

Preparation of Thermoplastic Cellulose Esters in [mTBNH][OAC] Ionic Liquid by Transesterification Reaction

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cellulose Dissolution and Esterification Procedure
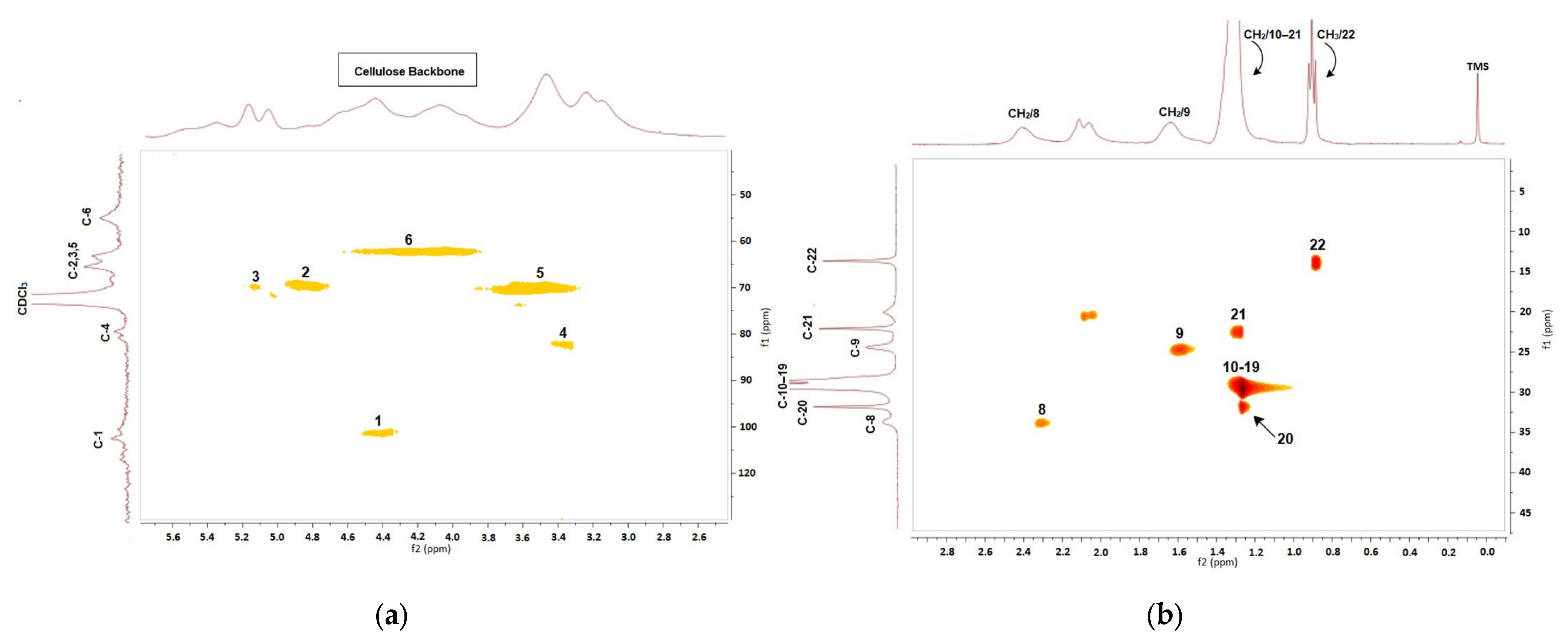
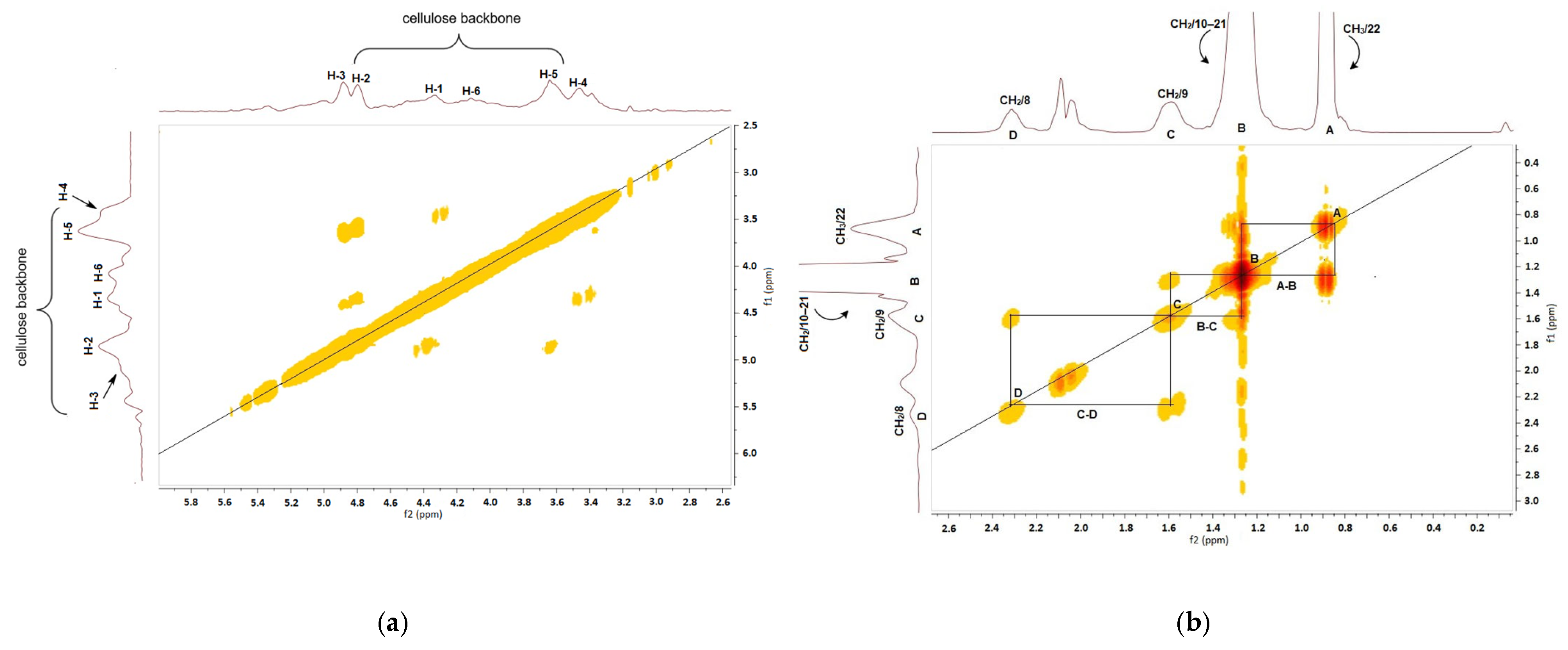
2.3. Characterization
3. Results and Discussion
3.1. Preparation of Cellulose Esters
Ionic Liquid Recycling
- Evaporation of water and ethanol through thin film or rotary distillation–waste pre-concentrate.
- Waste pre-concentrate mixed with acetone in a 1:8 ratio–hydrophilic by-products precipitate.
- Filtration of mixture and evaporation of acetone–waste concentrate obtained.
- Waste concentrate mixed with distilled water in 1:10–hydrophobic by-products precipitate.
- Filtration of mixture and evaporation of acetone–regenerated IL:DMSO binary solvent (recyclate) obtained.
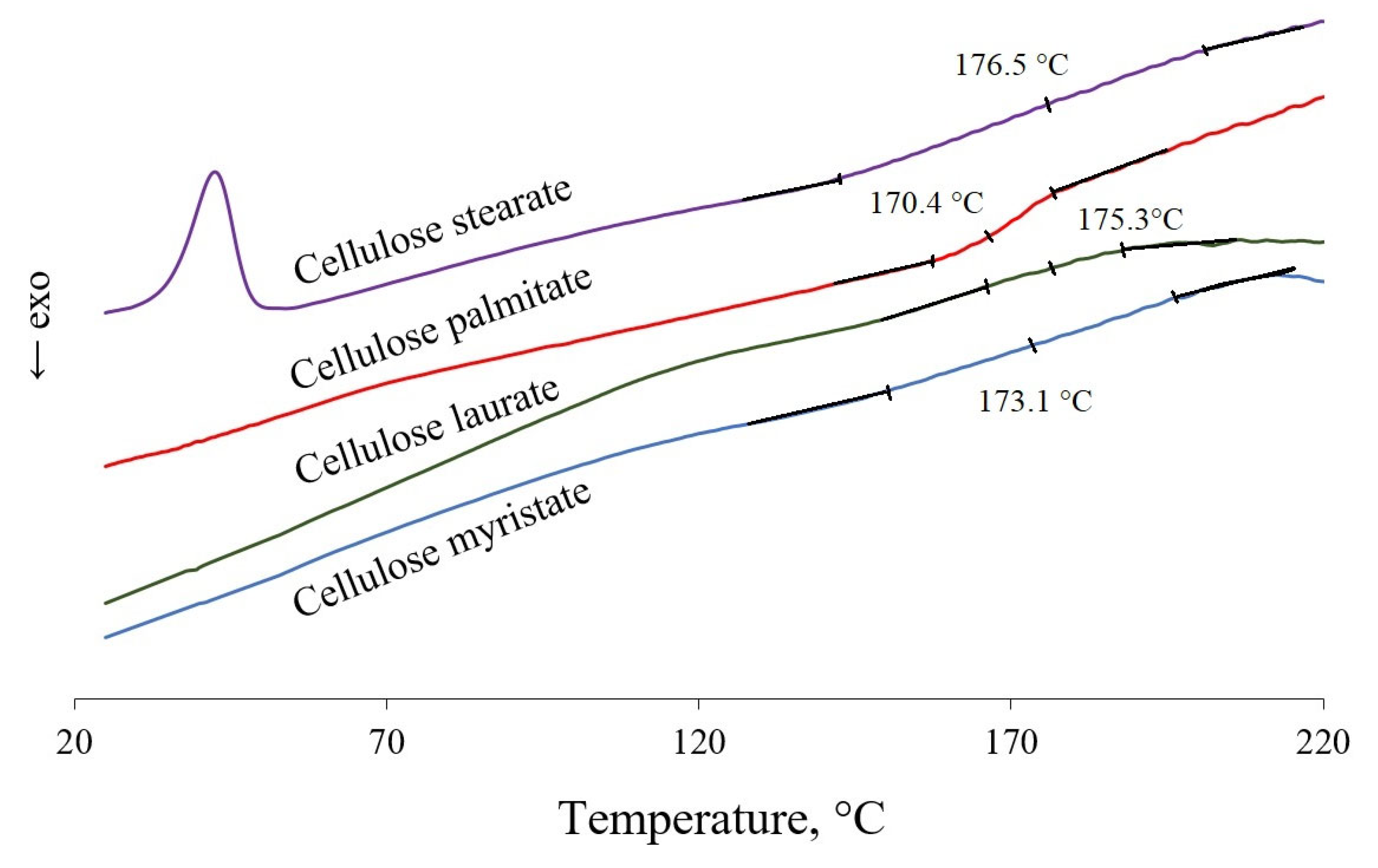
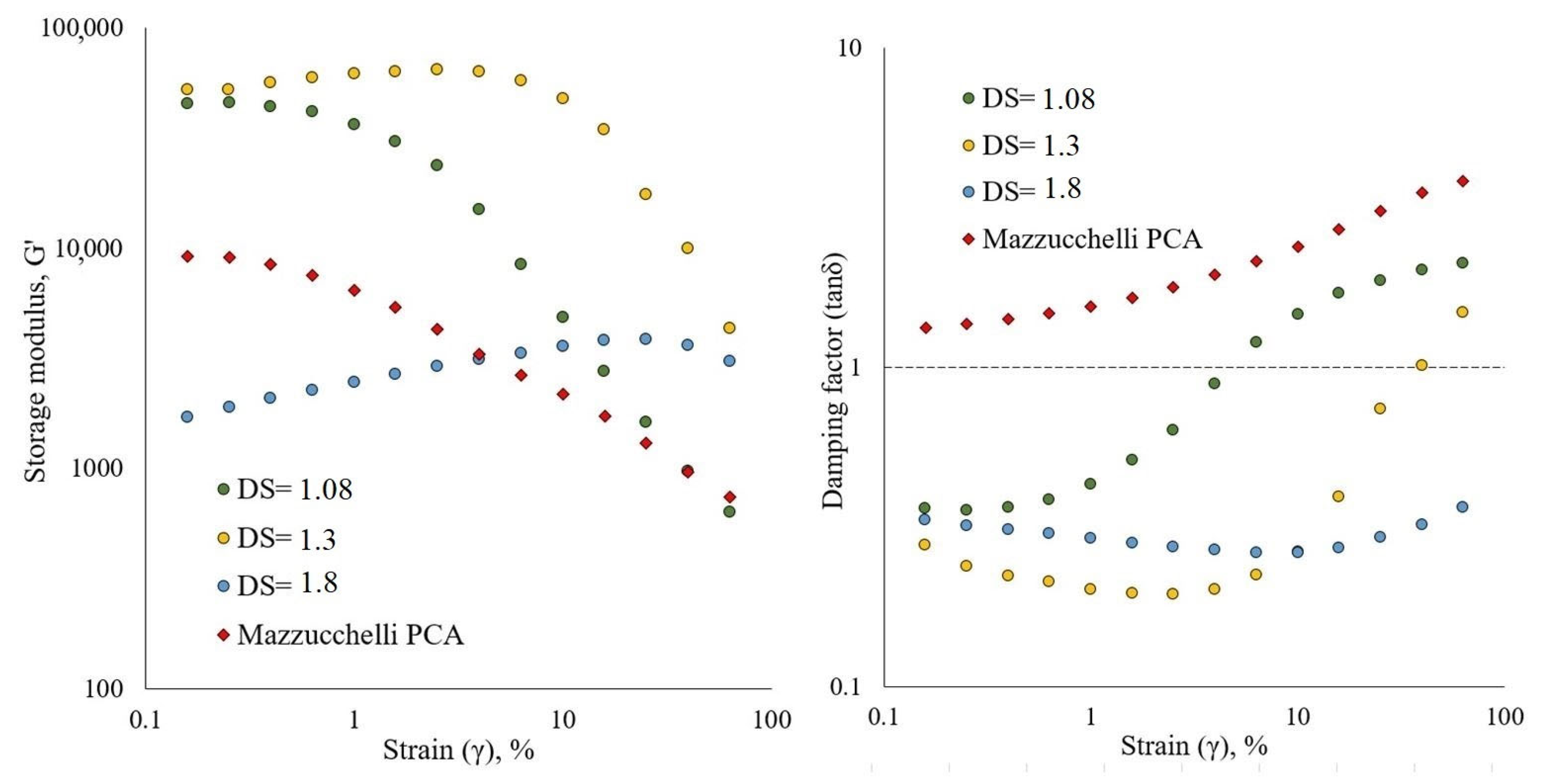
3.2. Physical Properties of Cellulose Esters
3.2.1. DSC
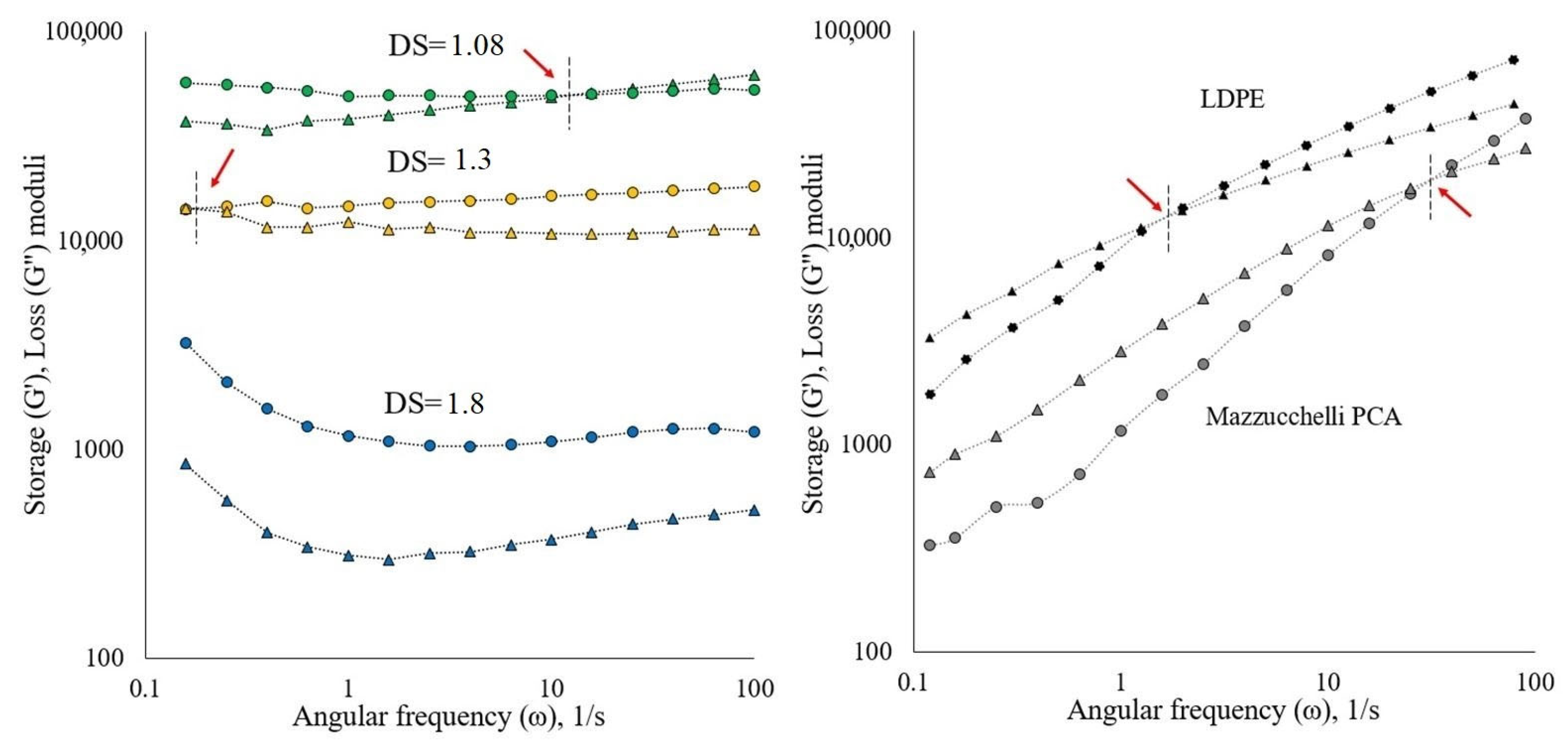
3.2.2. Rheology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qiu, X.; Hu, S. “Smart” Materials Based on Cellulose: A Review of the Preparations, Properties, and Applications. Materials 2013, 6, 738–781. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Li, J.; Zhu, S.; Chen, R.; Wu, Y.; Zhang, X.; Yu, Z. Potential applications of ionic liquids in wood related industries. Bioresources 2009, 4, 825–834. [Google Scholar] [CrossRef]
- Crépy, L.; Miri, V.; Joly, N.; Martin, P.; Lefebvre, J.-M. Effect of side chain length on structure and thermomechanical properties of fully substituted cellulose fatty esters. Carbohydr. Polym. 2011, 83, 1812–1820. [Google Scholar] [CrossRef]
- Heinze, T.; Dicke, R.; Koschella, A.; Kull, A.H.; Klohr, E.-A.; Koch, W. Effective preparation of cellulose derivatives in a new simple cellulose solvent. Macromol. Chem. Phys. 2000, 201, 627–631. [Google Scholar] [CrossRef]
- Young, R.J. Handbook of plastic materials and technology. In Polymer International; Rubin, I.I., Ed.; John Wiley & Sons Inc.: New York, NY, USA, 1991; Volume 25, p. 1745. ISBN 0-47-1-09634-2. [Google Scholar]
- Nawaz, H.; Zhang, J.; Tian, W.; Wu, J.; Zhang, J. Chemical Modification of Cellulose in Solvents for Functional Materials. In Green Chemistry and Chemical Engineering; Han, B., Wu, T., Eds.; Springer: New York, NY, USA, 2019; pp. 427–460. [Google Scholar]
- Zugenmaier, P. Materials of cellulose derivatives and fiber-reinforced cellulose-polypropylene composites: Characterization and application. Pure Appl. Chem. 2006, 78, 1843–1855. [Google Scholar] [CrossRef]
- Cumpstey, I. Chemical Modification of Polysaccharides. ISRN Org. Chem. 2013, 2013, 417672. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Belgacem, M.N.; Gandini, A. Controlled heterogeneous modification of cellulose fibers with fatty acids: Effect of reaction conditions on the extent of esterification and fiber properties. J. Appl. Polym. Sci. 2006, 100, 1093–1102. [Google Scholar] [CrossRef]
- Onwukamike, K.N.; Grelier, S.; Grau, E.; Cramail, H.; Meier, M.A.R. Sustainable Transesterification of Cellulose with High Oleic Sunflower Oil in a DBU-CO2 Switchable Solvent. ACS Sustain. Chem. Eng. 2018, 6, 8826–8835. [Google Scholar] [CrossRef]
- Barthel, S.; Heinze, T. Acylation and carbanilation of cellulose in ionic liquids. Green Chem. 2006, 8, 301–306. [Google Scholar] [CrossRef]
- Schenzel, A.; Hufendiek, A.; Barner-Kowollik, C.; Meier, M.A. Catalytic transesterification of cellulose in ionic liquids: Sustainable access to cellulose esters. Green Chem. 2014, 16, 3266–3271. [Google Scholar] [CrossRef]
- Antova, G.; Vasvasova, P.; Zlatanov, M. Studies upon the synthesis of cellulose stearate under microwave heating. Carbohydr. Polym. 2004, 57, 131–134. [Google Scholar] [CrossRef]
- Vicente, G.; Martínez, M.; Aracil, J. Integrated biodiesel production: A comparison of different homogeneous catalysts systems. Bioresour. Technol. 2004, 92, 297–305. [Google Scholar] [CrossRef]
- Cao, X.; Sun, S.; Peng, X.; Zhong, L.; Sun, R.; Jiang, D. Rapid Synthesis of Cellulose Esters by Transesterification of Cellulose with Vinyl Esters under the Catalysis of NaOH or KOH in DMSO. J. Agric. Food Chem. 2013, 61, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Gericke, M.; Fardim, P.; Heinze, T. Ionic Liquids—Promising but Challenging Solvents for Homogeneous Derivatization of Cellulose. Molecules 2012, 17, 7458–7502. [Google Scholar] [CrossRef] [PubMed]
- Fink, H.P.; Weigel, P.; Purz, H.J.; Ganster, J. Structure formation of regenerated cellulose materials from NMMO-solutions. Prog. Polym. Sci. 2001, 26, 1473–1524. [Google Scholar] [CrossRef]
- McCormick, C.L.; Dawsey, T.R. Preparation of Cellulose Derivatives via Ring-Opening Reactions with Cyclic Reagents in Lithium Chloride/N, N-Dimethylacetamide. Macromolecules 1990, 23, 3606–3610. [Google Scholar] [CrossRef]
- Heinze, T.; Liebert, T.; Koschella, A. Esterification of Polysaccharides, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Mäki-Arvela, P.; Anugwom, I.; Virtanen, P.; Sjöholm, R.; Mikkola, J.P. Dissolution of lignocellulosic materials and its constituents using ionic liquids—A review. Ind. Crops Prod. 2010, 32, 175–201. [Google Scholar] [CrossRef]
- Flieger, J.; Flieger, M. Ionic Liquids Toxicity-Benefits and Threats. Int. J. Mol. Sci. 2020, 21, 6267. [Google Scholar] [CrossRef] [PubMed]
- Isik, M.; Sardon, H.; Mecerreyes, D. Ionic Liquids and Cellulose: Dissolution, Chemical Modification and Preparation of New Cellulosic Materials. Int. J. Mol. Sci. 2014, 15, 11922–11940. [Google Scholar] [CrossRef]
- Ostonen, A.; Bervas, J.; Uusi-Kyyny, P.; Alopaeus, V.; Zaitsau, D.H.; Emel’yanenko, V.N.; Schick, C.; King, A.W.; Helminen, J.; Kilpelainen, I.; et al. Experimental and Theoretical Thermodynamic Study of Distillable Ionic Liquid 1,5-Diazabicyclo[4.3.0]non-5-enium Acetate. Ind. Eng. Chem. Res. 2016, 55, 10445–10454. [Google Scholar] [CrossRef]
- Köhler, S.; Liebert, T.; Schöbitz, M.; Schaller, J.; Meister, F.; Günther, W.; Heinze, T. Interactions of Ionic Liquids with Polysaccharides 1. Unexpected Acetylation of Cellulose with 1-Ethyl-3-ethylimidazolium Acetate. Macromol. Rapid Commun. 2007, 28, 2311–2317. [Google Scholar] [CrossRef]
- Hinner, L.P.; Wissner, J.L.; Beurer, A.; Nebel, B.A.; Hauer, B. Homogeneous vinyl ester-based synthesis of different cellulose derivatives in 1-ethyl-3-methyl-imidazolium acetate. Green Chem. 2016, 18, 6099–6107. [Google Scholar] [CrossRef]
- Szabó, L.; Milotskyi, R.; Sharma, G.; Takahashi, K. Cellulose processing in ionic liquids from a materials science perspective: Turning a versatile biopolymer into the cornerstone of our sustainable future. Green Chem. 2023, 25, 5338–5389. [Google Scholar] [CrossRef]
- Kostag, M.; Gericke, M.; Heinze, T.; El Seoud, O.A. Twenty-five years of cellulose chemistry: Innovations in the dissolution of the biopolymer and its transformation into esters and ethers. Cellulose 2019, 26, 139–184. [Google Scholar] [CrossRef]
- Xu, C.; Cheng, Z. Thermal Stability of Ionic Liquids: Current Status and Prospects for Future Development. Processes 2021, 9, 337. [Google Scholar] [CrossRef]
- Elsayed, S.; Hellsten, S.; Guizani, C.; Witos, J.; Rissanen, M.; Rantamäki, A.H.; Varis, P.; Wiedmer, S.K.; Sixta, H. Recycling of Superbase-Based Ionic Liquid Solvents for the Production of Textile-Grade Regenerated Cellulose Fibers in the Lyocell Process. ACS Sustain. Chem. Eng. 2020, 8, 14217–14227. [Google Scholar] [CrossRef]
- King, A.W.T.; Asikkala, J.; Mutikainen, I.; Järvi, P.; Kilpeläinen, I. Distillable Acid–Base Conjugate Ionic Liquids for Cellulose Dissolution and Processing. Angew. Chem. Int. Ed. 2011, 50, 6301–6305. [Google Scholar] [CrossRef]
- Martins, M.A.R.; Sosa, F.H.B.; Kilpeläinen, I.; Coutinho, J.A.P. Physico-chemical characterization of aqueous solutions of superbase ionic liquids with cellulose dissolution capability. Fluid Phase Equilibria 2022, 556, 113414. [Google Scholar] [CrossRef]
- Parviainen, A.; Wahlström, R.; Liimatainen, U.; Liitiä, T.; Rovio, S.; Helminen, J.K.J.; Hyväkkö, U.; King, A.W.T.; Suurnäkki, A.; Kilpeläinen, I. Sustainability of cellulose dissolution and regeneration in 1,5-diazabicyclo[4.3.0]non-5-enium acetate: A batch simulation of the IONCELL-F process. RSC Adv. 2015, 5, 69728–69737. [Google Scholar] [CrossRef]
- Elsayed, S.; Viard, B.; Guizani, C.; Hellstén, S.; Witos, J.; Sixta, H. Limitations of Cellulose Dissolution and Fiber Spinning in the Lyocell Process Using [mTBDH][OAc] and [DBNH][OAc] Solvents. Ind. Eng. Chem. Res. 2020, 59, 20211–20220. [Google Scholar] [CrossRef]
- Huang, F.Y. Synthesis and Properties of Cellulose Stearate. Adv. Mater. Res. 2011, 228–229, 919–924. [Google Scholar] [CrossRef]
- Crépy, L.; Chaveriat, L.; Banoub, J.; Martin, P.; Joly, N. Synthesis of Cellulose Fatty Esters as Plastics—Influence of the Degree of Substitution and the Fatty Chain Length on Mechanical Properties. ChemSusChem 2009, 2, 165–170. [Google Scholar] [CrossRef]
- Duchatel-Crépy, L.; Joly, N.; Martin, P.; Marin, A.; Tahon, J.-F.; Lefebvre, J.-M.; Gaucher, V. Substitution degree and fatty chain length influence on structure and properties of fatty acid cellulose esters. Carbohydr. Polym. 2020, 234, 115912. [Google Scholar] [CrossRef]
- Willberg-Keyriläinen, P.; Talja, R.; Asikainen, S.; Harlin, A.; Ropponen, J. The effect of cellulose molar mass on the properties of palmitate esters. Carbohydr. Polym. 2016, 151, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Baker, T.; Sacripante, G.G.; Lawton, D.J.W.; Marway, H.S.; Zhang, H.; Thompson, M.R. Solvent-free production of thermoplastic lignocellulose from wood pulp by reactive extrusion. Carbohydr. Polym. 2021, 270, 118361. [Google Scholar] [CrossRef]
- Virtanen, S.; Talja, R.; Vuoti, S. Synthesis and melt processing of cellulose esters for preparation of thermoforming materials and extended drug release tablets. Carbohydr. Polym. 2017, 177, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Todorov, A.R.; King, A.W.T.; Kilpeläinen, I. Transesterification of cellulose with unactivated esters in superbase–acid conjugate ionic liquids. RSC Adv. 2023, 13, 5983–5992. [Google Scholar] [CrossRef]
- Krasnou, I.; Gårdebjer, S.; Tarasova, E.; Larsson, A.; Westman, G.; Krumme, A. Permeability of water and oleic acid in composite films of phase separated polypropylene and cellulose stearate blends. Carbohydr. Polym. 2016, 152, 450–458. [Google Scholar] [CrossRef]
- Krasnou, I.; Tarasova, E.; Märtson, T.; Krumme, A. Thermoplastic Cellulose Stearate and Cellulose Laurate: Melt Rheology, Processing and Application Potential. Int. Polym. Process. 2015, 30, 210–216. [Google Scholar] [CrossRef]
- Lowman, D.W. Characterization of Cellulose Esters by Solution-State and Solid-State NMR Spectroscopy. In Cellulose Derivatives; American Chemical Society: Washington, DC, USA, 1998; Volume 688, pp. 131–162. [Google Scholar]
- ASTM D1795−13; Standard Test Method for Intrinsic Viscosity of Cellulose. ASTM International: West Conshohocken, PA, USA, 2013.
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Wen, X.; Wang, H.; Wei, Y.; Wang, X.; Liu, C. Preparation and characterization of cellulose laurate ester by catalyzed transesterification. Carbohydr. Polym. 2017, 168, 247–254. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.; Zhang, H.; He, J.; Ren, Q.; Guo, M. Homogeneous Acetylation of Cellulose in a New Ionic Liquid. Biomacromolecules 2004, 5, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xie, H.; Liu, E. Acylation of cellulose in reversible ionic liquids. Green Chem. 2014, 16, 3018–3023. [Google Scholar] [CrossRef]
- Kakko, T.; King, A.W.T.; Kilpeläinen, I. Homogenous esterification of cellulose pulp in [DBNH][OAc]. Cellulose 2017, 24, 5341–5354. [Google Scholar] [CrossRef]
- Chen, H.; Yang, F.; Du, J.; Xie, H.; Zhang, L.; Guo, Y.; Xu, Q.; Zheng, Q.; Li, N.; Liu, Y. Efficient transesterification reaction of cellulose with vinyl esters in DBU/DMSO/CO2 solvent system at low temperature. Cellulose 2018, 25, 6935–6945. [Google Scholar] [CrossRef]
- Xu, D.; Voiges, K.; Elder, T.; Mischnick, P.; Edgar, K.J. Regioselective Synthesis of Cellulose Ester Homopolymers. Biomacromolecules 2012, 13, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, G.; Glasser, W.G. Cellulose derivatives with low DS. I. A novel acylation system. Carbohydr. Polym. 1993, 22, 1–7. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IL:DMSO | DS | The Viscosity of Cellulose Solution at 60 °C, Pas |
|---|---|---|---|
| CL-1 | no DMSO | 0.70 | 2.56 |
| CL-2 | 3:1 | 0.60 | 1.10 |
| CL-3 | 1:1 | 0.60 | 0.75 |
| CL-4 | 1:2 | 0.38 | 0.47 |
| CL-5 | 1:3 | 0.35 | 0.40 |
| Sample | Reaction Conditions | DS | Mn, kDa | PDI | ||
|---|---|---|---|---|---|---|
| Time, h | Temperature, °C | AGU:VL | ||||
| CL-6 | 2 | 20 | 1:3 | 0 | insoluble | |
| CL-3 | 2 | 60 | 1:3 | 0.6 | 237.7 | 1.64 |
| CL-7 | 2 | 80 | 1:3 | 1.2 | 221.0 | 2.01 |
| CL-8 | 4 | 60 | 1:3 | 1.6 | 358.0 | 1.28 |
| CL-9 | 5 | 60 | 1:3 | 2.0 | 304.0 | 1.50 |
| CL-10 | 3 | 70 | 1:3 | 1.55 | 532.7 | 1.14 |
| CL-11 | 3 | 80 | 1:3 | 1.58 | 317.5 | 1.40 |
| Sample | Reaction Conditions | DS | Mn, kDa | PDI | ||
|---|---|---|---|---|---|---|
| Time, h | Temperature, °C | AGU:VE | ||||
| CL-10 | 3 | 70 | 1:3 | 1.55 | 532.7 | 1.14 |
| CM | 3 | 70 | 1:3 | 1.31 | 393.6 | 1.74 |
| CP-1 | 3 | 70 | 1:3 | 1.08 | 370.2 | 1.50 |
| CP-2 | 3 | 70 | 1:5 | 1.30 | 404.3 | 1.55 |
| CP-3 | 3 | 80 | 1:3 | 1.80 | 222.1 | 1.77 |
| CS-1 | 3 | 70 | 1:3 | 0.67 | insoluble | |
| CS-2 | 3 | 70 | 1:6 | 1.40 | 471.2 | 1.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarasova, E.; Savale, N.; Krasnou, I.; Kudrjašova, M.; Rjabovs, V.; Reile, I.; Vares, L.; Kallakas, H.; Kers, J.; Krumme, A. Preparation of Thermoplastic Cellulose Esters in [mTBNH][OAC] Ionic Liquid by Transesterification Reaction. Polymers 2023, 15, 3979. https://doi.org/10.3390/polym15193979
Tarasova E, Savale N, Krasnou I, Kudrjašova M, Rjabovs V, Reile I, Vares L, Kallakas H, Kers J, Krumme A. Preparation of Thermoplastic Cellulose Esters in [mTBNH][OAC] Ionic Liquid by Transesterification Reaction. Polymers. 2023; 15(19):3979. https://doi.org/10.3390/polym15193979
Chicago/Turabian StyleTarasova, Elvira, Nutan Savale, Illia Krasnou, Marina Kudrjašova, Vitalijs Rjabovs, Indrek Reile, Lauri Vares, Heikko Kallakas, Jaan Kers, and Andres Krumme. 2023. "Preparation of Thermoplastic Cellulose Esters in [mTBNH][OAC] Ionic Liquid by Transesterification Reaction" Polymers 15, no. 19: 3979. https://doi.org/10.3390/polym15193979
APA StyleTarasova, E., Savale, N., Krasnou, I., Kudrjašova, M., Rjabovs, V., Reile, I., Vares, L., Kallakas, H., Kers, J., & Krumme, A. (2023). Preparation of Thermoplastic Cellulose Esters in [mTBNH][OAC] Ionic Liquid by Transesterification Reaction. Polymers, 15(19), 3979. https://doi.org/10.3390/polym15193979