
Chemistry of Hydroxypropyl Cellulose Oxidized by Two Selective Oxidants

 , , ,
, , ,  and
and 
Abstract
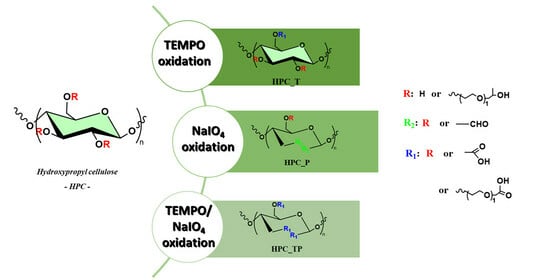
:
1. Introduction
2. Materials and Methods
2.1. Materials
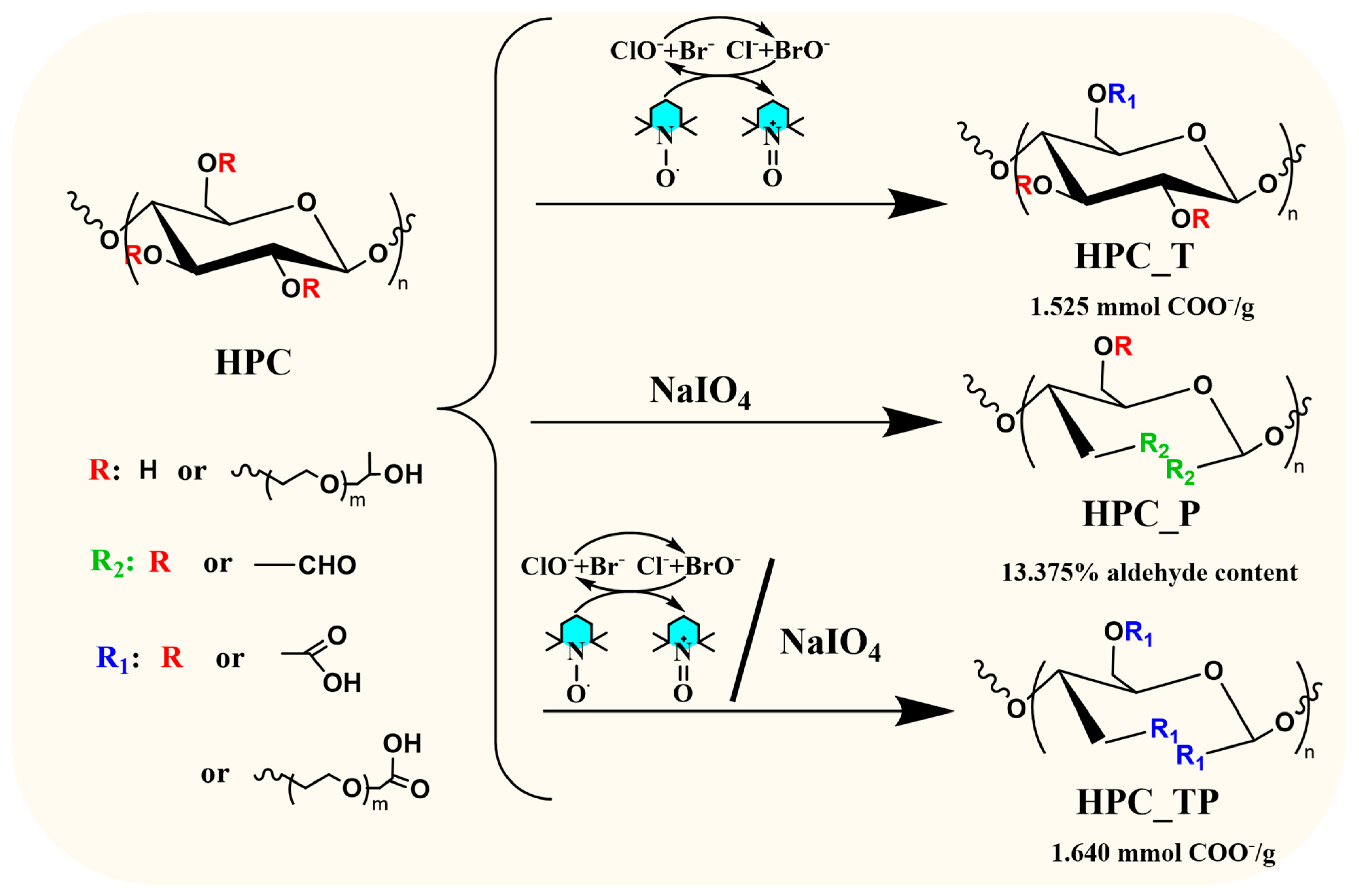
2.2. Preparation of Oxidized HPC
2.3. Characterization of Oxidized HPC
2.3.1. The Fourier-Transform Infrared (FTIR) Measurements
2.3.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.3.3. Environmental Scanning Electron Microscopy (SEM)
2.3.4. Crystallinity Determination using X-ray Diffraction (XRD)
2.3.5. Zeta Potential
2.3.6. Determination of Mass Yield
2.3.7. Conductometric and Potentiometric Titration
Determination of Carboxyl-Group Content in HPC_T and HPC_TP
Determination of Aldehyde-Group Content in HPC_P
2.3.8. Thermogravimetric Measurements
3. Results and Discussion
3.1. Mechanism of Reaction
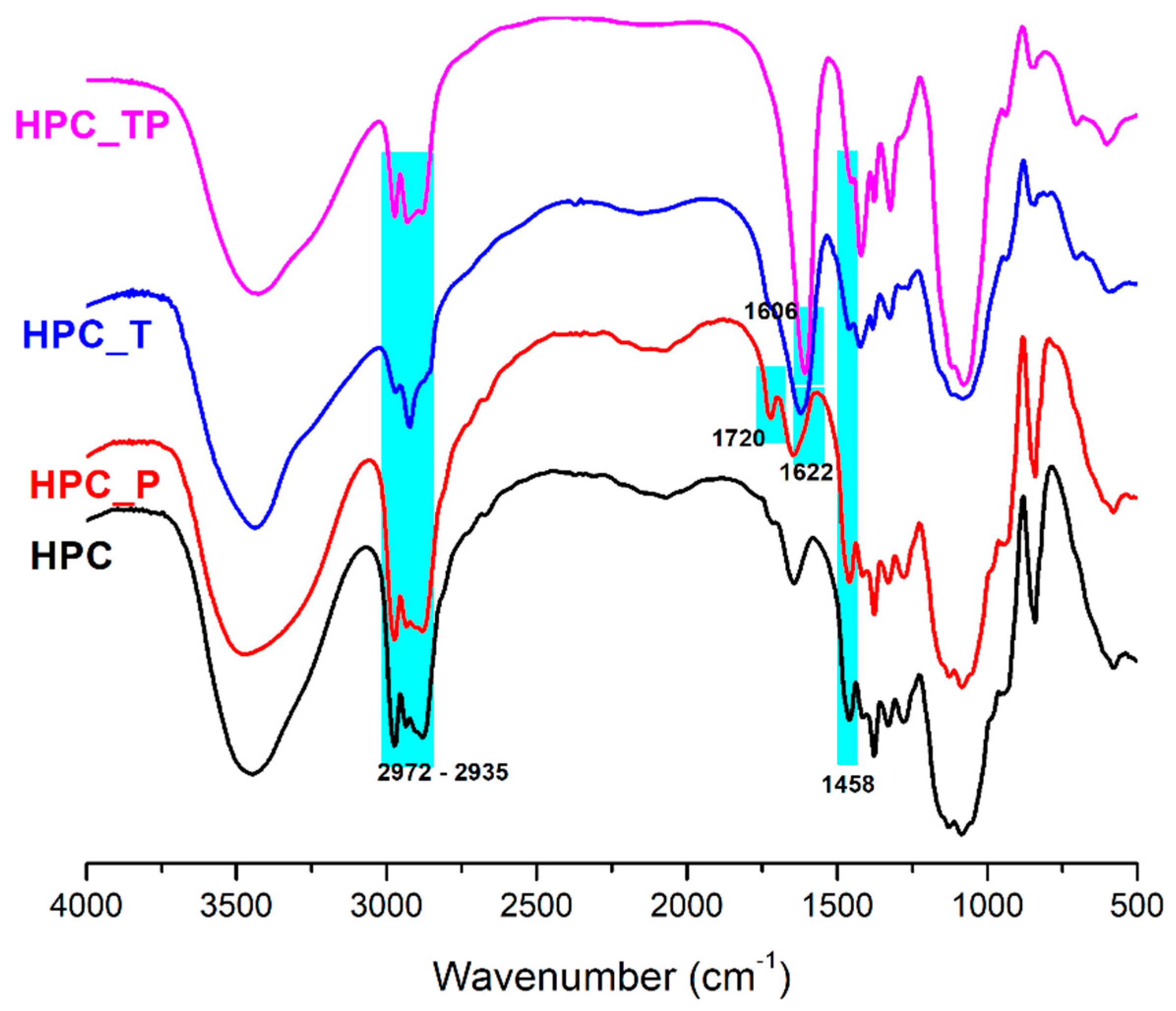
3.2. FTIR
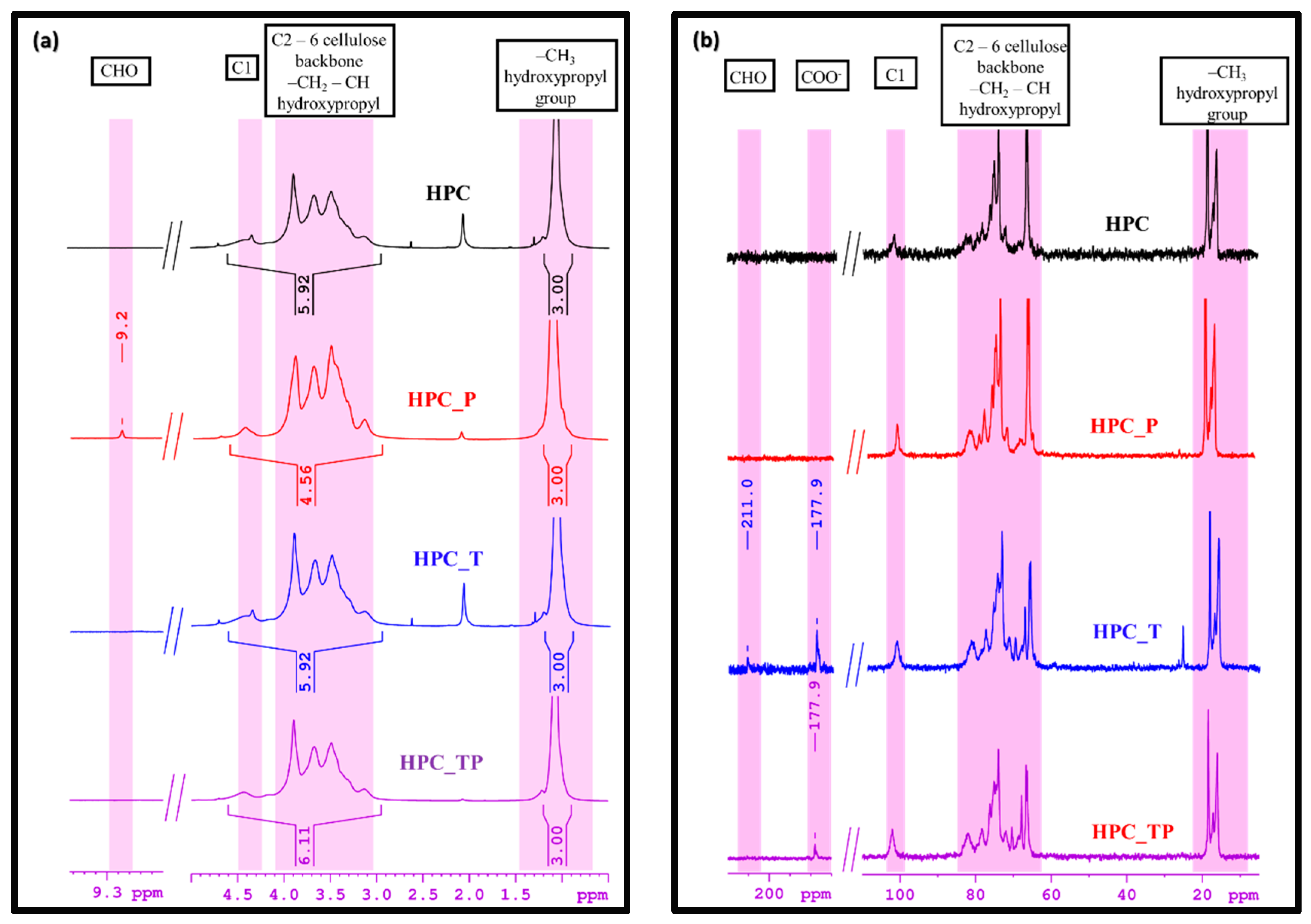
3.3. NMR
3.4. SEM
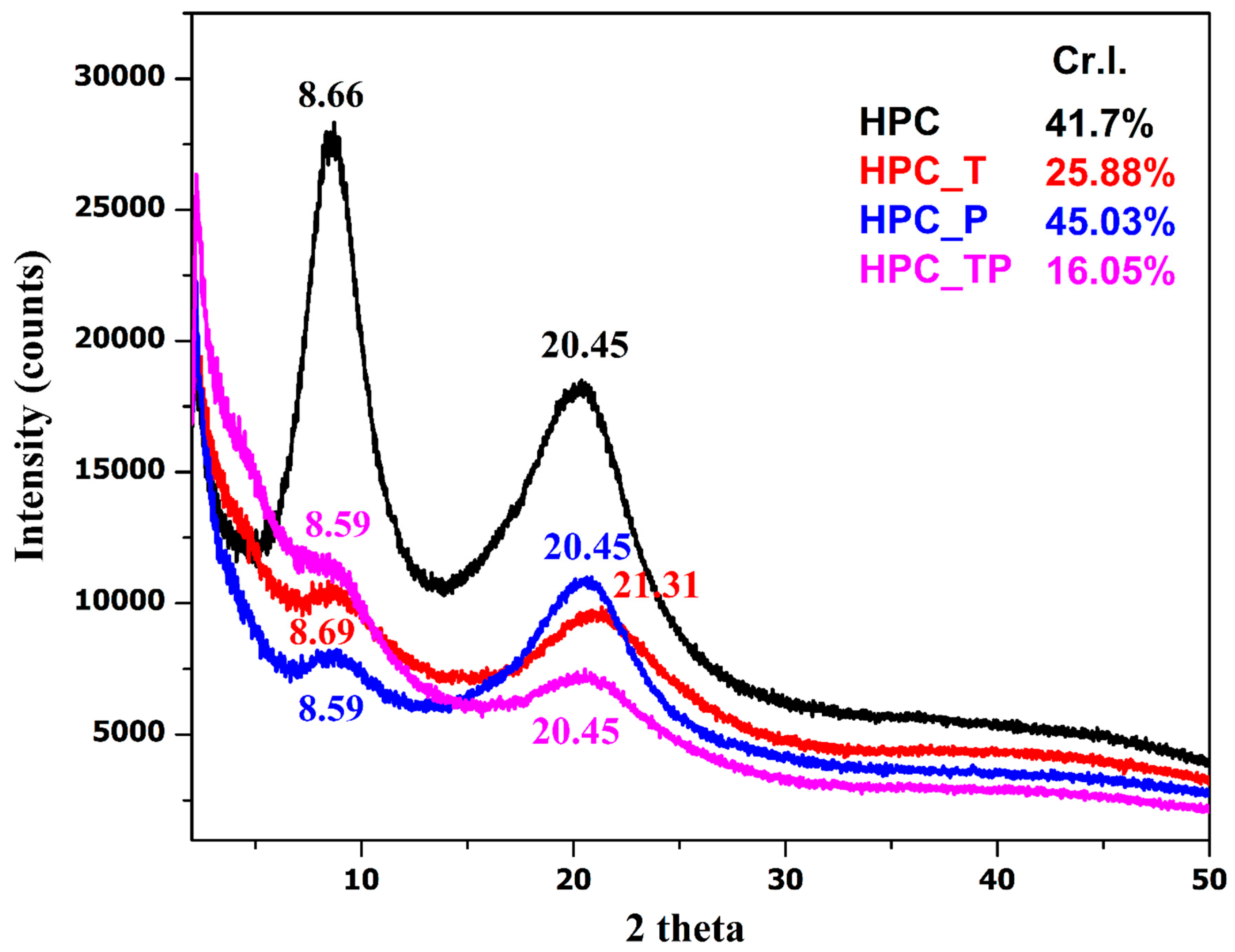
3.5. XRD
3.6. The Zeta Potential and the Quantity of the Carboxyl Content
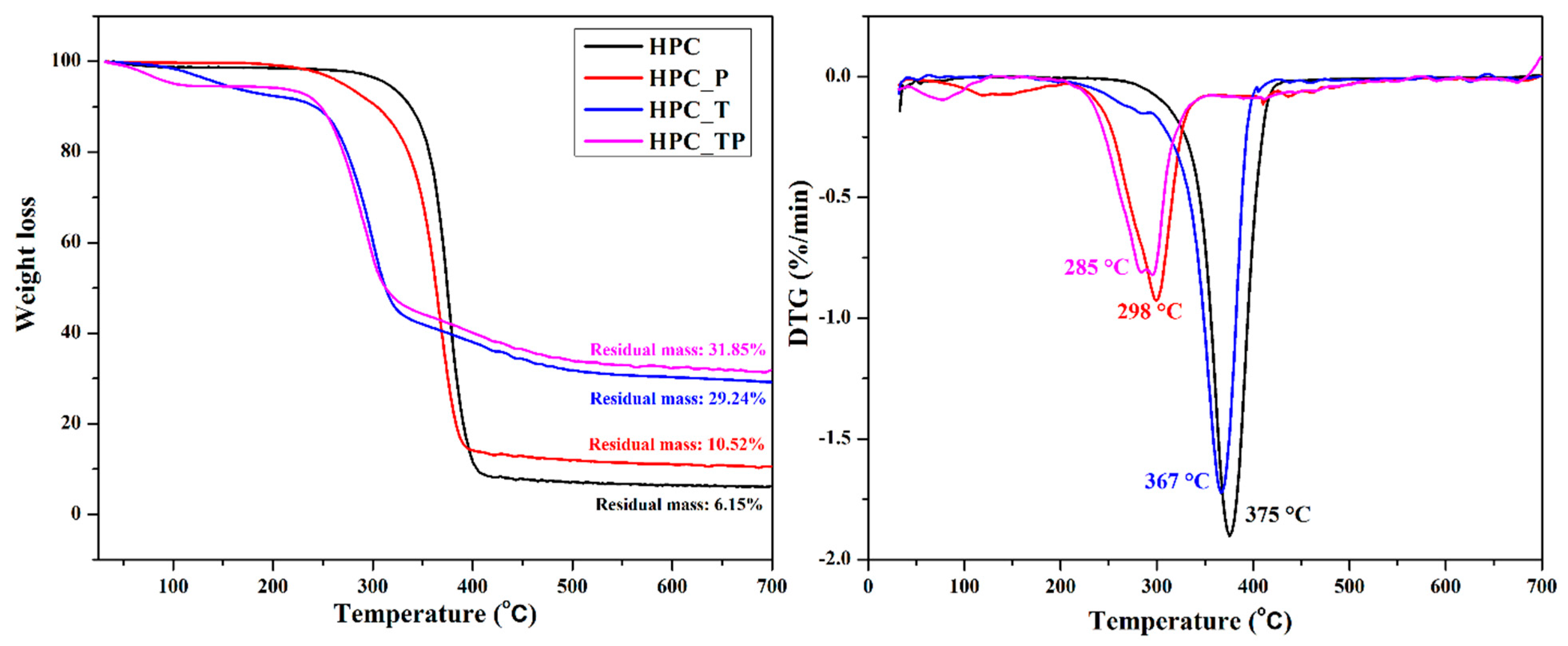
3.7. Thermal Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cooke, C.L.; An, H.J.; Kim, J.; Solnick, J.V.; Lebrilla, C.B. Method for Profiling Mucin Oligosaccharides from Gastric Biopsies of Rhesus Monkeys with and without Helicobacter Pylori Infection. Anal. Chem. 2007, 79, 8090–8097. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.H.; Bertozzi, C.R. Glycans in Cancer and Inflammation—Potential for Therapeutics and Diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ahmed, S.; Sameen, D.E.; Wang, Y.; Lu, R.; Dai, J.; Li, S.; Qin, W. A review of cellulose and its derivatives in biopolymer-based for food packaging application. Trends Food Sci. Technol. 2021, 112, 532–546. [Google Scholar] [CrossRef]
- Das, A.; Ringu, T.; Ghosh, S.; Pramanik, N. A Comprehensive Review on Recent Advances in Preparation, Physicochemical Characterization, and Bioengineering Applications of Biopolymers. Polym. Bull. 2023, 80, 7247–7312. [Google Scholar] [CrossRef]
- Seddiqi, H.; Oliaei, E.; Honarkar, H.; Jin, J.; Geonzon, L.C.; Bacabac, R.G.; Klein-Nulend, J. Cellulose and Its Derivatives: Towards Biomedical Applications. Cellulose 2021, 28, 1893–1931. [Google Scholar] [CrossRef]
- Joseph, B.; Sagarika, V.K.; Sabu, C.; Kalarikkal, N.; Thomas, S. Cellulose Nanocomposites: Fabrication and Biomedical Applications. J. Bioresour. Bioprod. 2020, 5, 223–237. [Google Scholar] [CrossRef]
- Cremer, G.; Danthine, S.; Van Hoed, V.; Dombree, A.; Laveaux, A.-S.; Damblon, C.; Karoui, R.; Blecker, C. Variability in the Substitution Pattern of Hydroxypropyl Cellulose Affects Its Physico-Chemical Properties. Heliyon 2023, 9, e13604. [Google Scholar] [CrossRef]
- Espinha, A.; Dore, C.; Matricardi, C.; Alonso, M.I.; Goñi, A.R.; Mihi, A. Hydroxypropyl Cellulose Photonic Architectures by Soft Nanoimprinting Lithography. Nat. Photonics 2018, 12, 343–348. [Google Scholar] [CrossRef]
- Arca, H.C.; Mosquera-Giraldo, L.I.; Bi, V.; Xu, D.; Taylor, L.S.; Edgar, K.J. Pharmaceutical Applications of Cellulose Ethers and Cellulose Ether Esters. Biomacromolecules 2018, 19, 2351–2376. [Google Scholar] [CrossRef]
- Nichols, B.L.B.; Chen, J.; Mischnick, P.; Edgar, K.J. Selective Oxidation of 2-Hydroxypropyl Ethers of Cellulose and Dextran: Simple and Efficient Introduction of Versatile Ketone Groups to Polysaccharides. Biomacromolecules 2020, 21, 4835–4849. [Google Scholar] [CrossRef]
- Basta, A.H.; Lotfy, V.F.; Micky, J.A.; Salem, A.M. Hydroxypropylcellulose-Based Liquid Crystal Materials. Carbohydr. Polym. Technol. Appl. 2021, 2, 100103. [Google Scholar] [CrossRef]
- Godinho, M.H.; Gray, D.G.; Pieranski, P. Revisiting (Hydroxypropyl) Cellulose (HPC)/Water Liquid Crystalline System. Liq. Cryst. 2017, 44, 2108–2120. [Google Scholar] [CrossRef]
- Seelinger, D.; Trosien, S.; Nau, M.; Biesalski, M. Tailored Oxidation of Hydroxypropyl Cellulose under Mild Conditions for the Generation of Wet Strength Agents for Paper. Carbohydr. Polym. 2021, 254, 117458. [Google Scholar] [CrossRef] [PubMed]
- Dragan, E.S.; Ghiorghita, C.A.; Dinu, M.V.; Duceac, I.A.; Coseri, S. Fabrication of Self-Antibacterial Chitosan/Oxidized Starch Polyelectrolyte Complex Sponges for Controlled Delivery of Curcumin. Food Hydrocoll. 2023, 135, 108147. [Google Scholar] [CrossRef]
- Baron, R.I.; Coseri, S. Preparation of Water-Soluble Cellulose Derivatives Using TEMPO Radical-Mediated Oxidation at Extended Reaction Time. React. Funct. Polym. 2020, 157, 104768. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose Nanofibers Prepared by TEMPO-Mediated Oxidation of Native Cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Coseri, S. Cellulose: To Depolymerize…or Not to? Biotechnol. Adv. 2017, 35, 251–266. [Google Scholar] [CrossRef]
- Biliuta, G.; Sacarescu, L.; Socoliuc, V.; Iacob, M.; Gheorghe, L.; Negru, D.; Coseri, S. Carboxylated Polysaccharides Decorated with Ultrasmall Magnetic Nanoparticles with Antibacterial and MRI Properties. Macromol. Chem. Phys. 2017, 218, 1700062. [Google Scholar] [CrossRef]
- Isogai, A.; Kato, Y. Preparation of Polyuronic Acid from Cellulose by TEMPO-Mediated Oxidation. Cellulose 1998, 4, 153–164. [Google Scholar] [CrossRef]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-Oxidized Cellulose Nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Shibata, I.; Isogai, A. Depolymerization of Cellouronic Acid during TEMPO-Mediated Oxidation. Cellulose 2003, 10, 151–158. [Google Scholar] [CrossRef]
- Okita, Y.; Saito, T.; Isogai, A. Entire Surface Oxidation of Various Cellulose Microfibrils by TEMPO-Mediated Oxidation. Biomacromolecules 2010, 11, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, D.J.; Browne, C.; Raghuwanshi, V.S.; Simon, G.P.; Garnier, G. One-Shot TEMPO-Periodate Oxidation of Native Cellulose. Carbohydr. Polym. 2019, 226, 115292. [Google Scholar] [CrossRef] [PubMed]
- Culica, M.E.; Rotaru, R.; Bejan, D.; Coroaba, A.; Mohan, T.; Coseri, S. Cellulose Surface Modification for Improved Attachment of Carbon Nanotubes. Cellulose 2022, 29, 6057–6076. [Google Scholar] [CrossRef]
- Baron, R.I.; Bercea, M.; Avadanei, M.; Lisa, G.; Biliuta, G.; Coseri, S. Green Route for the Fabrication of Self-Healable Hydrogels Based on Tricarboxy Cellulose and Poly(Vinyl Alcohol). Int. J. Biol. Macromol. 2019, 123, 744–751. [Google Scholar] [CrossRef]
- Ho, F.F.L.; Kohler, R.R.; Ward, G.A. Determination of Molar Substitution and Degree of Substitution of Hydroxypropyl Cellulose by Nuclear Magnetic Resonance Spectrometry. Anal. Chem. 1972, 44, 178–181. [Google Scholar] [CrossRef]
- Yang, X.J.; Zhi, Z.I.; Lu, L.D.; Wang, X. Determination of the Molar Substitution of Hydroxypropyl Celluloses by an NMR Method. Spectrosc. Lett. 2006, 31, 1279–1285. [Google Scholar] [CrossRef]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conrad, C.M. An Empirical Method for Estimating the Degree of Crystallinity of Native Cellulose Using the X-Ray Diffractometer. Text. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, M.Y.; Nie, S.P.; Li, C.; Wang, Y.X. Purification, Composition Analysis and Antioxidant Activity of a Polysaccharide from the Fruiting Bodies of Ganoderma Atrum. Food Chem. 2008, 107, 231–241. [Google Scholar] [CrossRef]
- Zai, Z.; Yan, M.; Shi, C.; Zhang, L.; Lu, H.; Xiong, Z.; Ma, J. Cellulose Nanofibrils (CNFs) in Uniform Diameter: Capturing the Impact of Carboxyl Group on Dispersion and Re-Dispersion of CNFs Suspensions. Int. J. Biol. Macromol. 2022, 207, 23–30. [Google Scholar] [CrossRef]
- Lai, C.; Zhang, S.; Sheng, L.; Liao, S.; Xi, T.; Zhang, Z. TEMPO-Mediated Oxidation of Bacterial Cellulose in a Bromide-Free System. Colloid. Polym. Sci. 2013, 291, 2985–2992. [Google Scholar] [CrossRef]
- Chen, J.; Edgar, K.J.; Frazier, C.E. Photo-curable, double-crosslinked, in situ-forming hydrogels based on oxidized hydroxypropyl cellulose. Cellulose 2021, 28, 3903–3915. [Google Scholar] [CrossRef]
- Chang, C.; Zhang, L. Cellulose-based hydrogels: Present status and application prospects. Carbohydr. Polym. 2011, 84, 40–53. [Google Scholar] [CrossRef]
- Coseri, S. A New and Efficient Heterogeneous System for the PhthalimideN-Oxyl (PINO) Radical Generation. Eur. J. Org. Chem. 2007, 2007, 1725–1729. [Google Scholar] [CrossRef]
- Biliuta, G.; Fras, L.; Harabagiu, V.; Coseri, S. Mild Oxidation of Cellulose Fibers Using Dioxygen as Ultimate Oxidizing Agent. Dig. J. Nanomater. Biostruct. 2011, 6, 293–299. [Google Scholar]
- Calvini, P.; Gorassini, A.; Luciano, G.; Franceschi, E. FTIR and WAXS Analysis of Periodate Oxycellulose: Evidence for a Cluster Mechanism of Oxidation. Vib. Spectrosc. 2006, 40, 177–183. [Google Scholar] [CrossRef]
- de Nooy, A.E.J.; Besemer, A.C.; van Bekkum, H.; van Dijk, J.A.P.P.; Smit, J.A.M. TEMPO-Mediated Oxidation of Pullulan and Influence of Ionic Strength and Linear Charge Density on the Dimensions of the Obtained Polyelectrolyte Chains. Macromolecules 1996, 29, 6541–6547. [Google Scholar] [CrossRef]
- Okada, T.; Asawa, T.; Sugiyama, Y.; Kirihara, M.; Iwai, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate (NaOCl·5H2O) Crystals as an Extra ordinary Oxidant for Primary and Secondary Alcohols. Synlett 2014, 25, 596–598. [Google Scholar] [CrossRef]
- Yixue, S.; Bin, C.; Yuan, G.; Chaoxi, W.; Lingmin, Z.; Peng, C.; Xiaoying, W.; Shunqing, T. Modification of Agarose with Carboxylation and Grafting Dopamine for Promotion of Its Cell-Adhesiveness. Carbohydr. Polym. 2013, 92, 2245–2251. [Google Scholar] [CrossRef]
- Sharma, R.; Varshney, V.K.; Chauhan, G.S.; Naithani, S.; Soni, P.L. Hydroxypropylation of Cellulose Isolated from Bamboo (Dendrocalamus Strictus) with Respect to Hydroxypropoxyl Content and Rheological Behavior of the Hydroxypropyl Cellulose. J. Appl. Polym. Sci. 2009, 113, 2450–2455. [Google Scholar] [CrossRef]
- Abdel-Halim, E.S.; Alanazi, H.H.; Al-Deyab, S.S. Utilization of Olive Tree Branch Cellulose in Synthesis of Hydroxypropyl Carboxymethyl Cellulose. Carbohydr. Polym. 2015, 127, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Kaldéus, T.; Larsson, P.T.; Boujemaoui, A.; Malmström, E. One-Pot Preparation of Bi-Functional Cellulose Nanofibrils. Cellulose 2018, 25, 7031–7042. [Google Scholar] [CrossRef]
- Zaltariov, M.F.; Filip, D.; Macocinschi, D.; Spiridon, I. Hydrohypropyl cellulose/polyurethane blends. The behavior after accelerated ageing. A ftir study. Cellul. Chem. Technol. 2020, 54, 903–914. [Google Scholar] [CrossRef]
- Rafidison, B.H.; Ramasawmy, H.; Chummun, J.; Florens, F.B.V. Using Infrared Spectrum Analyses to Predict Tensile Strength of Fibres in a Group of Closely Related Plant Species: Case of Mascarenes Pandanus spp. SN Appl. Sci. 2020, 2, 1922. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR Analysis of Cellulose Treated with Sodium Hydroxide and Carbon Dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef]
- Åkerholm, M.; Hinterstoisser, B.; Salmén, L. Characterization of the Crystalline Structure of Cellulose Using Static and Dynamic FT-IR Spectroscopy. Carbohydr. Res. 2004, 339, 569–578. [Google Scholar] [CrossRef]
- Smith, H.M.; Walker, L.P. Enzymatic Transformations of Cellulose Assessed by Quantitative High-Throughput Fourier Transform Infrared Spectroscopy (QHT-FTIR). Biotechnol. Bioeng. 2011, 108, 1509–1520. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Zhang, S.; Gao, Q.; Lu, Q.; Peng, R.; Xu, P.; Shang, H.; Yuan, Y.; Zou, H. Micro-FTIR Combined with Curve Fitting Method to Study Cellulose Crystallinity of Developing Cotton Fibers. Anal. Bioanal. Chem. 2021, 413, 1313–1320. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A. IR Study on Cellulose with the Varied Moisture Contents: Insight into the Supramolecular Structure. Materials 2020, 13, 4573. [Google Scholar] [CrossRef]
- Poletto, M.; Ornaghi Júnior, H.L.; Zattera, A.J.; Properties, T. Native Cellulose: Structure, Characterization and Thermal Properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef]
- Kimura, K.; Shigemura, T.; Kubo, M.; Maru, Y. 13C NMR Study of O-(2-Hydroxypropyl)Cellulose. Die Makromol. Chem. 1985, 186, 61–70. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, C.; Qin, Z.; Chen, G.; Xia, L.; Zhong, B. Preparation of Cauliflower Shaped Hp-Co/GNs Composite Microwave Absorbing Materials. Mater. Charact. 2022, 189, 111907. [Google Scholar] [CrossRef]
- Chan, C.L.C.; Bay, M.M.; Jacucci, G.; Vadrucci, R.; Williams, C.A.; van de Kerkhof, G.T.; Parker, R.M.; Vynck, K.; Frka-Petesic, B.; Vignolini, S. Visual Appearance of Chiral Nematic Cellulose-Based Photonic Films: Angular and Polarization Independent Color Response with a Twist. Adv. Mater. 2019, 31, 1905151. [Google Scholar] [CrossRef] [PubMed]
- Plappert, S.F.; Quraishi, S.; Pircher, N.; Mikkonen, K.S.; Veigel, S.; Klinger, K.M.; Potthast, A.; Rosenau, T.; Liebner, F.W. Transparent, Flexible, and Strong 2,3-Dialdehyde Cellulose Films with High Oxygen Barrier Properties. Biomacromolecules 2018, 19, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Barzic, A.I.; Soroceanu, M.; Rotaru, R.; Doroftei, F.; Asandulesa, M.; Tugui, C.; Dascalu, I.A.; Harabagiu, V. Cellulose derivative/barium titanate composites with high refractive index, conductivity and energy density. Cellulose 2022, 29, 863–878. [Google Scholar] [CrossRef]
- Echeverria, C.; Almeida, P.L.; Feio, G.; Figueirinhas, J.L.; Godinho, M.H. A Cellulosic Liquid Crystal Pool for Cellulose Nanocrystals: Structure and Molecular Dynamics at High Shear Rates. Eur. Polym. J. 2015, 72, 72–81. [Google Scholar] [CrossRef]
- Cheng, F.; Liu, C.; Wei, X.; Yan, T.; Li, H.; He, J.; Huang, Y. Preparation and Characterization of 2,2,6,6-Tetramethylpiperidine-1-Oxyl (TEMPO)-Oxidized Cellulose Nanocrystal/Alginate Biodegradable Composite Dressing for Hemostasis Applications. ACS Sustain. Chem. Eng. 2017, 5, 3819–3828. [Google Scholar] [CrossRef]
- Reischl, M.; Stana-Kleinschek, K.; Ribitsch, V. Electrokinetic Investigations of Oriented Cellulose Polymers. Macromol. Symp. 2006, 244, 31–47. [Google Scholar] [CrossRef]
- Sharma, P.R.; Varma, A.J. Functionalized Celluloses and Their Nanoparticles: Morphology, Thermal Properties, and Solubility Studies. Carbohydr. Polym. 2014, 104, 135–142. [Google Scholar] [CrossRef]
- do Nascimento, E.S.; Pereira, A.L.S.; Barros, M.d.O.; Barroso, M.K.d.A.; Lima, H.L.S.; Borges, M.d.F.; Feitosa, J.P.d.A.; de Azeredo, H.M.C.; Rosa, M.d.F. TEMPO Oxidation and High-Speed Blending as a Combined Approach to Disassemble Bacterial Cellulose. Cellulose 2019, 26, 2291–2302. [Google Scholar] [CrossRef]
- Sirvio, J.; Hyvakko, U.; Liimatainen, H.; Niinimaki, J.; Hormi, O. Periodate oxidation of cellulose at elevated temperatures using metal salts as cellulose activators. Carbohydr. Polym. 2011, 83, 1293–1297. [Google Scholar]
- Larsson, P.A.; Gimåker, M.; Wågberg, L. The influence of periodate oxidation on the moisture sorptivity and dimensional stability of paper. Cellulose 2008, 15, 837–847. [Google Scholar] [CrossRef]
- Wang, X.; Fang, G.; Hu, C.; Du, T. Application of Ultrasonic Waves in Activation of Microcrystalline Cellulose. J. Appl. Polym. Sci. 2008, 109, 2762–2767. [Google Scholar] [CrossRef]
- Pommerening, K.; Rein, H.; Bertram, D.; Muller, R. Estimation of dialdehyde groups in 2,3-dialdehyde bead-cellulose. Carbohydr. Polym. 1992, 233, 219–223. [Google Scholar] [CrossRef]
- Hofreiter, B.T.; Alexander, B.H.; Wolff, J.A. Rapid estimation of dialdehyde content of periodate oxystarch through quantitative alkali consumption. Anal. Chem. 1955, 27, 1930–1931. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TCI (A1377/A2879) | LOI (A1413/A943) | HBI (A3444/A1328) (%) |
|---|---|---|---|
| HPC | 1.32 | 0.39 | 9.2 |
| HPC_T | 1.45 | 1.15 | 6.6 |
| HPC_P | 1.30 | 0.39 | 9.3 |
| HPC_TP | 1.16 | 1.18 | 7.0 |
| Sample | a MS | b DS | c HC % |
|---|---|---|---|
| HPC | 4.93 | 2.49 | 63.8 |
| HPC_T | 2.40 | 1.55 | 45.59 |
| HPC_P | 4.49 | 1.75 | 46.18 |
| HPC_TP | 2.25 | 0.84 | 44.61 |
| Sample | a COO− Content (mmol/g HPC) | b Aldehyde Content (%) | c Mass Yield (%) | d Zeta Potential (ζ, mV) |
|---|---|---|---|---|
| HPC | - | - | - | −8.26 |
| HPC_T | 1.525 | - | 95 | −24.4 |
| HPC_P | 0.05 | 13.375 | 88 | −14.5 |
| HPC_PT | 1.640 | - | 70 | −26.3 |
| Sample | Samples Mass (mg) | W% | Total Weight Loss at 700 °C (Residue, %) | T Onset (°C) | T Endset (°C) | T Peak (DTG/°C) |
|---|---|---|---|---|---|---|
| HPC | 15.42 | 75.11 | 6.15 | 356.8 | 389.2 | 375 |
| HPC_P | 8.23 | 68.82 | 29.24 | 344.6 | 382.8 | 298 |
| HPC_T | 8.36 | 44.25 | 10.52 | 272.3 | 315.7 | 367 |
| HPC_TP | 8.11 | 45.31 | 31.85 | 255.2 | 311.5 | 285 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baron, R.I.; Biliuta, G.; Macsim, A.-M.; Dinu, M.V.; Coseri, S. Chemistry of Hydroxypropyl Cellulose Oxidized by Two Selective Oxidants. Polymers 2023, 15, 3930. https://doi.org/10.3390/polym15193930
Baron RI, Biliuta G, Macsim A-M, Dinu MV, Coseri S. Chemistry of Hydroxypropyl Cellulose Oxidized by Two Selective Oxidants. Polymers. 2023; 15(19):3930. https://doi.org/10.3390/polym15193930
Chicago/Turabian StyleBaron, Raluca Ioana, Gabriela Biliuta, Ana-Maria Macsim, Maria Valentina Dinu, and Sergiu Coseri. 2023. "Chemistry of Hydroxypropyl Cellulose Oxidized by Two Selective Oxidants" Polymers 15, no. 19: 3930. https://doi.org/10.3390/polym15193930
APA StyleBaron, R. I., Biliuta, G., Macsim, A.-M., Dinu, M. V., & Coseri, S. (2023). Chemistry of Hydroxypropyl Cellulose Oxidized by Two Selective Oxidants. Polymers, 15(19), 3930. https://doi.org/10.3390/polym15193930