
Biocompatible Polymer for Self-Humidification


Abstract
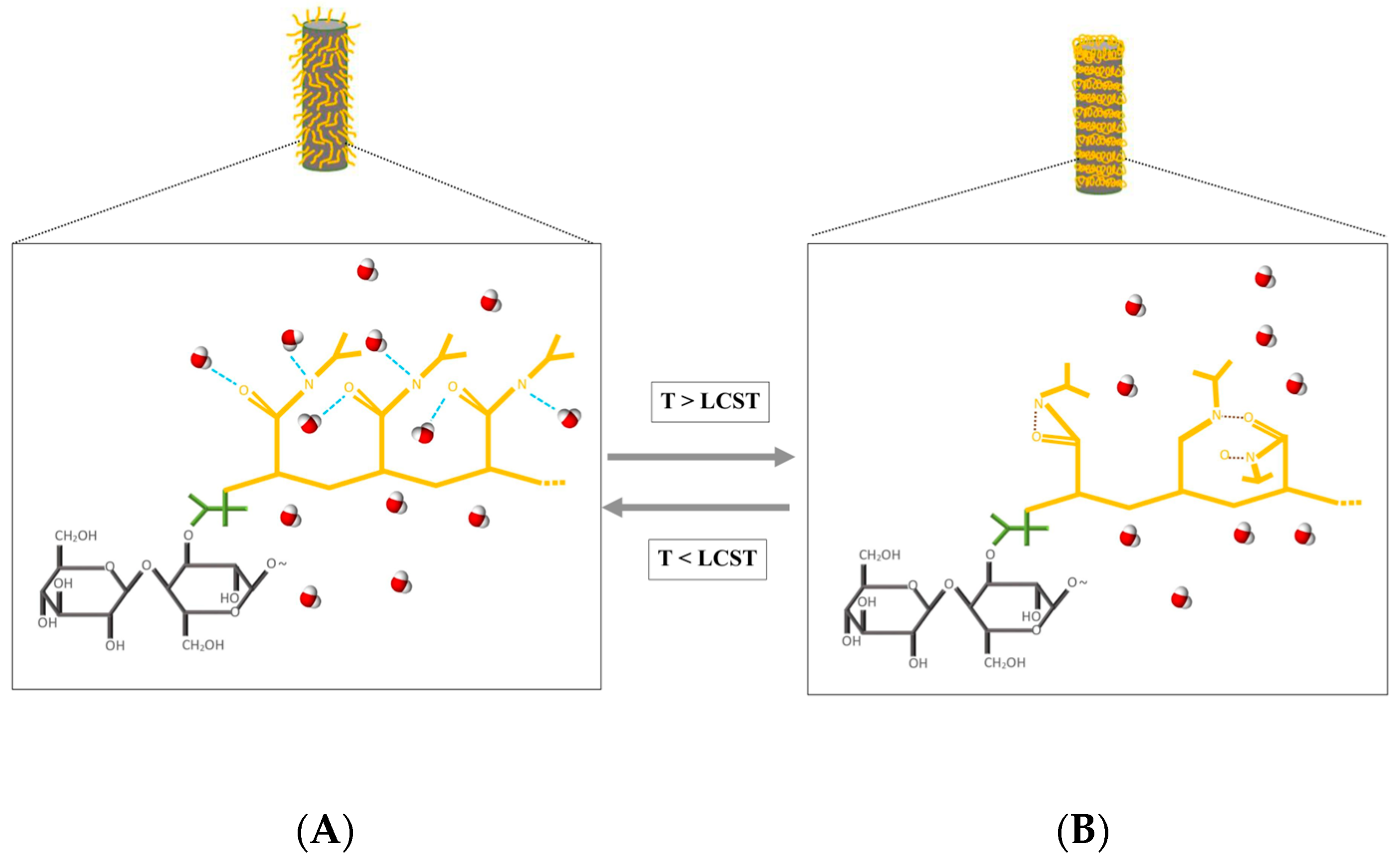
:1. Introduction
2. Materials and Methods
2.1. Substrate Materials
2.2. Chemicals
2.3. Methodology
2.3.1. Linear PNIPAM Synthesis
2.3.2. Branched PNIPAM Synthesis
2.3.3. Extended Purification Procedure
2.3.4. Synthesis of PNIPAM with Different Mw (5 kDa, 15 kDa, 50 kDa)
2.4. Smart Material Characterization
2.4.1. Proton Nuclear Magnetic Resonance (1H NMR)
2.4.2. Fourier Transform Infrared (FTIR) Spectroscopy
2.4.3. Energy Dispersive X-ray Spectroscopy (EDS)
2.4.4. Simultaneous Thermal Analysis
2.5. Statistical Analysis
3. Results and Discussion
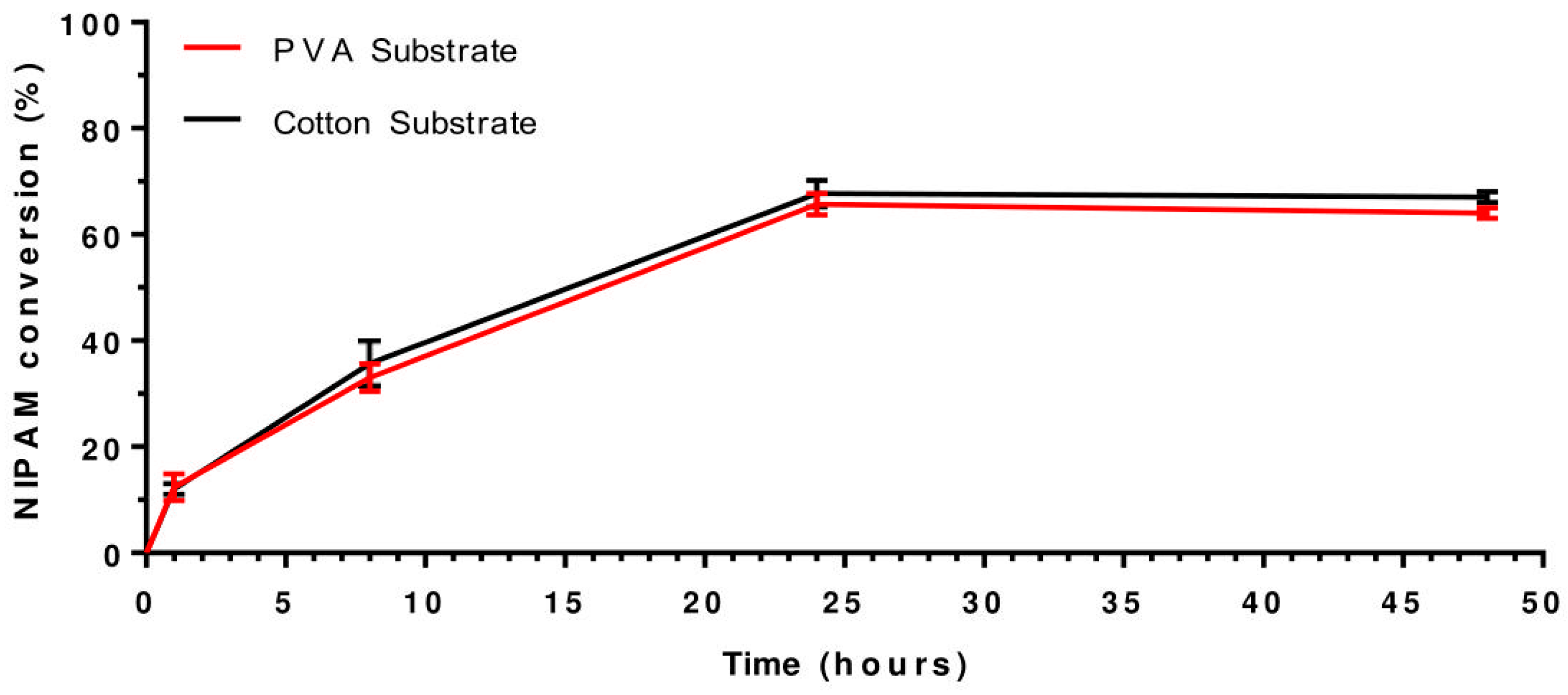
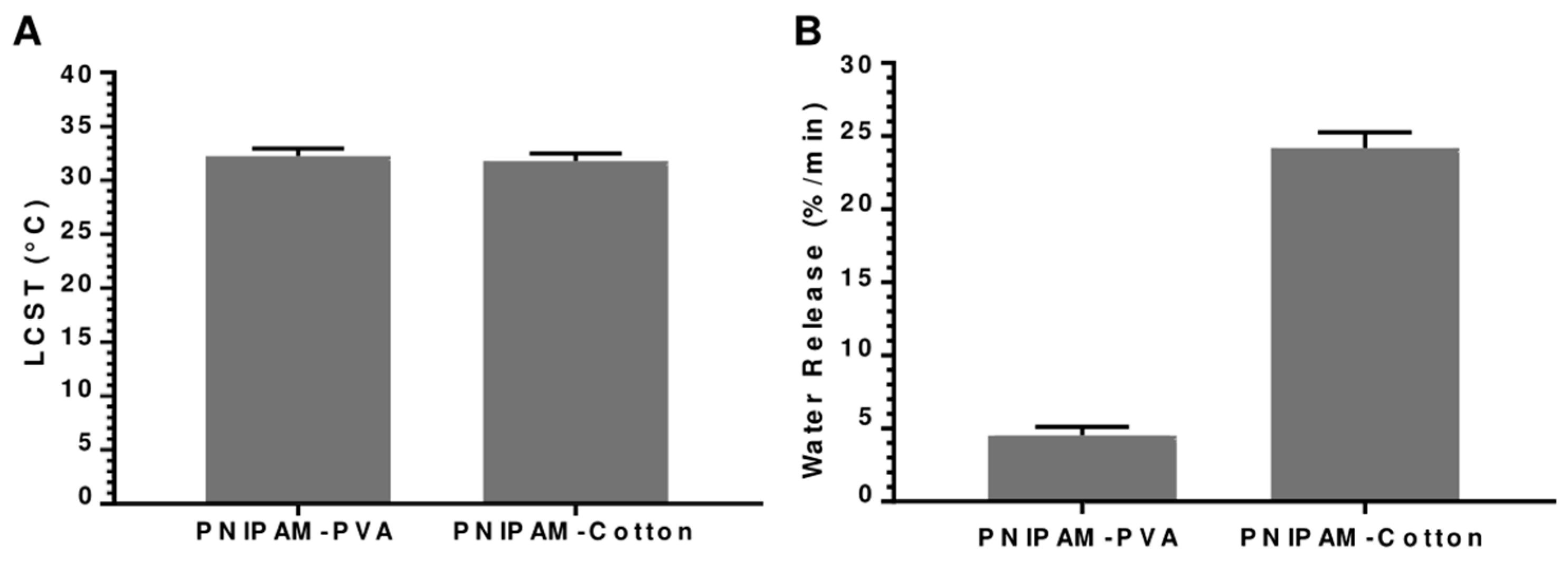
3.1. Analysis of Substrate Material: PVA vs. Cotton
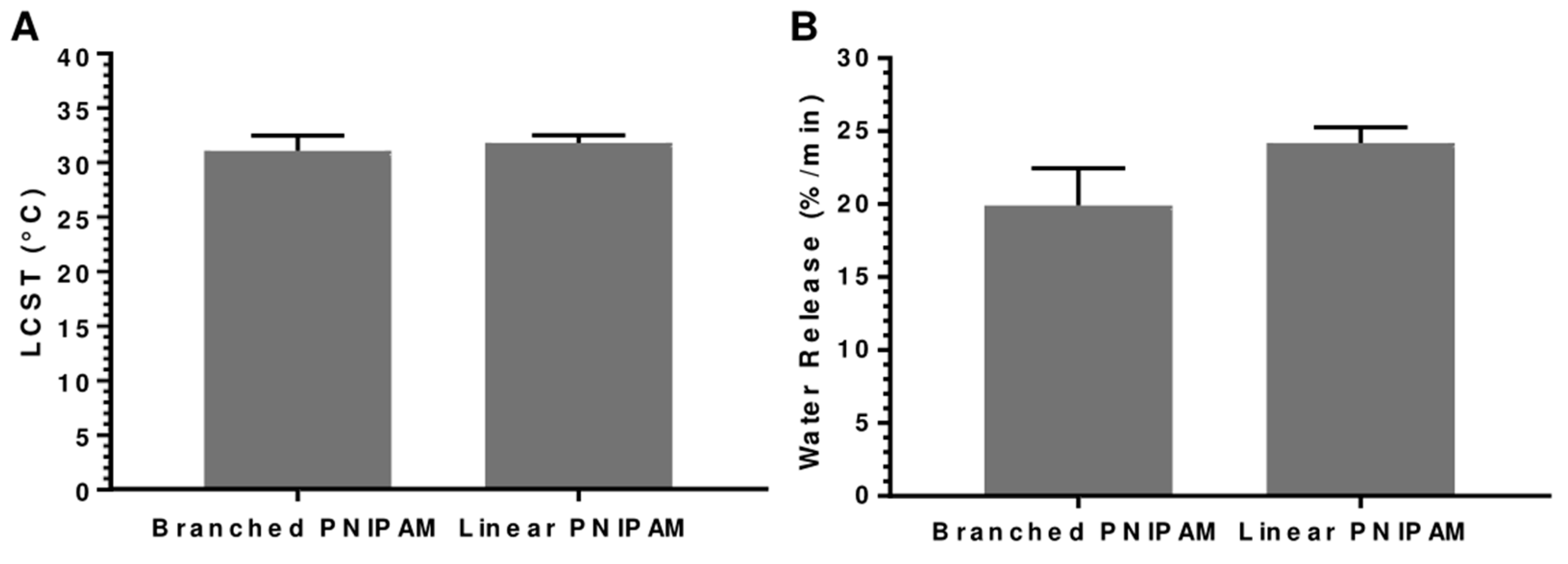
3.2. Analysis of Polymer Structure: Linear vs. Branched
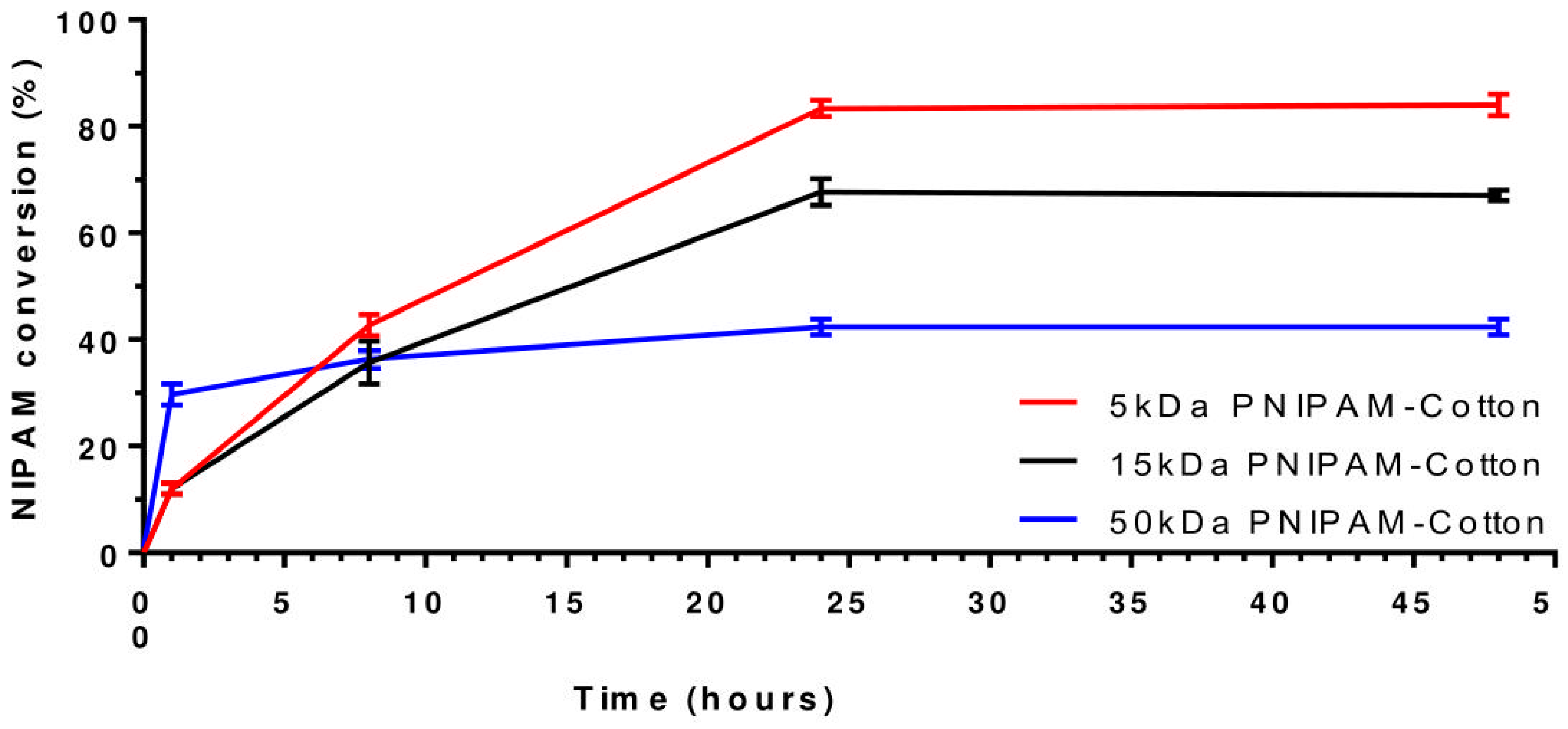
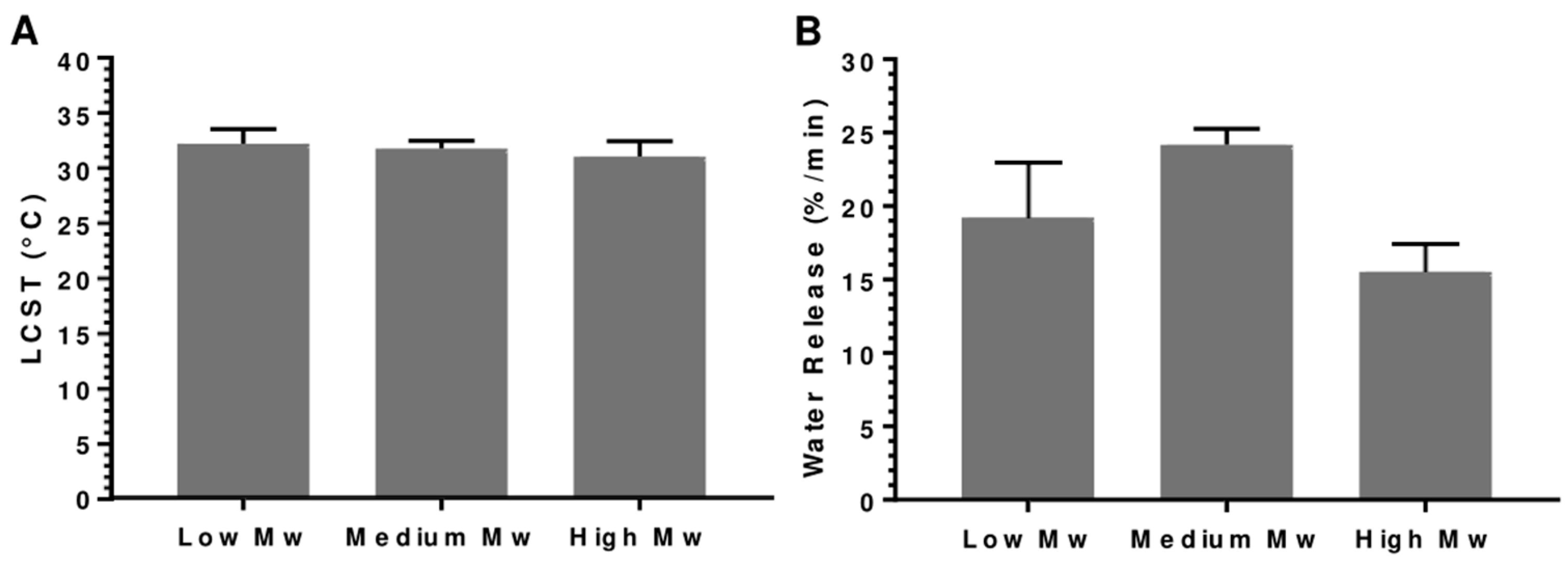
3.3. Analysis of Polymer Molecular Weight: 5, 15, 50 kDa
3.4. Assessment of the Optimized PNIPAM Smart Material
4. Conclusions
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Analysis of Substrate Material: PVA vs. Cotton
References
- Lima, L.H.; Morales, Y.; Cabral, T. Ocular biocompatibility of poly-N-isopropylacrylamide (pNIPAM). J. Ophthalmol. 2016, 2016, 5356371. [Google Scholar]
- Cooperstein, M.A.; Canavan, H.E. Assessment of cytotoxicity of (N-isopropyl acrylamide) and poly (N-isopropyl acrylamide)-coated surfaces. Biointerphases 2013, 8, 19. [Google Scholar]
- Patenaude, M.; Hoare, T. Injectable, degradable thermoresponsive poly (N-isopropylacrylamide) hydrogels. ACS Macro Lett. 2012, 1, 409–413. [Google Scholar]
- Lynden-Bell, R.; Rasaiah, J. From hydrophobic to hydrophilic behaviour: A simulation study of solvation entropy and free energy of simple solutes. J. Chem. Phys. 1997, 107, 1981–1991. [Google Scholar]
- Guilherme, M.; Silva, R.; Girotto, E.; Rubira, A.; Muniz, E. Hydrogels based on PAAm network with PNIPAAm included: Hydrophilic–hydrophobic transition measured by the partition of Orange II and Methylene Blue in water. Polymer 2003, 44, 4213–4219. [Google Scholar]
- Sun, S.; Wu, P. Tailoring the morphology of branched poly (N-isopropylacrylamide) via self-condensing atom-transfer radical copolymerization and its unique self-assembly behavior in alcohol. Soft Matter 2011, 7, 7526–7531. [Google Scholar]
- Wandera, D.; Himstedt, H.H.; Marroquin, M.; Wickramasinghe, S.R.; Husson, S.M. Modification of ultrafiltration membranes with block copolymer nanolayers for produced water treatment: The roles of polymer chain density and polymerization time on performance. J. Membr. Sci. 2012, 403, 250–260. [Google Scholar]
- Wandera, D.; Wickramasinghe, S.R.; Husson, S.M. Modification and characterization of ultrafiltration membranes for treatment of produced water. J. Membr. Sci. 2011, 373, 178–188. [Google Scholar]
- Wandera, D.; Wickramasinghe, S.R.; Husson, S.M. Stimuli-responsive membranes. J. Membr. Sci. 2010, 357, 6–35. [Google Scholar] [CrossRef]
- Yang, H.; Zhu, H.; Hendrix, M.M.; Lousberg, N.J.; de With, G.; Esteves, A.C.C.; Xin, J.H. Temperature-Triggered Collection and Release of Water from Fogs by a Sponge-Like Cotton Fabric. Adv. Mater. 2013, 25, 1150–1154. [Google Scholar] [CrossRef]
- Qiu, J.; Charleux, B.; Matyjaszewski, K. Controlled/living radical polymerization in aqueous media: Homogeneous and heterogeneous systems. Prog. Polym. Sci. 2001, 26, 2083–2134. [Google Scholar]
- Matyjaszewski, K.; Müller, A.H. 50 years of living polymerization. Prog. Polym. Sci. 2007, 32, 173. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Structural aspects of copper catalyzed atom transfer radical polymerization. Coord. Chem. Rev. 2005, 249, 1155–1184. [Google Scholar]
- Ran, J.; Wu, L.; Zhang, Z.; Xu, T. Atom transfer radical polymerization (ATRP): A versatile and forceful tool for functional membranes. Prog. Polym. Sci. 2014, 39, 124–144. [Google Scholar]
- DeMerlis, C.; Schoneker, D. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar]
- Karimi, A.; Navidbakhsh, M.; Faghihi, S. RETRACTED: Fabrication and mechanical characterization of a polyvinyl alcohol sponge for tissue engineering applications. Perfusion 2014, 29, 231–237. [Google Scholar] [CrossRef]
- Paradossi, G.; Cavalieri, F.; Chiessi, E.; Spagnoli, C.; Cowman, M.K. Poly (vinyl alcohol) as versatile biomaterial for potential biomedical applications. J. Mater. Sci. Mater. Med. 2003, 14, 687–691. [Google Scholar] [CrossRef]
- Ramaraj, B.; Poomalai, P. Ecofriendly poly (vinyl alcohol) and coconut shell powder composite films: Physico-mechanical, thermal properties, and swelling studies. J. Appl. Polym. Sci. 2006, 102, 3862–3867. [Google Scholar] [CrossRef]
- Karimi, A.; Navidbakhsh, M.; Haghi, A.M. An experimental study on the structural and mechanical properties of polyvinyl alcohol sponge using different stress–strain definitions. Adv. Polym. Technol. 2014, 33, 231. [Google Scholar]
- Wang, Y.; Chang, C.; Zhang, L. Effects of freezing/thawing cycles and cellulose nanowhiskers on structure and properties of biocompatible starch/PVA sponges. Macromol. Mater. Eng. 2010, 295, 137–145. [Google Scholar] [CrossRef]
- Choi, S.M.; Singh, D.; Kumar, A.; Oh, T.H.; Cho, Y.W.; Han, S.S. Porous three-dimensional PVA/gelatin sponge for skin tissue engineering. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 384–389. [Google Scholar]
- Trivedi, M.K.; Nayak, G.; Patil, S.; Tallapragada, R.M.; Mishra, R. Impact of biofield treatment on chemical and thermal properties of cellulose and cellulose acetate. J. Bioeng. Biomed. Sci. 2015, 5. [Google Scholar] [CrossRef]
- Szcześniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Kuckling, D.; Harmon, M.E.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 1. Synthesis and characterization of constrained temperature-responsive hydrogel layers. Macromolecules 2002, 35, 6377–6383. [Google Scholar]
- Carter, S.; Hunt, B.; Rimmer, S. Highly branched poly (N-isopropylacrylamide) s with imidazole end groups prepared by radical polymerization in the presence of a styryl monomer containing a dithioester group. Macromolecules 2005, 38, 4595–4603. [Google Scholar]
- Nastyshyn, S.; Stetsyshyn, Y.; Raczkowska, J.; Nastishin, Y.; Melnyk, Y.; Panchenko, Y.; Budkowski, A. Temperature-responsive polymer brush coatings for advanced biomedical applications. Polymers 2022, 14, 4245. [Google Scholar]
- Van Humbeeck, J. Simultaneous thermal analysis. In Handbook of Thermal Analysis and Calorimetry; Elsevier: Amsterdam, The Netherlands, 1998; Volume 1, pp. 497–508. [Google Scholar]
- Yang, H.; Esteves, A.C.C.; Zhu, H.; Wang, D.; Xin, J.H. In-situ study of the structure and dynamics of thermo-responsive PNIPAAm grafted on a cotton fabric. Polymer 2012, 53, 3577–3586. [Google Scholar]
- Xu, J.; Ye, J.; Liu, S. Synthesis of well-defined cyclic poly (N-isopropylacrylamide) via click chemistry and its unique thermal phase transition behavior. Macromolecules 2007, 40, 9103–9110. [Google Scholar]
- Bartual, S.G. An Investigation into the Human Upper Airways Humidification; Auckland University of Technology: Auckland, New Zealand, 2019. [Google Scholar]
- Plunkett, K.N.; Zhu, X.; Moore, J.S.; Leckband, D.E. PNIPAM chain collapse depends on the molecular weight and grafting density. Langmuir 2006, 22, 4259–4266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yan, C.; Winnik, F.; Leckband, D. End-grafted low-molecular-weight PNIPAM does not collapse above the LCST. Langmuir 2007, 23, 162–169. [Google Scholar]
- Lanzalaco, S.; Armelin, E. Poly (N-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y.; Fu, W.; Yao, M.; Ding, Z.; Xuan, J.; Li, D.; Wang, S.; Xia, Y.; Cao, M. Poly (N-isopropylacrylamide)-based thermoresponsive composite hydrogels for biomedical applications. Polymers 2020, 12, 580. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Nakayama, Y.; Matsuda, T. In vivo evaluation of poly (N-isopropylacrylamide)(PNIPAM)-grafted gelatin as an in situ-formable scaffold. J. Artif. Organs 2004, 7, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Ghosh, P.; Ghosh, A.; Haldar, C.; Dhara, S.; Panda, A.B.; Pal, S. Stimulus-responsive, biodegradable, biocompatible, covalently cross-linked hydrogel based on dextrin and poly (N-isopropylacrylamide) for in vitro/in vivo controlled drug release. ACS Appl. Mater. Interfaces 2015, 7, 14338–14351. [Google Scholar] [CrossRef]
- Wei, W.; Hu, X.; Qi, X.; Yu, H.; Liu, Y.; Li, J.; Zhang, J.; Dong, W. A novel thermo-responsive hydrogel based on salecan and poly (N-isopropylacrylamide): Synthesis and characterization. Colloids Surf. B Biointerfaces 2015, 125, 1–11. [Google Scholar] [CrossRef]
- Pooresmaeil, M.; Namazi, H.; Salehi, R. Photoluminescent folic acid functionalized biocompatible and stimuli-responsive nanostructured polymer brushes for targeted and controlled delivery of doxorubicin. Eur. Polym. J. 2021, 156, 110610. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Calculated BiB/PNIPAM from Different Methods | ||
|---|---|---|---|
| Weight Difference: | EDS Data: | NMR Data: | |
| Analysis 2: Presence of BiB on the substrate | |||
| PVA | 0.034 ± 0.015 | 0.028 ± 0.008 | N/A |
| Cotton | 0.158 ± 0.054 | 0.173 ± 0.026 | N/A |
| Analysis 4: PNIPAM synthesis on the substrate | |||
| PVA | 1.015 ± 0.125 | 1.114 ± 0.183 | 1.183 ± 0.182 |
| Cotton | 6.529 ± 0.107 | 7.110 ± 0.421 | 6.941 ± 0.851 |
| Analysis 6: Analysis of polymer structure | |||
| Branched | 3.815 ± 0.125 | 4.014 ± 0.583 | N/A |
| Linear | 6.529 ± 0.107 | 7.110 ± 0.421 | N/A |
| Analysis 8: Analysis of polymer MW. | |||
| 5 kDa | 1.931 ± 0.125 | 2.512 ± 1.583 | 2.329 ± 1.982 |
| 15 kDa | 6.529 ± 0.107 | 7.110 ± 1.421 | 6.941 ± 1.851 |
| 50 kDa | 10.627 ± 0.118 | 9.927 ± 1.219 | 11.784 ± 1.957 |
| Substrate/PNIPAM Structure/PNIPAM MW | Weight (%) of C | Weight (%) of O | Weight (%) of Br |
|---|---|---|---|
| Analysis 1: Presence of BiB on the substrate | |||
| PVA | 48.517 ± 1.231 | 44.700 ± 0.550 | 3.817 ± 0.685 |
| Cotton | 49.533 ± 1.978 | 43.613 ± 1.636 | 5.920 ± 0.387 |
| Analysis 3: PNIPAM synthesis on the substrate | |||
| PVA | 48.677 ± 0.956 | 37.803 ± 1.488 | 10.873 ± 0.646 |
| Cotton | 48.860 ± 1.010 | 37.231 ± 1.328 | 11.630 ± 1.324 |
| Analysis 5: Analysis of polymer structure | |||
| Branched | 48.657 ± 0.956 | 41.060 ± 1.488 | 10.283 ± 0.646 |
| Linear | 48.860 ± 1.010 | 37.231 ± 1.328 | 11.630 ± 1.324 |
| Analysis 7: Analysis of polymer MW | |||
| 5 kDa | 47.783 ± 2.252 | 37.230 ± 1.328 | 10.553 ± 1.072 |
| 15 kDa | 48.860 ± 1.010 | 37.231 ± 1.328 | 11.630 ± 1.324 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Jumaily, A.M.; Grau-Bartual, S.; Weerasinghe, N.T. Biocompatible Polymer for Self-Humidification. Polymers 2023, 15, 4101. https://doi.org/10.3390/polym15204101
Al-Jumaily AM, Grau-Bartual S, Weerasinghe NT. Biocompatible Polymer for Self-Humidification. Polymers. 2023; 15(20):4101. https://doi.org/10.3390/polym15204101
Chicago/Turabian StyleAl-Jumaily, Ahmed M., Sandra Grau-Bartual, and Nimesha T. Weerasinghe. 2023. "Biocompatible Polymer for Self-Humidification" Polymers 15, no. 20: 4101. https://doi.org/10.3390/polym15204101
APA StyleAl-Jumaily, A. M., Grau-Bartual, S., & Weerasinghe, N. T. (2023). Biocompatible Polymer for Self-Humidification. Polymers, 15(20), 4101. https://doi.org/10.3390/polym15204101