
Palladium Catalysts Supported in Microporous Phosphine Polymer Networks
 , , ,
, , ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Techniques
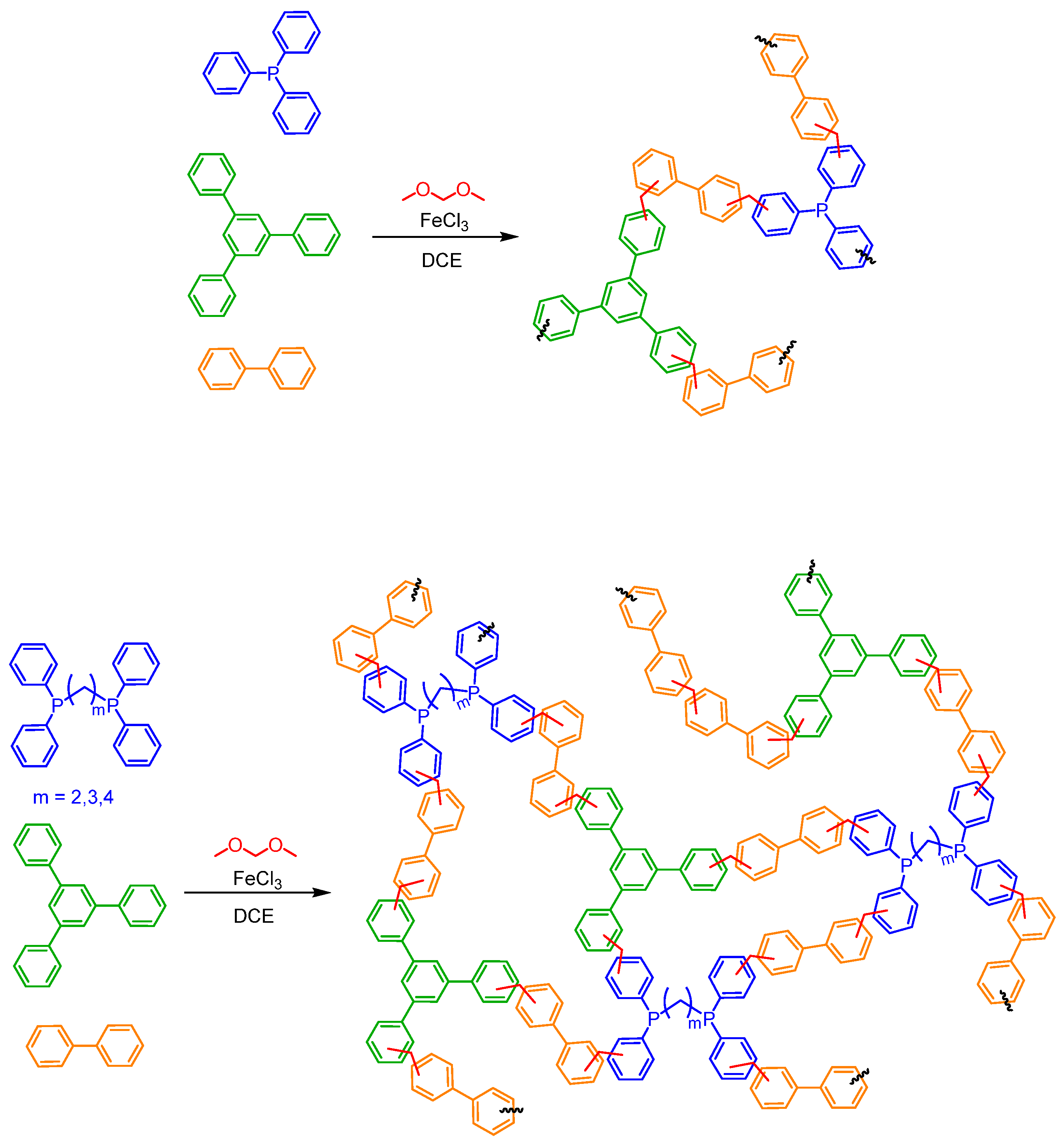
2.3. Synthesis of POPs
2.4. Synthesis of Supported Pd(II) Catalysts
2.5. General Procedure for Suzuki–Miyaura Reactions
2.6. Recycling Test
2.7. Leaching Test
2.8. Scale-Up of Suzuki–Miyaura Reaction
3. Results and Discussion
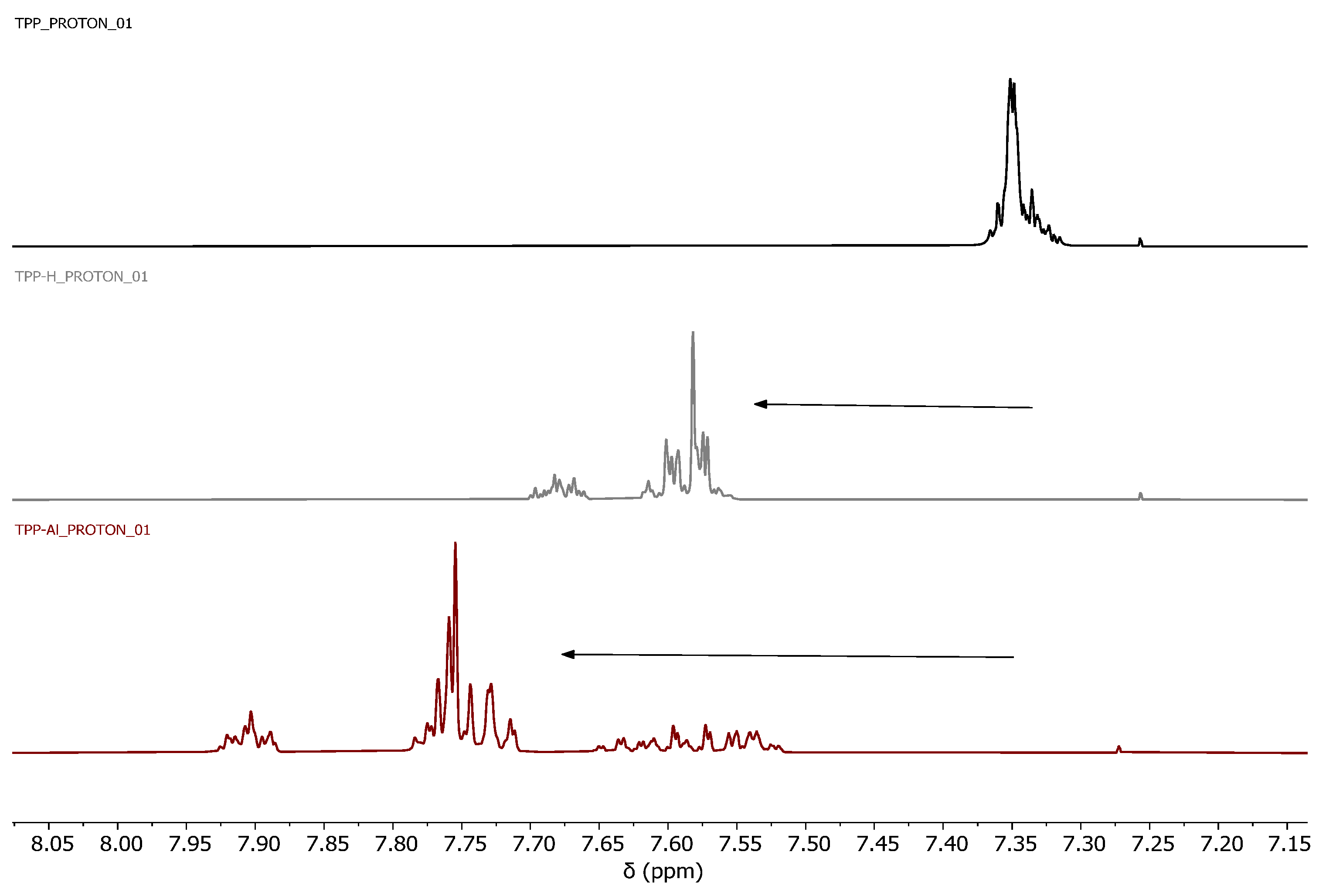
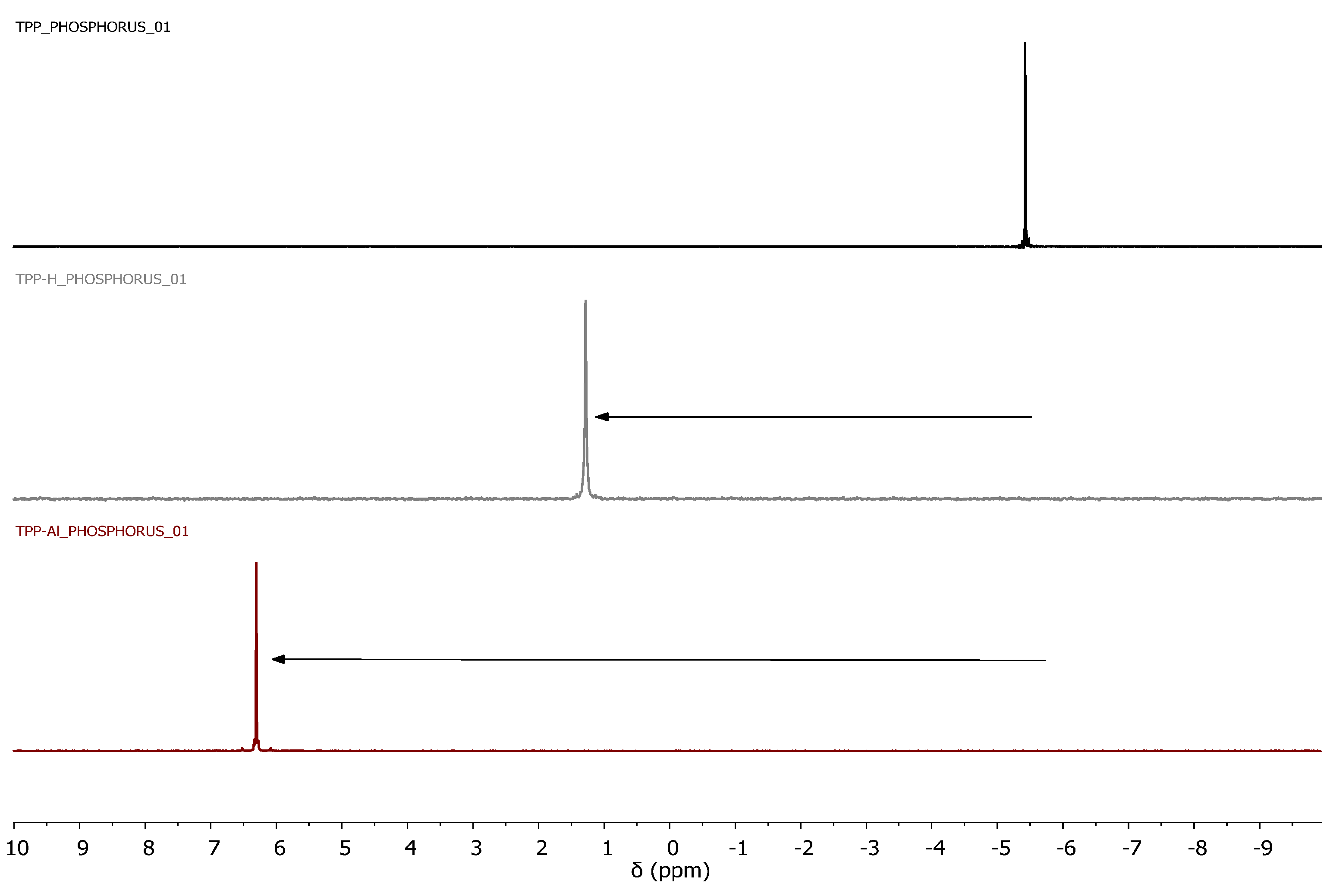
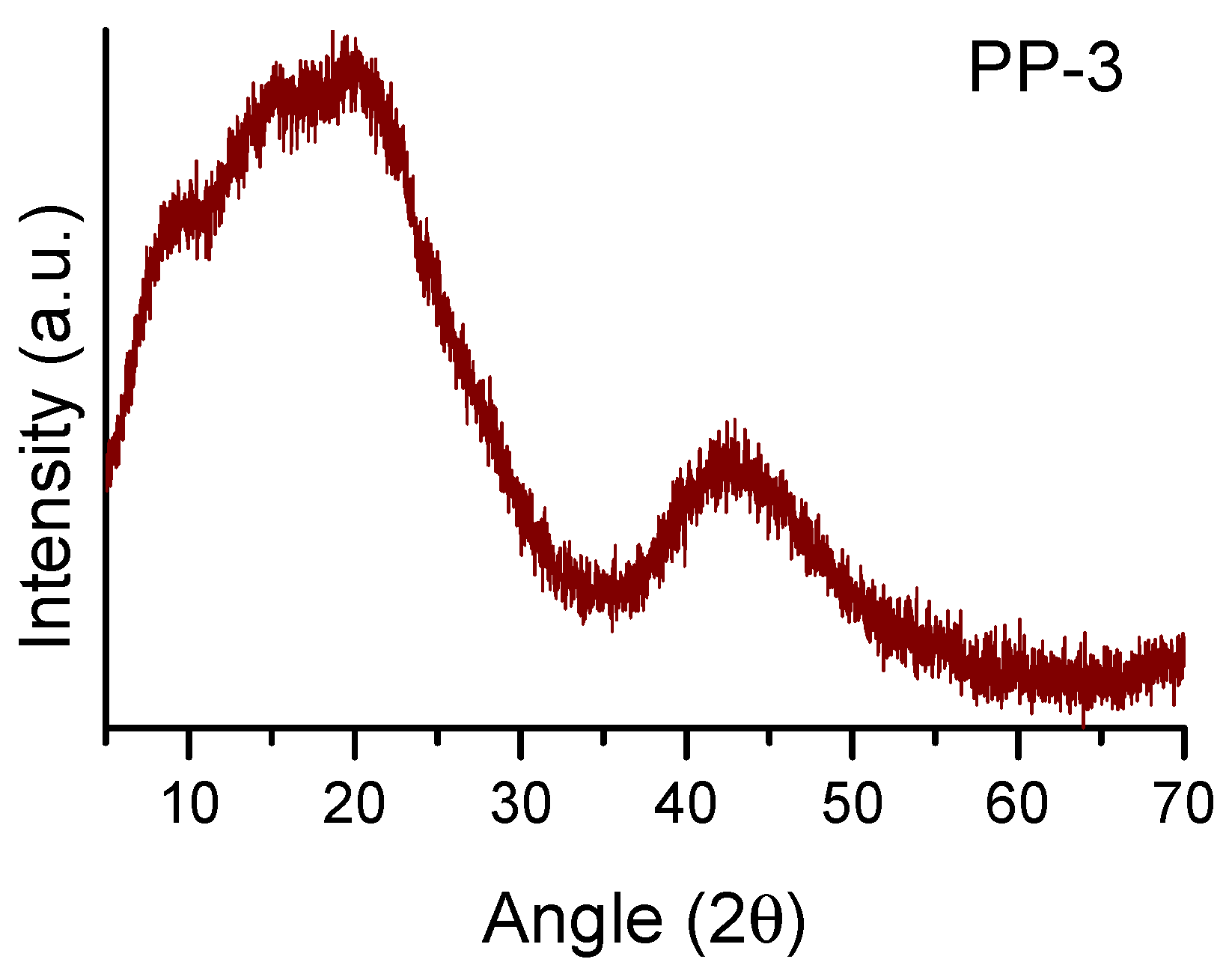
3.1. Synthesis and Characterization of POPs Based on Diphosphines
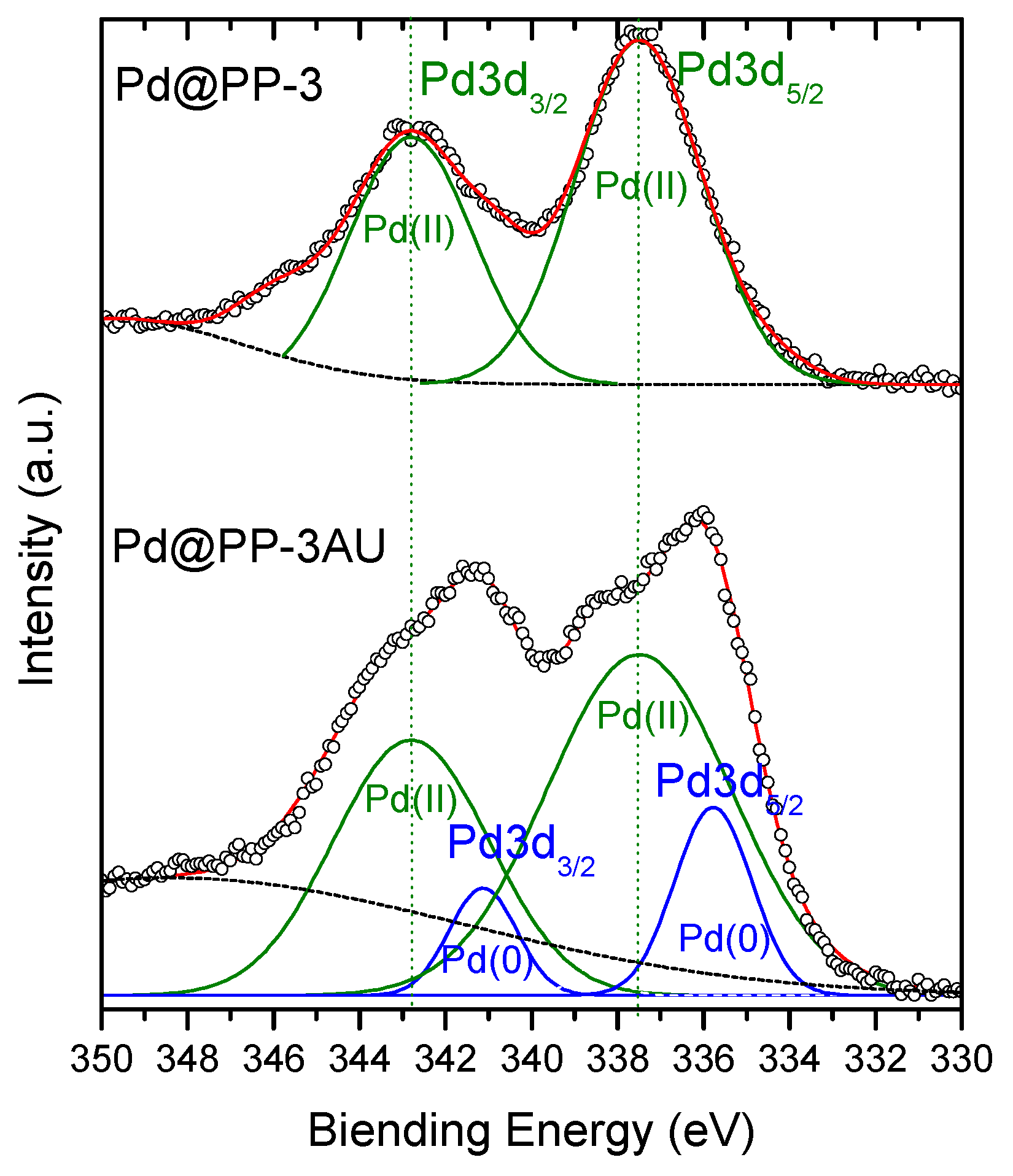
3.2. Synthesis and Characterization of Pd@PP-X Catalysts
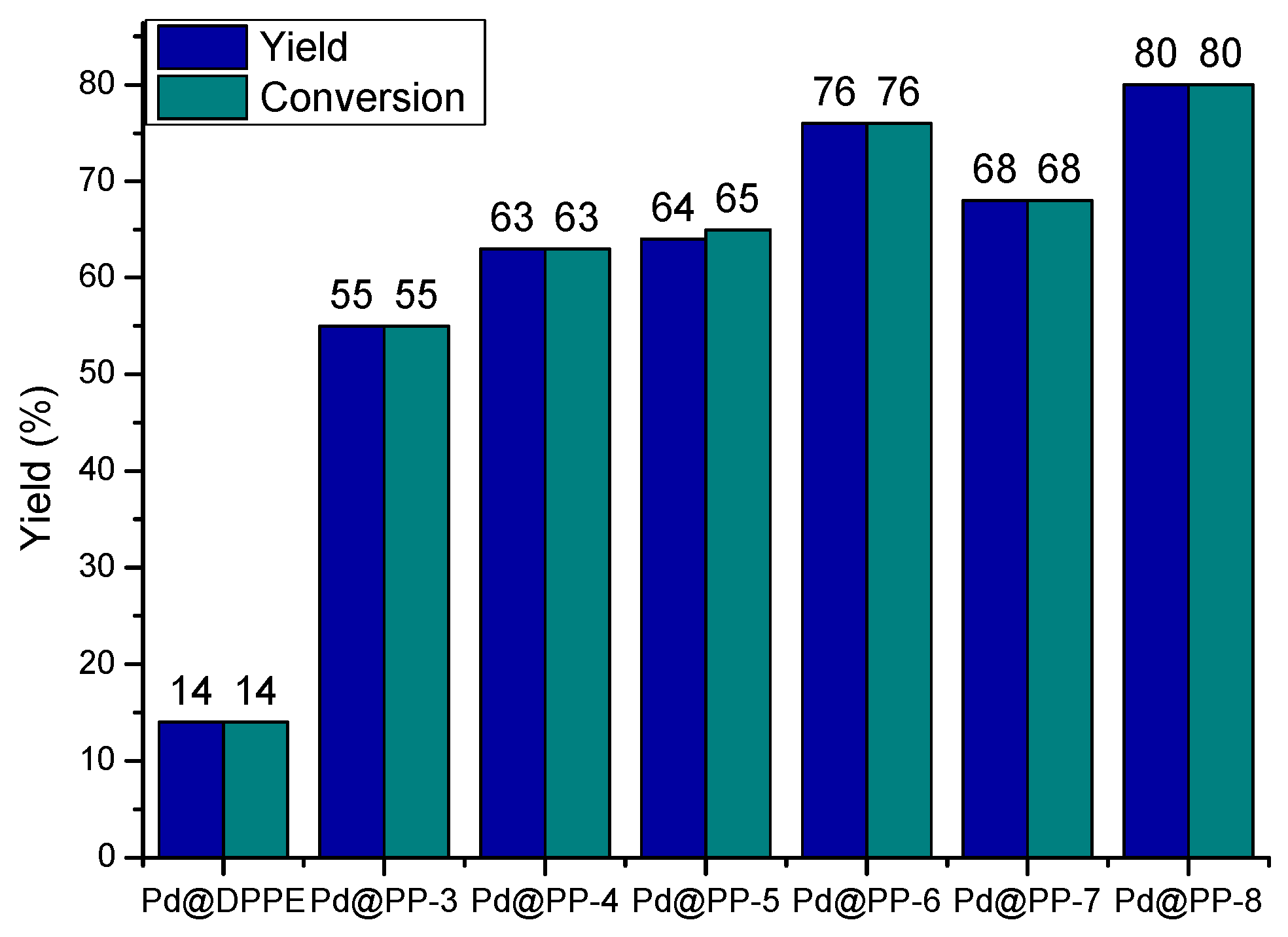
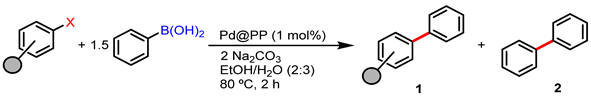
3.3. Catalytic Activity
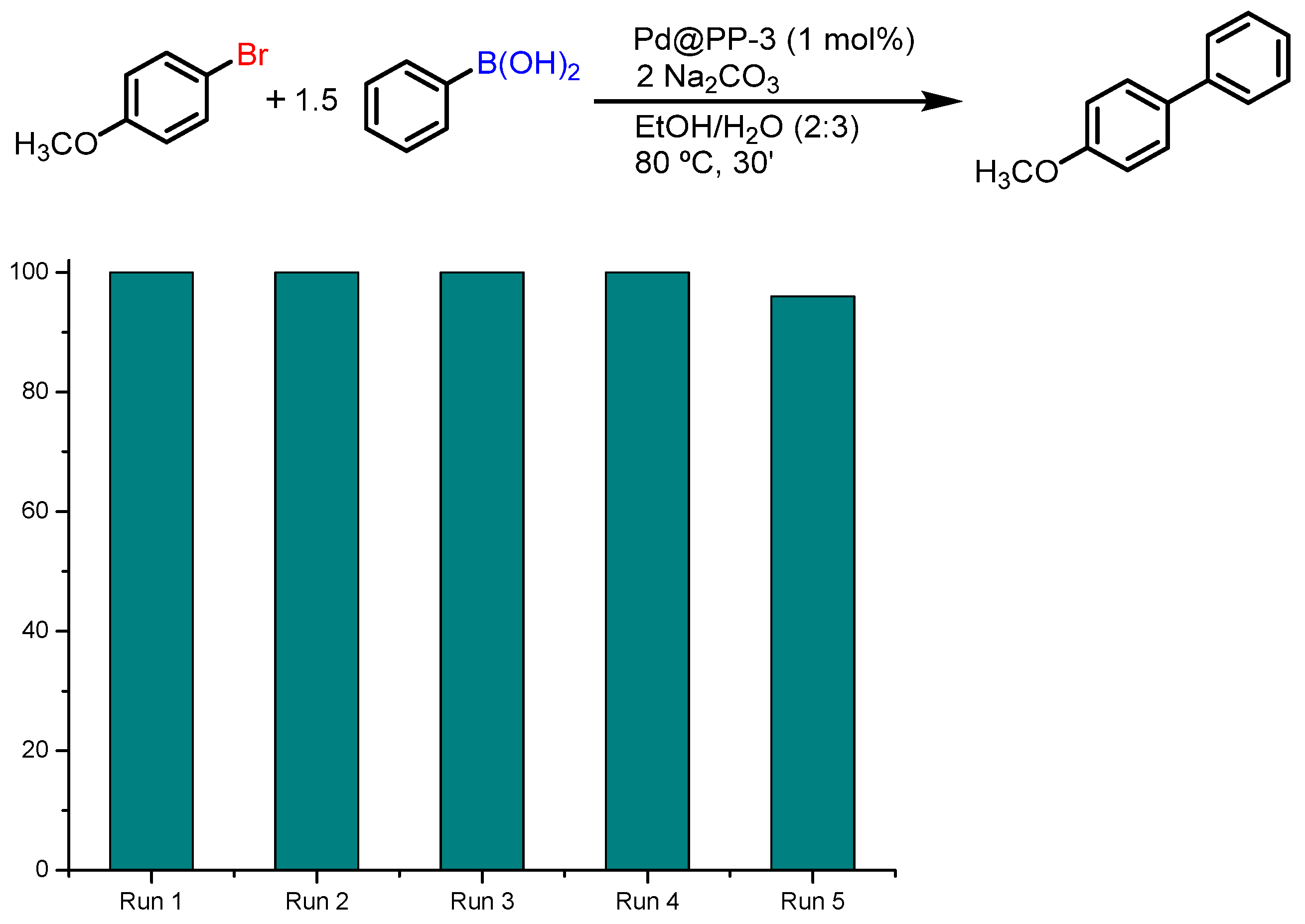
3.4. Recyclability of Pd@PPs
3.5. Leaching Test
3.6. Scale-Up Catalyzed Suzuki–Miyaura Reaction Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Z.; Xu, J.; Zheng, W.; Li, Y. Palladium Supported on Ethylenediaminetetraacetic Acid Functionalized Cellulose: Synthesis, Characterization, and Its Application in Carbon–Carbon Cross-Coupling Reactions. Cellulose 2022, 29, 2159–2173. [Google Scholar] [CrossRef]
- Wang, X.; Min, S.; Das, S.K.; Fan, W.; Huang, K.-W.; Lai, Z. Spatially Isolated Palladium in Porous Organic Polymers by Direct Knitting for Versatile Organic Transformations. J. Catal. 2017, 355, 101–109. [Google Scholar] [CrossRef]
- Tang, C.; Zou, Z.; Fu, Y.; Song, K. Highly Dispersed DPPF Locked in Knitting Hyper-Crosslinked Polymers as Efficient and Recyclable Catalyst. ChemistrySelect 2018, 3, 5987–5992. [Google Scholar] [CrossRef]
- Wang, X.; Ling, E.A.P.; Guan, C.; Zhang, Q.; Wu, W.; Liu, P.; Zheng, N.; Zhang, D.; Lopatin, S.; Lai, Z.; et al. Single-Site Ruthenium Pincer Complex Knitted into Porous Organic Polymers for Dehydrogenation of Formic Acid. ChemSusChem 2018, 11, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Valverde-González, A.; Marchal, G.; Maya, E.M.; Iglesias, M. A Step Forward in Solvent Knitting Strategies: Ruthenium and Gold Phosphine Complex Polymerization Results in Effective Heterogenized Catalysts. Catal. Sci. Technol. 2019, 9, 4552–4560. [Google Scholar] [CrossRef]
- Tao, R.; Ma, X.; Wei, X.; Jin, Y.; Qiu, L.; Zhang, W. Porous Organic Polymer Material Supported Palladium Nanoparticles. J. Mater. Chem. A 2020, 8, 17360–17391. [Google Scholar] [CrossRef]
- Verde-Sesto, E.; Maya, E.M.; Lozano, Á.E.; de la Campa, J.G.; Sánchez, F.; Iglesias, M. Novel Efficient Catalysts Based on Imine-Linked Mesoporous Polymers for Hydrogenation and Cyclopropanation Reactions. J. Mater. Chem. 2012, 22, 24637. [Google Scholar] [CrossRef]
- Merino, E.; Verde-Sesto, E.; Maya, E.M.; Corma, A.; Iglesias, M.; Sánchez, F. Mono-Functionalization of Porous Aromatic Frameworks to Use as Compatible Heterogeneous Catalysts in One-Pot Cascade Reactions. Appl. Catal. A Gen. 2014, 469, 206–212. [Google Scholar] [CrossRef]
- Kramer, S.; Bennedsen, N.R.; Kegnæs, S. Porous Organic Polymers Containing Active Metal Centers as Catalysts for Synthetic Organic Chemistry. ACS Catal. 2018, 8, 6961–6982. [Google Scholar] [CrossRef]
- Gu, Y.; Son, S.U.; Li, T.; Tan, B. Low-Cost Hypercrosslinked Polymers by Direct Knitting Strategy for Catalytic Applications. Adv. Funct. Mater. 2021, 31, 2008265. [Google Scholar] [CrossRef]
- Valverde-Gonzalez, A.; Iglesias, M.; Maya, E.M. Metal Catalysis with Knitting Aryl Polymers: Design, Catalytic Applications, and Future Trends. Chem. Mater. 2021, 33, 6616–6639. [Google Scholar] [CrossRef]
- Bi, J.; Dong, Y.; Meng, D.; Zhu, D.; Li, T. The Study and Application of Three Highly Porous Hyper-Crosslinked Catalysts Possessing Similar Catalytic Centers. Polymer 2019, 164, 183–190. [Google Scholar] [CrossRef]
- Li, B.; Gong, R.; Wang, W.; Huang, X.; Zhang, W.; Li, H.; Hu, C.; Tan, B. A New Strategy to Microporous Polymers: Knitting Rigid Aromatic Building Blocks by External Cross-Linker. Macromolecules 2011, 44, 2410–2414. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, L.; Xiang, D.; Liu, S.; Xu, W.; Luo, Q.; Shu, Y.; Ouyang, Y.; Lin, H. Palladium Supported on Triazolyl-Functionalized Hypercrosslinked Polymers as a Recyclable Catalyst for Suzuki-Miyaura Coupling Reactions. RSC Adv. 2020, 10, 17123–17128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Das, A.; Maji, B. Phosphorus Containing Porous Organic Polymers: Synthetic Techniques and Applications in Organic Synthesis and Catalysis. Org. Biomol. Chem. 2021, 19, 4174–4192. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, W.; Xiang, D.; Luo, Q.; Zeng, S.; Zheng, L.; Tan, Y.; Ouyang, Y.; Lin, H. Palladium Immobilized on 2,2′-Dipyridyl-Based Hypercrosslinked Polymers as a Heterogeneous Catalyst for Suzuki–Miyaura Reaction and Heck Reaction. Catal. Lett. 2020, 150, 2558–2565. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Tan, B. Porosity Engineering of Hyper-Cross-Linked Polymers Based on Fine-Tuned Rigidity in Building Blocks and High-Pressure Methane Storage Applications. Macromolecules 2023, 56, 1213–1222. [Google Scholar] [CrossRef]
- Xu, W.; Liu, C.; Xiang, D.; Luo, Q.; Shu, Y.; Lin, H.; Hu, Y.; Zhang, Z.; Ouyang, Y. Palladium Catalyst Immobilized on Functionalized Microporous Organic Polymers for C-C Coupling Reactions. RSC Adv. 2019, 9, 34595–34600. [Google Scholar] [CrossRef]
- Gäumann, P.; Cartagenova, D.; Ranocchiari, M. Phosphine-Functionalized Porous Materials for Catalytic Organic Synthesis. Eur. J. Org. Chem. 2022, 2022, e202201006. [Google Scholar] [CrossRef]
- Shen, L.; Han, X.; Dong, B.; Yang, Y.; Yang, J.; Li, F. Phosphine-Based Hyper-Cross-Linked Polymer-Supported Neutral Gold(I) Complex as a Recyclable Catalyst for the Regioselective Hydration of Alkynes to Ketones. ACS Appl. Polym. Mater. 2022, 4, 7408–7416. [Google Scholar] [CrossRef]
- Li, B.; Guan, Z.; Yang, X.; Wang, W.D.; Wang, W.; Hussain, I.; Song, K.; Tan, B.; Li, T. Multifunctional Microporous Organic Polymers. J. Mater. Chem. A 2014, 2, 11930. [Google Scholar] [CrossRef]
- Li, B.; Guan, Z.; Wang, W.; Yang, X.; Hu, J.; Tan, B.; Li, T. Highly Dispersed Pd Catalyst Locked in Knitting Aryl Network Polymers for Suzuki-Miyaura Coupling Reactions of Aryl Chlorides in Aqueous Media. Adv. Mater. 2012, 24, 3390–3395. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, H.M.; Zolotukhin, M.G.; Khalilov, L.M.; Dzhemilev, U.M. Superelectrophiles in Aromatic Polymer Chemistry. Macromolecules 2001, 34, 1122–1124. [Google Scholar] [CrossRef]
- Nieto, D.R.; Fomine, S.; Zolotukhin, M.G.; Fomina, L.; del Carmen Gutiérrez Hernandez, M. Superelectrophilic Activation of N -Substituted Isatins: Implications for Polymer Synthesis, a Theoretical Study. Macromol. Theory Simul. 2009, 18, 138–144. [Google Scholar] [CrossRef]
- Hernández-Cruz, O.; Zolotukhin, M.G.; Fomine, S.; Alexandrova, L.; Aguilar-Lugo, C.; Ruiz-Treviño, F.A.; Ramos-Ortíz, G.; Maldonado, J.L.; Cadenas-Pliego, G. High- T g Functional Aromatic Polymers. Macromolecules 2015, 48, 1026–1037. [Google Scholar] [CrossRef]
- Lopez-Iglesias, B.; Suárez-García, F.; Aguilar-Lugo, C.; González Ortega, A.; Bartolomé, C.; Martínez-Ilarduya, J.M.; De La Campa, J.G.; Lozano, Á.E.; Álvarez, C. Microporous Polymer Networks for Carbon Capture Applications. ACS Appl. Mater. Interfaces 2018, 10, 26195–26205. [Google Scholar] [CrossRef]
- Aguilar-Lugo, C.; Suárez-García, F.; Hernández, A.; Miguel, J.A.; Lozano, Á.E.; de la Campa, J.G.; Álvarez, C. New Materials for Gas Separation Applications: Mixed Matrix Membranes Made from Linear Polyimides and Porous Polymer Networks Having Lactam Groups. Ind. Eng. Chem. Res. 2019, 58, 9585–9595. [Google Scholar] [CrossRef]
- Rico-Martínez, S.; Álvarez, C.; Hernández, A.; Miguel, J.A.; Lozano, Á.E. Mixed Matrix Membranes Loaded with a Porous Organic Polymer Having Bipyridine Moieties. Membranes 2022, 12, 547. [Google Scholar] [CrossRef]
- Esteban, N.; Ferrer, M.L.; Ania, C.O.; de la Campa, J.G.; Lozano, Á.E.; Álvarez, C.; Miguel, J.A. Porous Organic Polymers Containing Active Metal Centers for Suzuki–Miyaura Heterocoupling Reactions. ACS Appl. Mater. Interfaces 2020, 12, 56974–56986. [Google Scholar] [CrossRef]
- Vargas, E.L.; Esteban, N.; Cencerrero, J.; Francés, V.; Álvarez, C.; Miguel, J.A.; Gallardo, A.; Lozano, A.E.; Cid, M.B. Pyrrolidine-Based Catalytic Microporous Polymers in Sustainable C N and C C Bond Formation via Iminium and Enamine Activation. Mater. Today Chem. 2022, 24, 100966. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, X.; Gu, Y.; Hu, J.; Li, G.; Shi, K. Palladium Supported on Triphenylphosphine-Functionalized Porous Organic Polymer: An Efficient Heterogeneous Catalyst for Aminocarbonylation. Transit. Met. Chem. 2016, 41, 1–7. [Google Scholar] [CrossRef]
- Xu, C.; Wang, H.; Wang, Q.; Wang, Y.; Zhang, Y.; Fan, G. Ruthenium Coordinated with Triphenylphosphine-Hyper-Crosslinked Polymer: An Efficient Catalyst for Hydrogen Evolution Reaction and Hydrolysis of Ammonia Borane. Appl. Surf. Sci. 2019, 466, 193–201. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, Q.; Yang, X.; Ye, X.; Duan, H.; Zhang, J.; Zhao, B.; Huang, Y. Knitting Aryl Network Polymers-Incorporated Ag Nanoparticles: A Mild and Efficient Catalyst for the Fixation of CO 2 as Carboxylic Acid. ACS Sustain. Chem. Eng. 2017, 5, 9634–9639. [Google Scholar] [CrossRef]
- Wang, Z.; Hasegawa, S.; Motokura, K.; Kuang, S.; Yang, Y. A Single-Atom Pd Catalyst Anchored on a Porous Organic Polymer for Highly Efficient Telomerization of 1,3-Butadiene with Methanol. Ind. Eng. Chem. Res. 2023, 62, 3151–3156. [Google Scholar] [CrossRef]
- Lei, Y.; Wu, L.; Zhang, X.; Mei, H.; Gu, Y.; Li, G. Palladium supported on triphenylphosphine functionalized porous organic polymer: A highly active and recyclable catalyst for alkoxycarbonylation of aryl iodides. J. Mol. Catal. A Chem. 2015, 398, 164–169. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Silvestre-Albero, A.M.; Juárez-Galán, J.M.; Silvestre-Albero, J.; Rodríguez-Reinoso, F. Low-Pressure Hysteresis in Adsorption: An Artifact? J. Phys. Chem. C 2012, 116, 16652–16655. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers (Acronyms) | R (%) | |
|---|---|---|
| PP-1 | 135TPB-TPP-DMM (1:1:3) | 75 |
| PP-2 | 135TPB-TPP-BP-DMM (1:1:3:6) | 99 |
| PP-3 | 135TPB-DPPE-DMM (4:3:12) | 79 |
| PP-4 | 135TPB-DPPE-BP-DMM (4:3:12:24) | 92 |
| PP-5 | 135TPB-DPPP-DMM (4:3:12) | 90 |
| PP-6 | 135TPB-DPPP-BP-DMM (4:3:12:24) | 99 |
| PP-7 | 135TPB-DPPB-DMM (4:3:12) | 86 |
| PP-8 | 135TPB-DPPB-BP-DMM (4:3:12:24) | 100 |
| Pd@PP | %Pd Content ICP-OES |
|---|---|
| Pd@PP-3 | 4.96 |
| Pd@PP-4 | 1.06 * |
| Pd@PP-5 | 5.08 |
| Pd@PP-6 | 1.71 * |
| Pd@PP-7 | 5.86 |
| Pd@PP-8 | 2.83 |
 | ||||
|---|---|---|---|---|
| Entry | Ar-Br | Pd-PP | Conversion (%) | Relative Yield Hetero: Homocoupling (%) |
| 1 | 4-Bromoanisole | Pd@PP-3 | 100 | 100:0 |
| 2 | Pd@PP-4 | 100 | 100.0 | |
| 3 | Pd@PP-5 | 100 | 100:0 | |
| 4 | Pd@PP-6 | 100 | 100:0 | |
| 5 | Pd@PP-7 | 100 | 100:0 | |
| 6 | Pd@PP-8 | 100 | 100:0 | |
| 7 | 4-Bromobiphenyl | Pd@PP-3 | 94 | 87:8 |
| 8 | Pd@PP-4 | 100 | 98:2 | |
| 9 | Pd@PP-5 | 100 | 98:2 | |
| 10 | Pd@PP-6 | 100 | 98:2 | |
| 11 | Pd@PP-7 | 100 | 97:3 | |
| 12 | Pd@PP-8 | 100 | 98:2 | |
| 13 | 1-Bromonaphtalene | Pd@PP-3 | 97 | 96:1 |
| 14 | Pd@PP-4 | 100 | 99:0 | |
| 15 | Pd@PP-5 | 100 | 99:0 | |
| 16 | Pd@PP-6 | 100 | 99:0 | |
| 17 | Pd@PP-7 | 100 | 99:0 | |
| 18 | Pd@PP-8 | 100 | 98:0 | |
| 19 | 9-Bromophenanthrene | Pd@PP-3 | 100 | 96:2 |
| 20 | Pd@PP-4 | 100 | 100:0 | |
| 21 | Pd@PP-5 | 100 | 100:0 | |
| 22 | Pd@PP-6 | 100 | 100:0 | |
| 23 | Pd@PP-7 | 97 | 94:3 | |
| 24 | Pd@PP-8 | 98 | 98:0 | |
 | ||||
|---|---|---|---|---|
| Entry | Aryl Halide | Pd-PP | Conversion (%) | Relative Yield Hetero: Homocoupling (%) |
| 25 a | 4-Bromoanisole | PP-3 | 0 | 0:0 |
| 26 b | 4-Bromoanisole | Pd@PP-3 | 90 | 82:8 |
| 27 | Bromobenzene c | Pd@PP-3 | 100 | 100:0 |
| 28 | 4-Bromotoluene | Pd@PP-3 | 100 | 100:0 |
| 29 | 1-Bromo-4-(tert-butyl)benzene | Pd@PP-3 | 100 | 98:1 |
| 30 | Bromopentamethylbenzene | Pd@PP-3 | 100 | 15:3 |
| 31 | 2-Bromo-1,3,5-triisopropilbenceno | Pd@PP-3 | 43 | 10:3 |
| 32 | 4-Chloroanisole | Pd@PP-3 | 100 | 85:0 |
| 33 | Chlorobenzene c | Pd@PP-3 | 100 | 92:0 |
| 34 | 4-Chlorotoluene | Pd@PP-3 | 81 | 59:12 |
| 35 | 1-Chloronaphthalene | Pd@PP-3 | 30 | 22:6 |
| TON | TOF (h−1) | |
|---|---|---|
| Pd@PP-3 | 548 | 2192 |
| Pd@PP-4 | 625 | 2500 |
| Pd@PP-5 | 647 | 2588 |
| Pd@PP-6 | 762 | 3048 |
| Pd@PP-7 | 677 | 2708 |
| Pd@PP-8 | 802 | 3208 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteban, N.; Claros, M.; Álvarez, C.; Lozano, Á.E.; Bartolomé, C.; Martínez-Ilarduya, J.M.; Miguel, J.A. Palladium Catalysts Supported in Microporous Phosphine Polymer Networks. Polymers 2023, 15, 4143. https://doi.org/10.3390/polym15204143
Esteban N, Claros M, Álvarez C, Lozano ÁE, Bartolomé C, Martínez-Ilarduya JM, Miguel JA. Palladium Catalysts Supported in Microporous Phosphine Polymer Networks. Polymers. 2023; 15(20):4143. https://doi.org/10.3390/polym15204143
Chicago/Turabian StyleEsteban, Noelia, Miguel Claros, Cristina Álvarez, Ángel E. Lozano, Camino Bartolomé, Jesús M. Martínez-Ilarduya, and Jesús A. Miguel. 2023. "Palladium Catalysts Supported in Microporous Phosphine Polymer Networks" Polymers 15, no. 20: 4143. https://doi.org/10.3390/polym15204143
APA StyleEsteban, N., Claros, M., Álvarez, C., Lozano, Á. E., Bartolomé, C., Martínez-Ilarduya, J. M., & Miguel, J. A. (2023). Palladium Catalysts Supported in Microporous Phosphine Polymer Networks. Polymers, 15(20), 4143. https://doi.org/10.3390/polym15204143