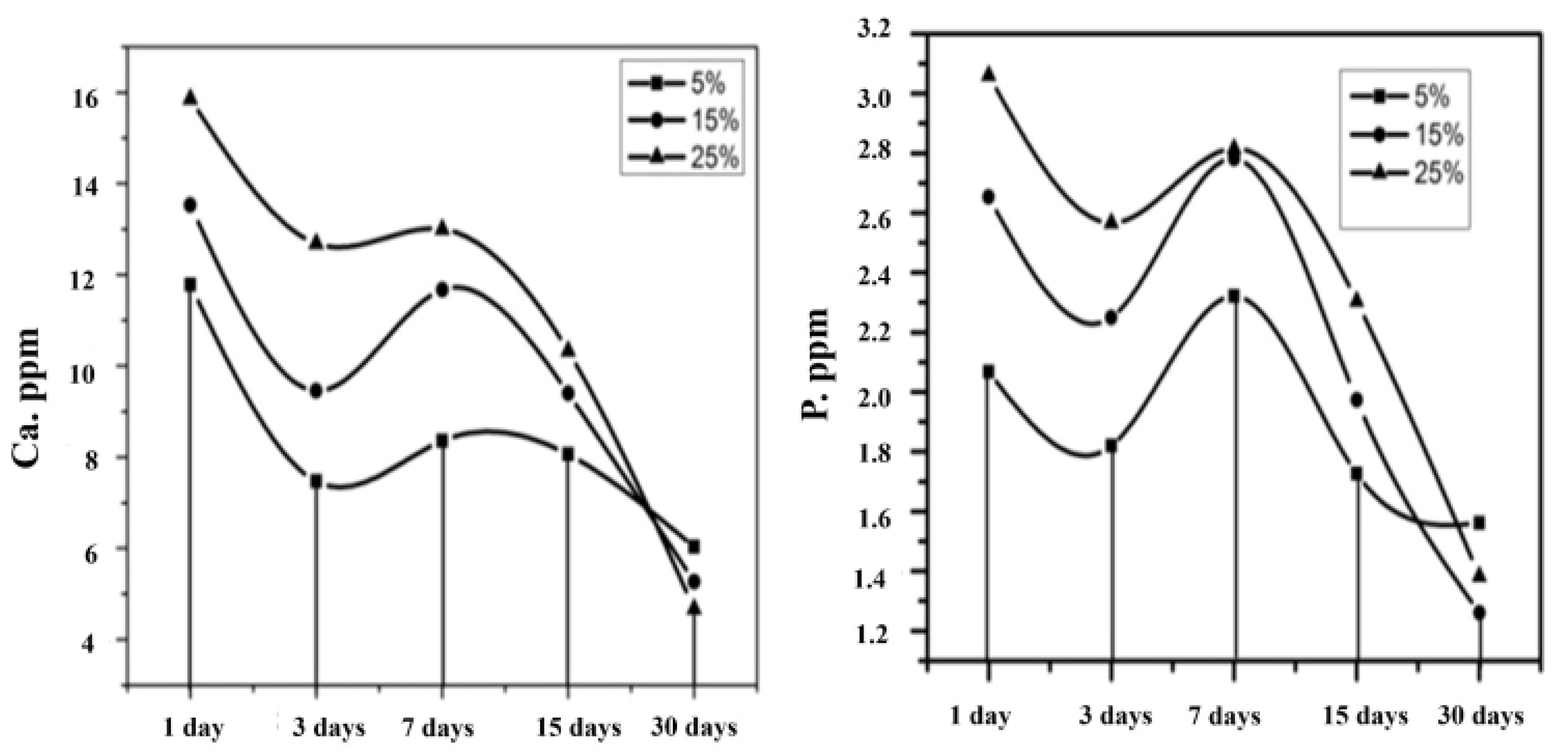
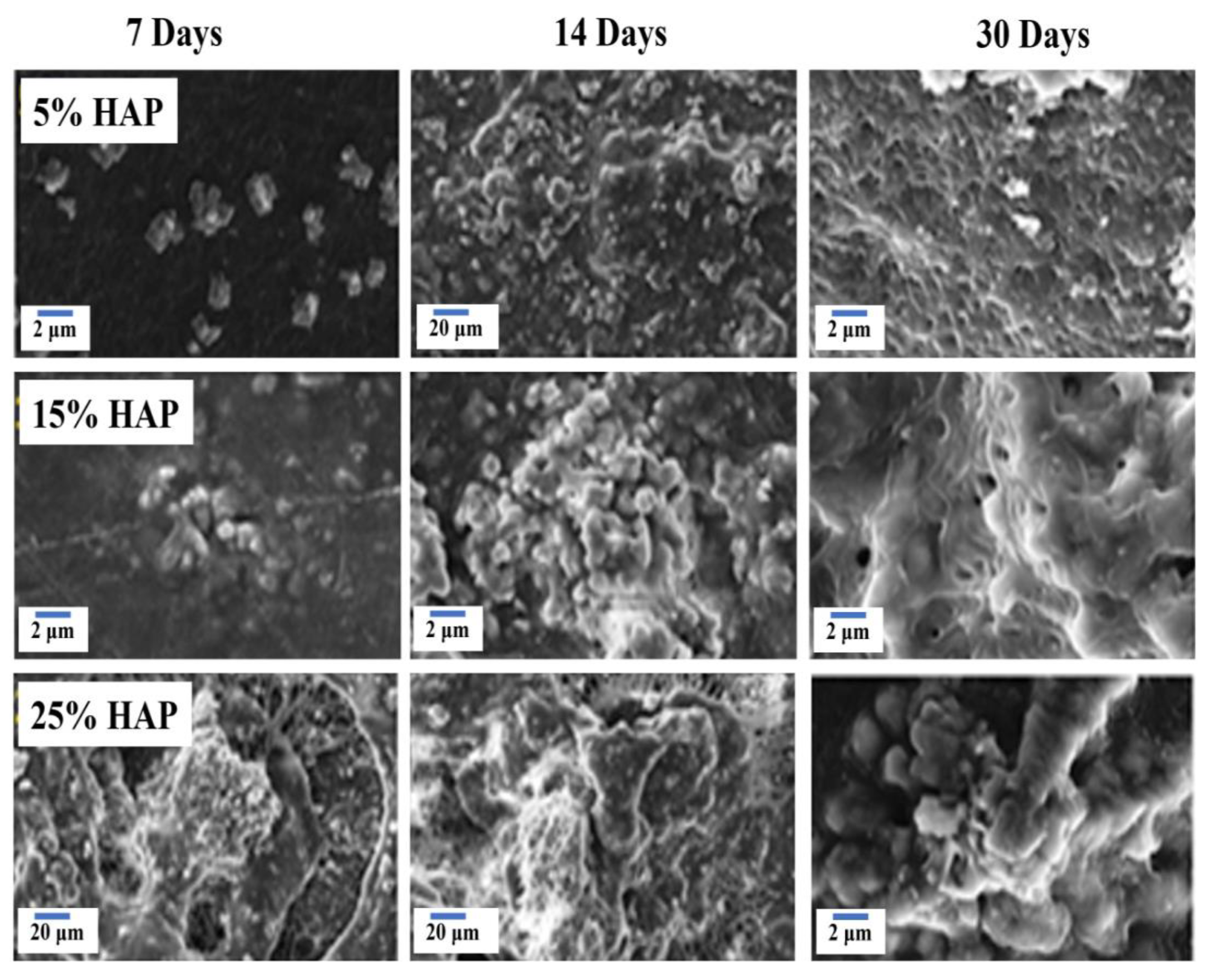
3.7. Bioactivity Analysis by SBF Immersion
3.8. X-ray Photoelectron Spectroscopy (XPS)
3.9. Hemolytic Assay
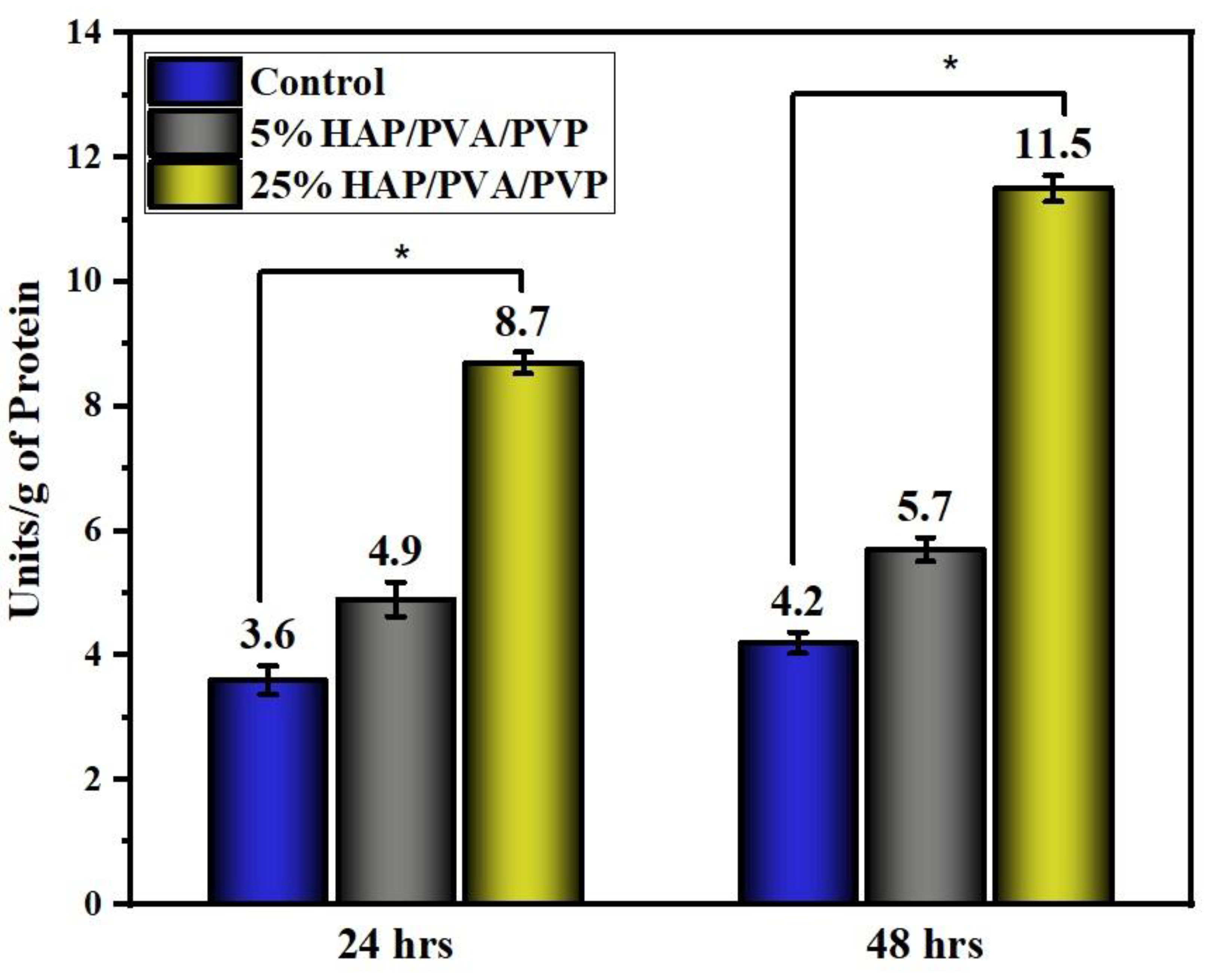
3.10. Alkaline Phosphate Activity (ALP)
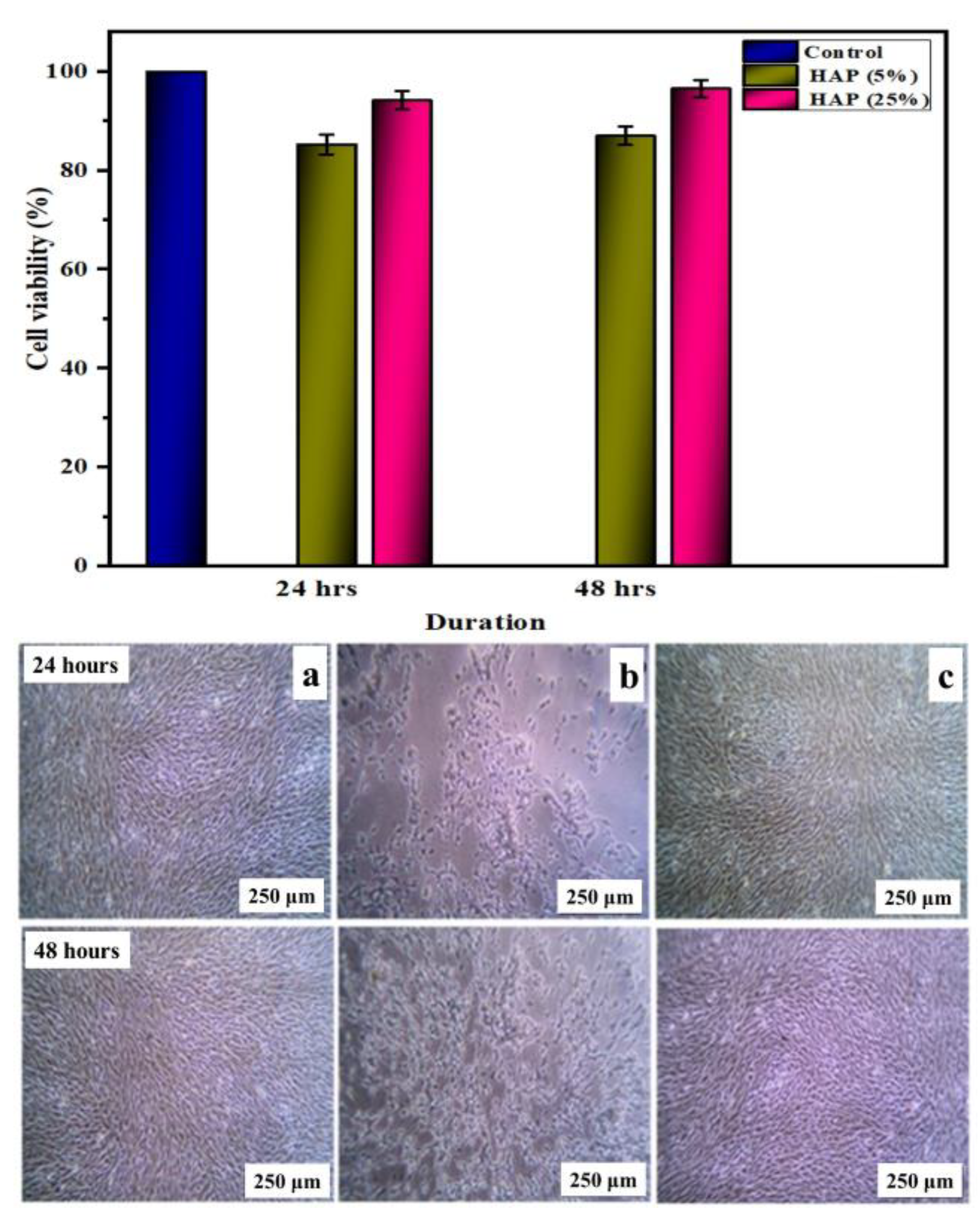
3.11. Biocompatibility Assessment in Osteoblast Cell Lines
4. Conclusions
- ➢
- The phase purity was confirmed by XRD diffraction, and a triplet peak corresponding to HAP was found to be well pronounced, with different concentrations of HAP. When the concentration of HAP increased, the fiber diameter was found to be decreasing, and the pore size of the nanofiber increased from 6 μm for the 5% HAP matrix to 12 μm for the 25% HAP matrix; these findings show an enrichment in the bioactivity of the fabricated nanofibrous scaffold when treated with SBF.
- ➢
- By optimizing the parameters of the electrospinning of nanofibrous scaffolds, successful fabrication was obtained that exhibited a porous structure with the desired pore size and cumulative pore volume, which can help to increase the bioactivity of the fabricated matrix, and the results were further analyzed using SEM-EDAX and ICP-OES analysis.
- ➢
- The XPS analysis confirmed the formation of apatite by the dissolution of the polymer during SBF immersion. The absence of “N” binding energy with the reduction in the “C” binding energy and the existence of P-O-P, P-O, and P-OH binding energy confirmed the formation of an apatite layer over the scaffold.
- ➢
- In vitro hemocompatibility testing showed a lower percentage of hemolytic activity and, hence, proved that the matrices were more hemocompatible. In addition, both the alkaline phosphatase activity and MTT assay of the nanofibrous matrix on MG-63 cell lines exhibited good cytocompatibility, and from microscopic images, we observed that the cells adhered well, with an elongated spindle morphology. Hence, from the observed results, the fabricated multicomponent HAP-PVA-PVP nanofibrous scaffold could provide a potential matrix for bone-regenerative applications.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.P.; Chang, Y.S. Preparation and characterization of composite nanofibers of polycaprolactone and nanohydroxyapatite for osteogenic differentiation of mesenchymal stem cells. Colloids Surf. B Biointerfaces 2011, 86, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Farokhi, M.; Mottaghitalab, F.; Shokrgozar, M.A.; Ou, K.L.; Mao, C.; Hosseinkhani, H. Importance of dual delivery systems for bone tissue engineering. J. Control. Release 2016, 225, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.; Kanjwal, M.; Macossay, J.; Barakat, N.; Kim, H.Y. A simple approach for synthesis, characterization and bioactivity of bovine bones to fabricate the polyurethane nanofiber containing hydroxyapatite nanoparticles. Express Polym. Lett. 2012, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Stella, M.; Vijayalakshmi, U. Influence of Polymer Based Scaffolds with Genipin Cross Linker for the Effective (Pore Size) Usage in Biomedical Applications. Trends Biomater. Artif. Organs 2018, 32, 83–92. [Google Scholar]
- Priyadarshini, B.; Rama, M.; Chetan; Vijayalakshmi, U. Bioactive coating as a surface modification technique for biocompatible metallic implants: A review. J. Asian Ceram. Soc. 2019, 7, 397–406. [Google Scholar] [CrossRef]
- Tancred, D.C.; Mccormack, B.O.; Carr, J. A quantitative study of the sintering and mechanical properties of hydroxyapatite/phosphate glass composites. Biomaterials 1998, 19, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Hibbeler, R. Force-System Resultants and Equilibrium. In The Engineering Handbook, Electrical Engineering Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Vijayalakshmi, U.; Rajeswari, S. Influence of process parameters on the sol–gel synthesis of nano hydroxyapatite using various phosphorus precursors. J. Sol-Gel Sci. Technol. 2012, 63, 45–55. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, L.; Zheng, H.; Wang, Z. Hydroxyapatite nanocrystals for biomedical applications. J. Phys. Chem. C 2010, 114, 18352–18357. [Google Scholar] [CrossRef]
- Chen, F.; Tang, Q.L.; Zhu, Y.J. Hydroxyapatite nanorods/poly(vinyl pyrolidone) composite nanofibers, arrays and three-dimensional fabrics: Electrospun preparation and transformation to hydroxyapatite nanostructures. Acta Biomater. 2010, 6, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Uma Maheshwari, S.; Samuel, V.K.; Nagiah, N. Fabrication and evaluation of (PVA/HAp/PCL) bilayer composites as potential scaffolds for bone tissue regeneration application. Ceram. Int. 2014, 40, 8469–8477. [Google Scholar] [CrossRef]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric scaffolds in tissue engineering application: A review. Int. J. Polym. Sci. 2011, 2011, 290602. [Google Scholar] [CrossRef]
- Chen, L.J.; Wang, M. Production and evaluation of biodegradable composites based on PHB-PHV copolymer. Biomaterials 2002, 23, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.X. Scaffolds for tissue fabrication. Mater. Today 2004, 7, 30–40. [Google Scholar] [CrossRef]
- Hou, Q.; Grijpma, D.W.; Feijen, J. Preparation of Porous Poly(e-caprolactone) Structures. Macromol. Rapid Commun. 2002, 23, 247–252. [Google Scholar] [CrossRef]
- Mary Stella, S.; Vijayalakshmi, U. Influence of chemically modified Luffa on the preparation of nanofiber and its biological evaluation for biomedical applications. J. Biomed. Mater. Res. Part A 2019, 107, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Chahal, S.; Hussain, F.S.J.; Kumar, A.; Rasad, M.S.B.A.; Yusoff, M.M. Fabrication, characterization and in vitro biocompatibility of electrospun hydroxyethyl cellulose/poly (vinyl) alcohol nanofibrous composite biomaterial for bone tissue engineering. Chem. Eng. Sci. 2016, 144, 17–29. [Google Scholar] [CrossRef]
- Chaudhuri, B.; Mondal, B.; Ray, S.K.; Sarkar, S.C. A novel biocompatible conducting polyvinyl alcohol (PVA)-polyvinylpyrrolidone (PVP)-hydroxyapatite (HAP) composite scaffolds for probable biological application. Colloids Surf. B Biointerfaces 2016, 143, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Anandan, D.; Mary Stella, S.; Arunai Nambiraj, N.; Vijayalakshmi, U.; Amit Kumar, J. Development of mechanically compliant 3D composite scaffolds for bone tissue engineering applications. J. Biomed. Mater. Res. Part A 2018, 106, 3267–3274. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Ouriemchi, E.M.; Vergnaud, J.M. Processes of drug transfer with three different polymeric systems with transdermal drug delivery. Comput. Theor. Polym. Sci. 2000, 10, 391–401. [Google Scholar] [CrossRef]
- Mukasheva, F.; Moazzam, M.; Yernaimanova, B.; Shehzad, A.; Zhanbassynova, A.; Berillo, D.; Akilbekova, D. Design and characterization of 3D printed pore gradient hydrogel scaffold for bone tissue engineering. Bioprinting 2024, 39, 00341. [Google Scholar] [CrossRef]
- Ratner, B.; Hoffman, A.S.; Schosen, F.J.; Lemons, J.E. Biomaterials Science—An introduction to Materials in Medicine. Academic Press: New York, NY, USA, 2004; pp. 1–12. [Google Scholar]
- Zhou, Z.; Feng, W.; Moghadas, B.K.; Baneshi, N.; Noshadi, B.; Baghaei, S.; Dehkordi, D.A. Review of recent advances in bone scaffold fabrication methods for tissue engineering for treating bone diseases and injuries. Tissue Cell 2024, 88, 102390. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W. Scaffold design and fabrication technologies for engineering tissues—State of the art and future perspectives. J. Biomater. Sci. Polym. Ed. 2001, 12, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Wendorff, J.H.; Greiner, A. Use of electrospinning technique for biomedical applications. Polymer 2008, 49, 5603–5621. [Google Scholar] [CrossRef]
- Rama, M.; Simona, C.; Vijayalakshmi, U. Fabrication and in vitro drug delivery evaluation of cephalexin monohydrate-loaded PLA: PVA/HAP: TiO2 fibrous scaffolds for bone regeneration. ACS Omega 2023, 8, 5017–5032. [Google Scholar]
- Ito, Y.; Hasuda, H.; Kamitakahara, M. A composite of hydroxyapatite with electrospun biodegradable nanofibers as a tissue engineering material. J. Biosci. Bioeng. 2005, 100, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Demirtaş, T.T.; Kaynak, G.; Gumuşderelioʇlu, M. Bone-like hydroxyapatite precipitated from 10×SBF-like solution by microwave irradiation. Mater. Sci. Eng. C 2015, 49, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W3. J. Biomed. Mater Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Rama, M.; Vijayalakshmi, U. Influence of silk fibroin on the preparation of nanofibrous scaffolds for the effective use in osteoregenerative applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102182. [Google Scholar] [CrossRef]
- Stella, S.M.; Sridhar, T.M.; Ramprasath, R.; Gimbun, J.; Vijayalakshmi, U. Physio-Chemical and Biological Characterization of Novel HPC (Hydroxypropylcellulose): HAP (Hydroxyapatite): PLA (Poly Lactic Acid) Electrospun Nanofibers as Implantable Material for Bone Regenerative Application. Polymers 2022, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Rama, M.; Sundara Ganeasan, M.; Sridhar, T.M.; Arunai Nambi Raj, N.; Vijayalakshmi, U. Comparative Analysis of Electrospun Silk Fibroin/Chitosan Sandwich-Structured Scaffolds for Osteo Regeneration: Evaluating Mechanical Properties, Biological Performance, and Drug Release. ACS Omega 2024, 26, 28072. [Google Scholar]
- Wu, S.; He, M.; Yang, M.; Zhang, B.; Wang, F.; Li, Q. Near-infrared spectroscopy study of serpentine minerals and assignment of the OH group. Crystals 2021, 11, 1130. [Google Scholar] [CrossRef]
- Serret, A.; Cabanas, M.V.; Vallet-Regi, M. Stabilization of calcium oxyapatites with lanthanum (III)-created anionic vacancies. Chem. Mater. 2000, 12, 3836–3841. [Google Scholar] [CrossRef]
- Ramesan, M.T.; Varghese, M.; Periyat, P. Silver-doped zinc oxide as a nanofiller for development of poly (vinyl alcohol)/poly (vinyl pyrrolidone) blend nanocomposites. Adv. Polym. Technol. 2018, 37, 137–143. [Google Scholar] [CrossRef]
- Lai, G.J.; Shalumon, K.T.; Chen, S.H.; Chen, J.P. Composite chitosan/silk fibroin nanofibers for modulation of osteogenic differentiation and proliferation of human mesenchymal stem cells. Carbohydr. Polym. 2014, 111, 288–297. [Google Scholar] [CrossRef]
- Toskas, G.; Cherif, C.; Hund, R.D. Chitosan(PEO)/silica hybrid nanofibers as a potential biomaterial for bone regeneration. Carbohydr. Polym. 2013, 94, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Bharath, G.; Naldoni, A.; Ramsait, K.H. Enhanced electrocatalytic activity of gold nanoparticles on hydroxyapatite nanorods for sensitive hydrazine sensors. J. Mater. Chem. A 2016, 4, 6385–6394. [Google Scholar] [CrossRef]
- Oudadesse, H.; Mostafa, A.; Bui, X.V. Studies of doped biomimetic nano-hydroxyapatite/polymer matrix. Composites for applications in biomedical field. Recent Res. Morden Sci. 2011, 368–374. [Google Scholar]
- Sooksaen, P.; Pengsuwan, N.; Karawatthanaworrakul, S.; Pianpraditkul, S. Formation of porous apatite layer during in vitro study of hydroxyapatite-AW based glass composites. Adv. Condens. Matter Phys. 2015, 2015, 158582. [Google Scholar] [CrossRef]
- Nagakane, K.; Yoshida, Y.; Hirata, I. Analysis of chemical interaction of 4-MET with hydroxyapatite using XPS. Dent. Mater. J. 2006, 25, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Nagakane, K.; Fukuda, R.; Nakayama, Y.; Okazaki, M.; Shintani, H.; Inoue, S.; Tagawa, Y.; Suzuki, K.; De Munck, J.; et al. Comparative study on adhesive performance of functional monomers. J. Dent. Res. 2004, 83, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shansky, J.; Borselli, C.; Mooney, D.; Vandenburgh, H. Design and Fabrication of a Biodegradable, Covalently With and without genipinShape-Memory Alginate Scaffold for Cell and Growth Factor Delivery. Tissue Eng. Part A 2012, 18, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, B.; Ramya, S.; Shinyjoy, E.; Kavitha, L.; Gopi, D.; Vijayalakshmi, U. Structural, morphological and biological evaluations of cerium incorporated hydroxyapatite sol–gel coatings on Ti–6Al–4V for orthopaedic applications. J. Mater. Res. Technol. 2021, 12, 1319–1338. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Concentration of PVA (%) | Concentration of PVP (%) | Concentration of HAP (wt%) |
|---|---|---|---|
| 1 | 5 | 5 | 0 |
| 2 | 5 | 5 | 5 (0.5 g/10 mL) |
| 3 | 5 | 5 | 10 (1.0 g/10 mL) |
| 4 | 5 | 5 | 15 (1.5 g/10 mL) |
| 5 | 5 | 5 | 20 (2.0 g/10 mL) |
| 6 | 5 | 5 | 25 (2.5 g/10 mL) |
| S.No | Sample | Step 1 Weight Loss (°C) | Step 2 Weight Loss (°C) | Step 3 Weight Loss (°C) | % of Weight Loss |
|---|---|---|---|---|---|
| 1 | PVA-PVP | 80 | 340 | 390 | 92.808 |
| 2 | 5% HAP-PVA-PVP | 105 | 340 | 470 | 93.057 |
| 3 | 10% HAP-PVA-PVP | 130 | 350 | 480 | 95.661 |
| 4 | 15% HAP-PVA-PVP | 130 | 350 | 490 | 81.246 |
| 5 | 20% HAP-PVA-PVP | 140 | 360 | 470 | 78.059 |
| 6 | 25% HAP-PVA-PVP | 150 | 410 | 470 | 71.301 |
| S.No | Sample | Tg | Tm | Decomposition Temperature |
|---|---|---|---|---|
| 1 | PVA-PVP | 56.82 | 194 | 330 |
| 2 | 5% HAP-PVA-PVP | 93.46 | 322 | 413 |
| 3 | 10% HAP-PVA-PVP | 65.33 | 304 | 436 |
| 4 | 15% HAP-PVA-PVP | 72.54 | 319 | 417 |
| 5 | 20% HAP-PVA-PVP | 66.49 | 325 | 447 |
| 6 | 25% HAP-PVA-PVP | 119.42 | 318 | 450 |
| S.No | Sample | Ca 2p | P 2p | C1s | O1s | N | ||
|---|---|---|---|---|---|---|---|---|
| 2P3/2 | 2P1/2 | 2P3/2 | 2P1/2 | |||||
| 1 | HAP-PVA-PVP (Before immersion) | 347.50 | 351.02 | 133.66 | 134.14 | (a) 284.75-(C-C) (b) 285.57-(C-N) (c) 286.63(C=O/COH) (d) 289.67(O-C=O) | 532.32 533.27 | 400.02 |
| 2 | HAP-PVA-PVP (After immersion) | 349.10 | 352.63 | 134.94 | 135.65 | 286.2 | 532.56 533.27 | Nil |
| S.No | Sample | Hemolytic Ratio (% ±SD) |
|---|---|---|
| 1 | PVA-PVP | 1.82 ± 0.045 |
| 2 | 5% HAP-PVA-PVP | 2.19 ± 0.038 |
| 3 | 10% HAP-PVA-PVP | 2.45 ± 0.062 |
| 4 | 15% HAP-PVA-PVP | 2.71 ± 0.054 |
| 5 | 20% HAP-PVA-PVP | 2.95 ± 0.065 |
| 6 | 25% HAP-PVA-PVP | 3.11 ± 0.079 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, S.M.M.; Rama, M.; Sridhar, T.M.; Vijayalakshmi, U. Optimization of Biologically Inspired Electrospun Scaffold for Effective Use in Bone Regenerative Applications. Polymers 2024, 16, 2023. https://doi.org/10.3390/polym16142023
Stella SMM, Rama M, Sridhar TM, Vijayalakshmi U. Optimization of Biologically Inspired Electrospun Scaffold for Effective Use in Bone Regenerative Applications. Polymers. 2024; 16(14):2023. https://doi.org/10.3390/polym16142023
Chicago/Turabian StyleStella, Susai Mani Mary, Murugapandian Rama, T. M. Sridhar, and Uthirapathy Vijayalakshmi. 2024. "Optimization of Biologically Inspired Electrospun Scaffold for Effective Use in Bone Regenerative Applications" Polymers 16, no. 14: 2023. https://doi.org/10.3390/polym16142023
APA StyleStella, S. M. M., Rama, M., Sridhar, T. M., & Vijayalakshmi, U. (2024). Optimization of Biologically Inspired Electrospun Scaffold for Effective Use in Bone Regenerative Applications. Polymers, 16(14), 2023. https://doi.org/10.3390/polym16142023