

Synthesis and Characterization of ABA-Type Triblock Copolymers Using Novel Bifunctional PS, PMMA, and PCL Macroinitiators Bearing p-xylene-bis(2-mercaptoethyloxy) Core

Abstract
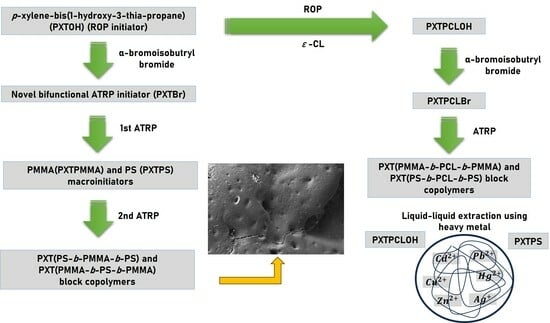
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Measurement
2.3. Synthesis of p-xylene-bis(1-hydroxy-3-thia-propane) (PXTOH)
2.4. Synthesis of 1,4-phenylenebis(methylenethioethane-2,1-diyl)bis(2-bromo-2-methylpropanoate) (PXTBr)
2.5. Synthesis of PXTPMMA and PXTPS via ATRP
2.6. Synthesis of PXTPCLOH and PXTPCLBr
2.7. The Synthesis of Block Copolymers
2.8. Measurement of the Heavy–Metal–Binding Properties of the PCL and PS
3. Results and Discussion
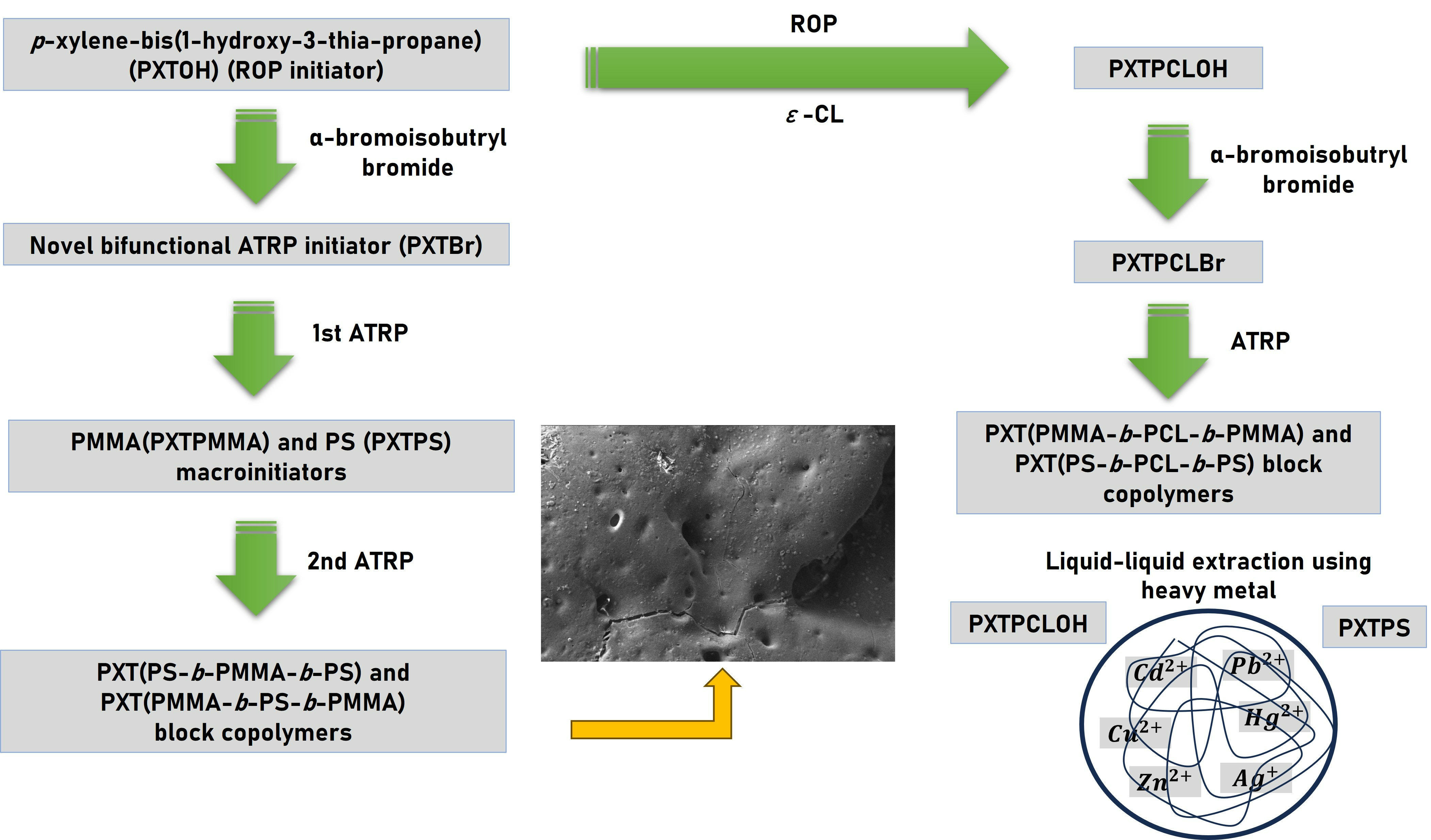
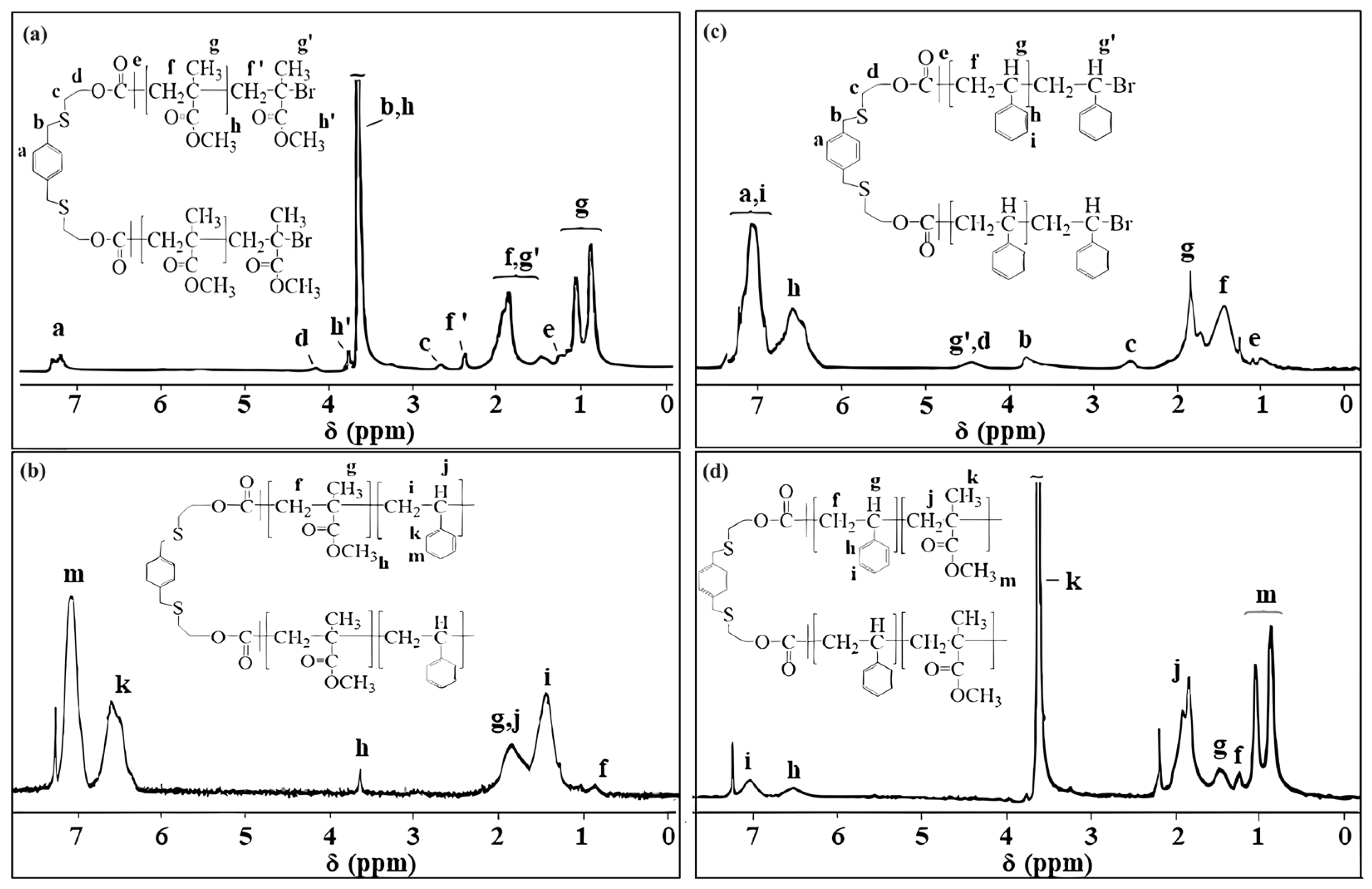
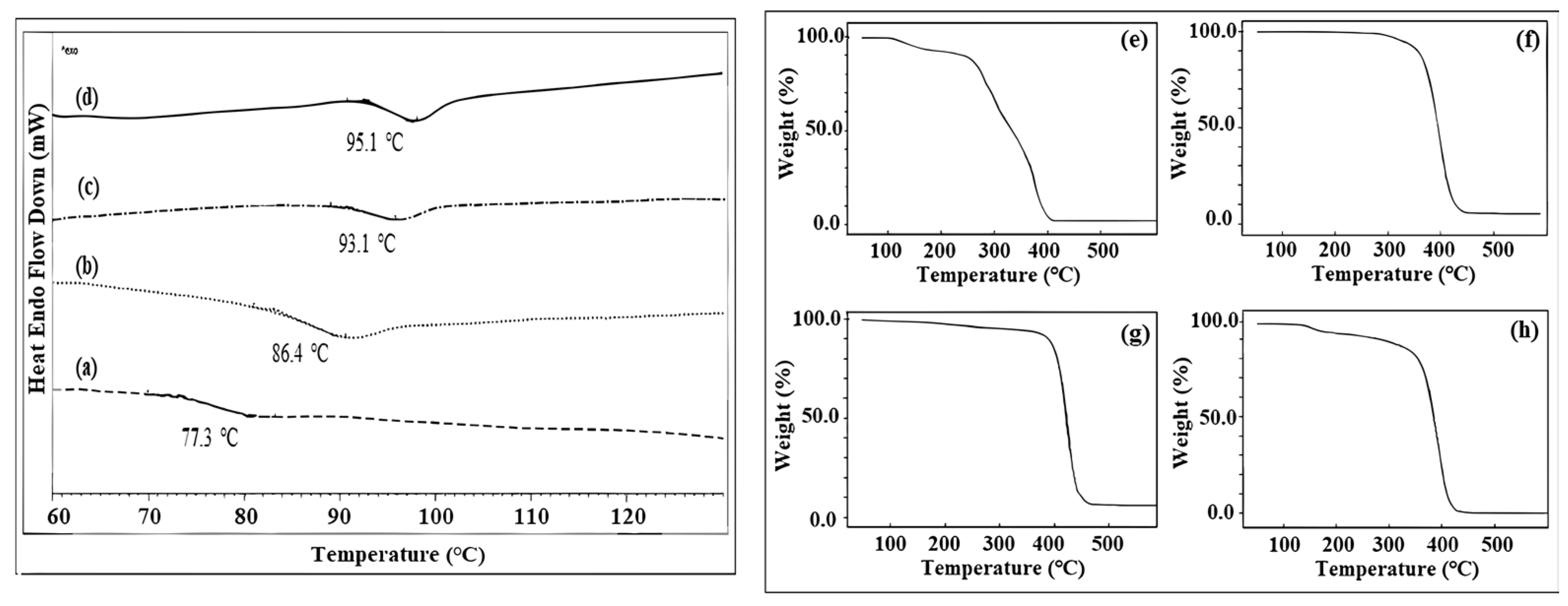
3.1. The Characterization of PXTBr (ATRP Initiator)
3.2. The Characterization of Macroinitiators (PXTPMMA and PXTPS) and Their Block Copolymers
3.3. The Characterization of PXTPCLOH and PXTPCLBr
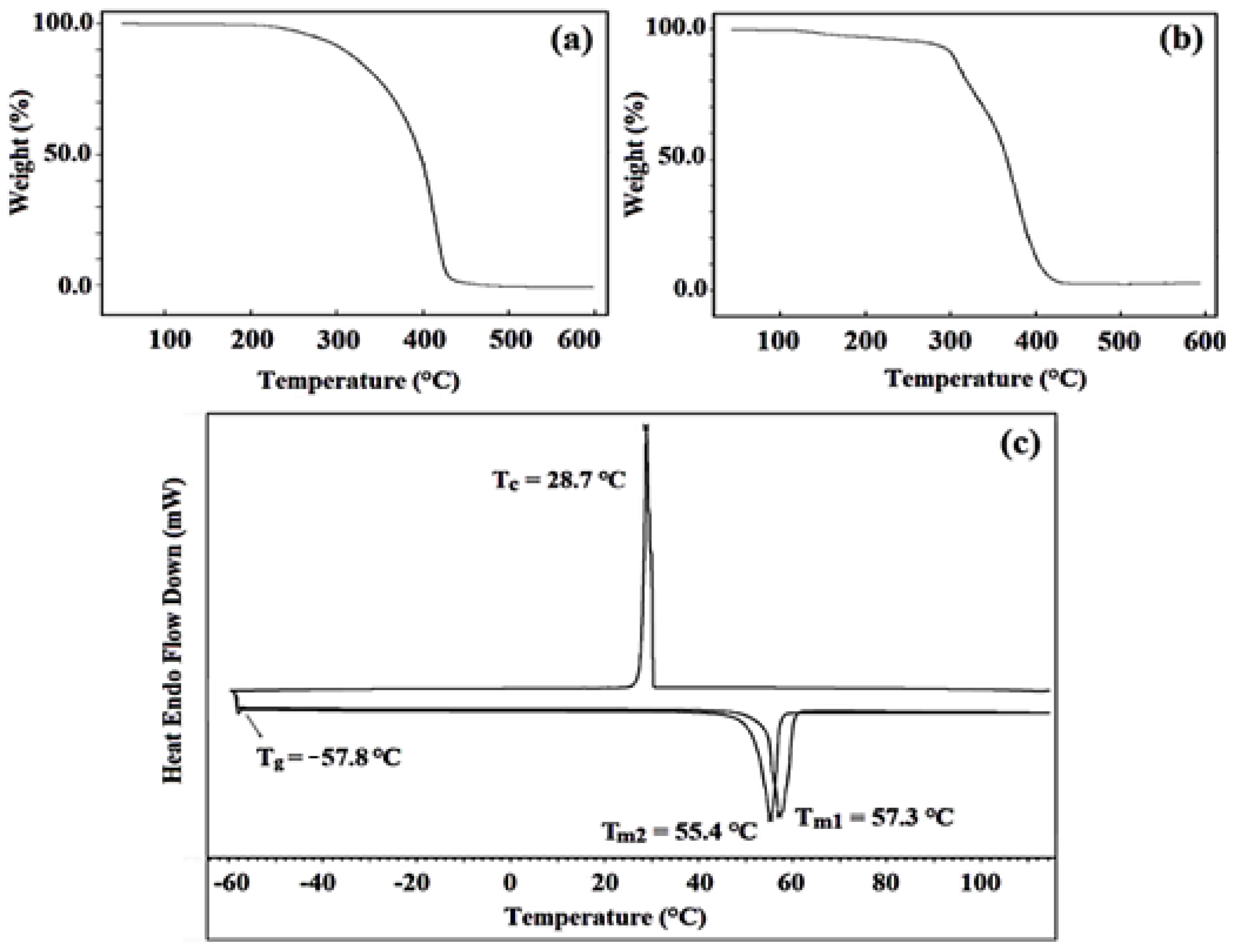
3.4. The Characterization of Block Copolymers Using PXTPCLBr via ATRP
3.5. Measurement of the Heavy–Metal–Binding Properties of PXTPCLOH and PXTPS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matyjaszewski, K.; Tsarevsky, N.V. Macromolecular Engineering by Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 6513–6533. [Google Scholar] [CrossRef] [PubMed]
- Jankova, K.; Bednarek, M.; Hvilsted, S. Star polymers by ATRP of styrene and acrylates employing multifunctional initiators. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 3748–3759. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Wang, J.S.; Matyjaszewski, K. Controlled Living Radical Polymerization. atom transfer radical polymerization in the presence of transition–metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441. [Google Scholar] [CrossRef]
- Tang, W.; Matyjaszewski, K. Effects of Initiator Structure on Activation Rate Constants in ATRP. Macromolecules 2007, 40, 1858–1863. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef]
- Kopping, J.T.; Tolstyka, Z.P.; Maynard, H.D. Telechelic Aminooxy Polystyrene Synthesized by ATRP and ATR Coupling. Macromolecules 2007, 40, 8593–8599. [Google Scholar] [CrossRef]
- Tong, Y.Y.; Dong, Y.Q.; Du, F.S.; Li, Z.C. Synthesis of Well–Defined Poly(Vinyl Acetate)–b–Polystyrene by Combination of ATRP and RAFT Polymerization. Macromolecules 2008, 41, 7339–7346. [Google Scholar] [CrossRef]
- Hegewald, J.; Pionteck, J.; Häußler, L.; Komber, H.; Voit, B. End-functionalized polystyrene by ATRP: A facile approach to primary amino and carboxylic acid terminal groups. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 3845–3859. [Google Scholar] [CrossRef]
- Van Camp, W.; Gao, H.; Prez, F.E.; Matyjaszewski, K. Effect of crosslinker multiplicity on the gel point in ATRP. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2016–2023. [Google Scholar] [CrossRef][Green Version]
- Lammens, M.; Fournier, D.; Fijten, M.W.; Hoogenboom, R.; Du Prez, F. Star–shaped polyacrylates: Highly functionalized architectures via CuAAC click Hoogenboom conjugation. Macromol. Rapid Commun. 2009, 30, 2049–2055. [Google Scholar] [CrossRef]
- Mespouille, L.; Vachaudez, M.; Suriano, F.; Gerbaux, P.; Van Camp, W.; Coulembier, O.; Degee, P.; Flammang, R.; Du Prez, F.; Dubois, P. Controlled synthesis of amphiphilic block copolymers based on polyester and poly(amino methacrylate): Comprehensive study of reaction mechanisms. React. Funct. Polym. 2008, 68, 990–1003. [Google Scholar] [CrossRef]
- Aliyev, E.; Shishatskiy, S.; Abetz, C.; Lee, Y.J.; Neumann, S.; Emmler, T.; Filiz, V. SI–ATRP Polymer–Functionalized Graphene Oxide for Water Vapor Separation. Adv. Mater. Interfaces 2020, 7, 2000443. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Miller, P.J.; Pyun, J.; Kickelbick, G.; Diamanti, S. Synthesis and characterization of star polymers with varying arm number, length, and composition from organic and hybrid inorganic/organic multifunctional initiators. Macromolecules 1999, 32, 6526–6535. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal–Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3746. [Google Scholar] [CrossRef]
- Ritz, P.; Látalová, P.; Janata, M.; Toman, L.; Kříž, J.; Genzer, J.; Vlček, P. Synthesis of amphiphilic copolymers by ATRP initiated with a bifunctional initiator containing trichloromethyl groups. React. Funct Polym. 2007, 67, 1027–1039. [Google Scholar] [CrossRef]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional polymers by atom transfer radical polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Yagci, Y.; Nuyken, O.; Graubner, V.M. Telechelic Polymers, Encyclopedia of Polymer Science and Technology; Kroschwitz, J.I., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2005; Volume 12, pp. 57–130. [Google Scholar]
- Feng, H.; Lu, X.; Wang, W.; Kang, N.G.; Mays, J.W. Block Copolymers: Synthesis, Self–assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef]
- Savaşkan, S.; Beşirli, N.; Hazer, B. Synthesis of some new cation-exchanger resins. J. Appl. Polym. Sci. 1996, 59, 1515–1524. [Google Scholar] [CrossRef]
- Yilmaz, S.S.; Kul, D.; Erdöl, M.; Özdemir, M.; Abbasoglu, R. Synthesis of a novel crosslinked superabsorbent copolymer with diazacyclooctadecane crown ether and its sorption capability. Eur. Polym. J. 2007, 43, 1923–1932. [Google Scholar] [CrossRef]
- Misir, M.; Ozturk, T.; Emirik, M.; Yilmaz, S.S. Synthesis of novel tetrahydrofuran–epichlorohydrin [poly(THF–b–ECH)] macromonomeric peroxy initiators by cationic copolymerization and the quantum chemically investigation of initiation system effects. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 2896–2909. [Google Scholar] [CrossRef]
- Yagci, Y. Handbook of Vinyl Polymers: Radical Polymerization, Process, and Technology; Mishra, M., Ed.; KCRC Press: Grand Rapids, MI, USA, 2008; Chapter 9; ISBN 9781420015133. [Google Scholar] [CrossRef]
- Huang, G.C.; Ji, S.X. Effect of Halogen Chain End Fidelity on the Synthesis of Poly(methylmethacrylate–b–styrene) by ATRP. Chin. J. Polym. Sci. 2018, 36, 1217–1224. [Google Scholar] [CrossRef]
- Aimi, J.; Komura, M.; Iyoda, T.; Saeki, A.; Seki, S.; Takeuchi, M.; Nakanishi, T. Synthesis and self–assembly of phthalocyanine–tethered block copolymers. J. Mater. Chem. C 2015, 3, 2484–2490. [Google Scholar] [CrossRef]
- Spruell, J.M.; Levy, B.A.; Sutherland, A.; Dichtel, W.R.; Cheng, J.Y.; Stoddart, J.F.; Nelson, A. Facile postpolymerization end–modification of RAFT polymers. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 346–356. [Google Scholar] [CrossRef]
- Xiong, L.; Wang, R.; Liang, H.; Pang, Y.; Guan, J. Synthesis and Characterization of Poly(methyl methacrylate)–b–Polystyrene/TiO2 Nanocomposites Via Reversible Addition–Fragmentation Chain Transfer Polymerization. J. Macromol. Sci. Part A Pure Appl. Chem. 2010, 47, 903–908. [Google Scholar] [CrossRef]
- Lee, J.; Lee, K.; Park, J.; Kim, J.K.; Chang, T. Characterization and fractionation of PS–b–PMMA diblock copolymer synthesized via click chemistry. Polymer 2015, 80, 46–51. [Google Scholar] [CrossRef]
- Zhao, B.; Brittain, W.J. Synthesis of Tethered Polystyrene–Block–Poly(methyl Methacrylate) Monolayer on a Silicate Substrate by Sequential Carbocationic Polymerization and Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 1999, 121, 3557–3558. [Google Scholar] [CrossRef]
- Khaydarov, A.A.; Hamley, I.W.; Legge, T.M.; Perrier, S. Surface structure of thin asymmetric PS–b–PMMA diblock copolymers investigated by atomic force microscopy. Eur. Polym. J. 2007, 43, 789–796. [Google Scholar] [CrossRef][Green Version]
- Bolton, J.; Rzayev, J. Tandem RAFT-ATRP Synthesis of Polystyrene–Poly(Methyl Methacrylate) Bottlebrush Block Copolymers and Their Self-Assembly into Cylindrical Nanostructures. ACS Macro Lett. 2012, 1, 15–18. [Google Scholar] [CrossRef]
- Temel, G.; Aydogan, B.; Arsu, N.; Yagci, Y. Synthesis of block and star copolymers by photoinduced radical coupling process. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 2938–2947. [Google Scholar] [CrossRef]
- Aitchison, T.J.; Ginic-Markovic, M.; Clarke, S.; Valiyaveettil, S. Polystyrene–block–poly(methyl methacrylate): Initiation issues with block copolymer formation using ARGET ATRP. Macromol. Chem. Phys. 2012, 213, 79–86. [Google Scholar] [CrossRef]
- Couthouis, J.; Keul, H.; Möller, M. MALDI–TOF analysis of halogen telechelic poly(methyl methacrylate)s and poly(methyl acrylate)s prepared by atom transfer radical polymerization (ATRP) or single electron transfer–living radical polymerization (SET–LRP). Macromol. Chem. Phys. 2015, 216, 1791–1800. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Kahveci, M.U.; Yagci, Y. Telechelic Polymers by Living and Controlled/Living Polymerization Methods. Prog. Polym. Sci. 2011, 36, 455–567. [Google Scholar] [CrossRef]
- Simula, A.; Nikolaou, V.; Anastasaki, A.; Alsubaie, F.; Nurumbetova, G.; Wilson, P.; Kempea, K.; Haddleton, D.M. Synthesis of well–defined α,ω–tlechelic multiblock copolymers in aqueous medium: In situ generation of α,ω–diols. Polym. Chem. 2015, 6, 2226–2233. [Google Scholar] [CrossRef]
- Neumann, H.; Keul, H.; Höcker, H. Atom transfer radical polymerization (ATRP) of styrene and methyl methacrylate with α,α–dichlorotoluene as initiator; a kinetic study. Macromol. Chem. Phys. 2000, 201, 980–984. [Google Scholar] [CrossRef]
- Dayananda, K.; Ramakrishnan, A.; Dhamodharan, R.J. Synthesis and Characterization of Block Copolymers of P(MMA–b–BA–b–MMA) via Ambient Temperature ATRP of MMA. J. Macromol. Sci. A 2005, 42, 471–484. [Google Scholar] [CrossRef]
- Durmaz, Y.Y.; Yılmaz, G.; Yagci, Y. N–alkoxy pyridinium ion terminated polystyrenes: A facile route to photoinduced block copolymerization. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 423–428. [Google Scholar] [CrossRef]
- Davis, K.A.; Matyjaszewski, K. Atom transfer radical polymerization of tert–butyl acrylate and preparation of block copolymers. Macromolecules 2000, 33, 4039–4047. [Google Scholar] [CrossRef]
- Hirao, A.; Matsuo, Y.; Oie, T.; Goseki, R.; Ishizone, T.; Sugiyama, K.; Gröschel, A.H.; Müller, A.H.E. Facile Synthesis of Triblock Co- and Terpolymers of Styrene, 2–Vinylpyridine, and Methyl Methacrylate by a New Methodology Combining Living Anionic Diblock Copolymers with a Specially Designed Linking Reaction. Macromolecules 2011, 44, 6345–6355. [Google Scholar] [CrossRef]
- Karkare, P.; Kumar, S.; Murthy, C.N. ARGET–ATRP using β–CD as reducing agent for the synthesis of PMMA–b–PS–b–PMMA triblock copolymers. J. Appl. Polym. Sci. 2018, 136, 47117. [Google Scholar] [CrossRef]
- Wu, C.; Xie, Z.W.; Zhang, G.Z.; Zi, G.F.; Tu, Y.F.; Yang, Y.L.; Cai, P.; Nie, T. Self–assemly assisted polymerization (SAAP):approaching long multi–block copolymers with ordered chain sequence and controllable block length. Chem.Commun. 2002, 23, 2898–2899. [Google Scholar] [CrossRef] [PubMed]
- Toman, L.; Janata, M.; Spěváček, J.; Vlček, P.; Látalová, P.; Masař, B.; Sikora, A. Sequential synthesis of multiblock copolymers by atom transfer radical and cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 6098–6108. [Google Scholar] [CrossRef]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef]
- Guillaume, S. Recent advances in ring–opening polymerization strategies toward α,ω–hydroxy telechelic polyesters and resulting copolymers. Eur. Polym. J. 2013, 49, 768–779. [Google Scholar] [CrossRef]
- Soni, V.; Pandey, V.; Asati, S.; Gour, V.; Tekade, R.K. Basic Fundamentals of Drug Delivery. In Advances in Pharmaceutical Product Development and Research for Drug Delivery; Rakesh, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; p. 407. [Google Scholar] [CrossRef]
- Patton, D.L.; Advincula, R.C. A Versatile Synthetic Route to Macromonomers via RAFT Polymerization. Macromolecules 2006, 39, 8674–8683. [Google Scholar] [CrossRef]
- Biela, T.; Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Progress in polymerization of cyclic esters: Mechanisms and synthetic applications. Macromol. Symp. 2006, 240, 47–55. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Bilgin, A.; Yagci, C. Octa–armed star–shaped poly(ε–caprolactone)s with a phthalocyanine core by ring–opening polymerization: Synthesis and characterization. Eur. Polym. J. 2014, 61, 240–252. [Google Scholar] [CrossRef]
- Navarro-Baena, I.; Marcos-Fernández, A.; Fernández-Torres, A.; Kenny, J.M.; Peponi, L. Synthesis of PLLA–b–PCL–b–PLLA linear tri–block copolymers and their corresponding poly(ester–urethane)s: Effect of the molecular weight on their crystallization and mechanical properties. RSC Adv. 2014, 4, 8510–8524. [Google Scholar] [CrossRef]
- Dhanasekaran, N.P.D.; Muthuvelu, K.S.; Arumugasamy, S.K. Recent Advancement in Biomedical Applications of Polycaprolactone and Polycaprolactone–Based Materials. In Encyclopedia of Materials: Plastics and Polymers; Elsevier: Amsterdam, The Netherlands, 2022; pp. 795–809. [Google Scholar]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε–caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef]
- Yilmaz, G. One-Pot Synthesis of Star Copolymers by the Combination of Metal–Free ATRP and ROP Processes. Polymers 2019, 11, 1577. [Google Scholar] [CrossRef] [PubMed]
- Schappacher, M.; Fur, N.; Guillaume, S.M. Poly(methyl methacrylate)–poly(caprolactone) AB and ABA block copolymers by combined ring–opening polymerization and atom transfer radical polymerization. Macromolecules 2007, 40, 8887–8896. [Google Scholar] [CrossRef]
- Yuan, W.; Huang, X.; Tang, X. Synthesis of star–shaped PCL–b–PMMA/PSt from cyclotriphosphazene initiator by ring opening polymerization and atom transfer polymerization. Polym. Bull. 2005, 55, 225–233. [Google Scholar] [CrossRef]
- Liu, R.X.; Shi, Y.; Fu, Z.F. Synthesis of PCL–PS Star–Block Copolymer by Combination of Lipase–Catalyzed Ring–Opening Polymerization and ATRP. Adv. Mat. Res. 2006, 11–12, 749–752. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Muller, A.J.; Lin, M.-C.; Chen, H.-L.; Jeng, U.-S.; Priftis, D.; Pitsikalis, M.; Hadjichristidis, N. Influence of Macromolecular Architecture on the Crystallization of (PCL2)–b–(PS2) 4–Miktoarm Star Block Copolymers in Comparison to Linear PCL-b-PS Diblock Copolymer Analogues. Macromolecules 2009, 42, 8353–8364. [Google Scholar] [CrossRef]
- Heise, A.; Trollsas, M.; Magbitang, T.; Hedrick, J.L.; Frank, C.W.; Miller, R.D. Star Polymers with Alternating Arms from Miktofunctional μ–Initiators Using Consecutive Atom Transfer Radical Polymerization and Ring–Opening Polymerization. Macromolecules 2001, 34, 2798–2804. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Wang, W.; Wang, J.; Chen, X. ABA2–type triblock copolymer composed of PCL and PSt:synthesis and characterization. Polym. Bull. 2011, 67, 1507–1518. [Google Scholar] [CrossRef]
- Motala–Timol, S.; Jhurry, D. Synthesis of PDMAEMA–PCL–PDMAEMA triblock copolymers. Eur. Polym. J. 2007, 43, 3042–3049. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, W.-H.; Aimi, J.; Huang, Y.-S.; Venkatesan, S.; Chiang, Y.-W.; Huang, S.-H.; Kuo, S.-W.; Chen, T. Synthesis of well–defined PCL-b-PnBA-b-PMMA ABC–type triblock copolymers: Toward the construction of nanostructures in epoxy thermosets. Polym. Chem. 2018, 9, 5644–5654. [Google Scholar] [CrossRef]
- Glaied, O.; Delaite, C.; Riess, G. Synthesis of PCL-b-PVAc block copolymers by combination of click chemistry, ROP, and RAFT polymerization. Polym. Bull. 2012, 68, 607–621. [Google Scholar] [CrossRef]
- Bilgin, A.; Mendi, A.; Yildiz, U. Novel Phthalocyanine Polymers with Very Flexible Pentathiatetraethylene Units. Polymer 2006, 47, 8462–8473. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 2nd ed.; Pergamon Press: Oxford, UK, 1989. [Google Scholar]
- Escriche, L.; Almajano, M.P.; Casabó, J.; Teixidor, F.; Lockhart, J.C.; Forsyth, G.A.; Kivekäs, R.; Sundberg, M. Synthesis and molecular dynamics studies of the new ditopic para–xylyl containing macrocycle 2,5,8,17,20,23-hexathia[9,9]-p-cyclophane(p–S6). X-ray crystal structure of the dicopper(I) complex [Cu2(p–S6)CH3CN)2](BF4)2. Polyhedron 1996, 15, 4203–4209. [Google Scholar] [CrossRef]
- Bilgin, A.; Yağcı, C.; Yıldız, U. Novel network polymeric phthalocyanines: Synthesis and characterization. Macromol. Chem. Phys. 2005, 206, 2257–2268. [Google Scholar] [CrossRef]
- Hill, L.; Sims, H.; Nguyen, N.; Collins, C.; Palmer, J.; Wasson, F.A. Degradable Difunctional Initiator for ATRP That Responds to Hydrogen Peroxide. Polymers 2022, 14, 1733. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Introduction to Living Polymeriz. Living and/or Controlled Polymerization. J. Phys. Org. Chem. 1995, 8, 197–207. [Google Scholar] [CrossRef]
- Guan, X.; Ma, X.; Zhou, H.; Chen, F.; Li, Z. Synthesis and thermal decomposition kinetics of poly(methylmethacrylate)–b–poly(styrene) block copolymers. J. Thermoplast. Compos. Mat. 2017, 30, 691–706. [Google Scholar] [CrossRef]
- Elzein, T.; Nasser–Eddine, M.; Delaite, C.; Bistac, S.; Dumas, P. FTIR study of polycaprolactone chain organization at interfaces. J. Colloid Interface Sci. 2004, 273, 381–387. [Google Scholar] [CrossRef]
- Zain, M.N.; Ahmad, S.H.; Ali, E.S.; Zubir, S.A.; Ahad, N.A. Characteristics of Hydrolysis Resistant Polycaprolactone/Palm Kernel Oil Based Polyol. Adv. Mater. Res. 2012, 576, 334–337. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Ye, D.; Zhang, J.; Guo, Y.; You, R.; Yan, S.; Li, M.; Qu, J. Fabrication and characterization of electrospun PCL/Antheraea pernyi silk fibrion nanofibrous scafolds. Polym. Eng. Sci. 2017, 57, 206–213. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.F. Physical properties and biodegradation of acrylic acid grafted poly (ε-caprolactone)/chitosan blends. J. Polym. Res. 2010, 17, 221–232. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, A.; Marínez-Richa, A. One–step route to α–hydroxyl–ω–(carboxylic acid) polylactones using catalysis by decamolybdate anion. Macromolecules 2005, 38, 1599–1608. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, Y.; Lang, M. Synthesis and self–assembly behavior of six–armed block copolymers with pH and thermo–responsive properties. Macromol. Res. 2011, 19, 113–121. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Wang, W.; Chen, L.; Wang, S.; Wang, J. Chemoenzymatic synthesis of Y-shaped diblock copolymer. Polym. Bull. 2009, 62, 643–655. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, C.; Li, A.; Yang, X.; Lu, L.; Wang, X. Preparation and characterization of mesoporous zirconia made by using a poly (methyl methacrylate) template. Nanoscale Res. Lett. 2008, 3, 118–122. [Google Scholar] [CrossRef]
- Simões, M.C.R.; Cragg, S.M.; Barbu, E.; De Sousa, F.B. The potential of electrospun poly (methyl methacrylate)/polycaprolactone core–sheath fibers for drug delivery applications. J. Mater. Sci. 2019, 54, 5712–5725. [Google Scholar] [CrossRef]
- He, X.; Liang, L.; Xie, M.; Zhang, Y.; Lin, S.; Yan, D. Synthesis of Novel Linear PEO-b-PS-b-PCL Triblock Copolymers by the Combination of ATRP, ROP, and a Click Reaction. Macromol. Chem. Phys. 2007, 208, 1797–1802. [Google Scholar] [CrossRef]
- Degirmenci, M.; Gokkaya, C.; Durgun, M. One–Step Synthesis of Mid–Chain Functional Macrophotoinitiator of Polystyrene–Poly(ε–Caprolactone) Diblock Copolymer via Simultaneous ATRP and ROP Using a Dual–Functional Photoinitiator. Polym. J. 2016, 48, 139–145. [Google Scholar] [CrossRef]
- Fukushima, K.; Tabuani, D.; Abbate, C.; Arena, M.; Rizzarelli, P. Preparation, Characterization and Biodegradation of Biopolymer Nanocomposites Based on Fumed Silica. Eur. Polym. J. 2011, 47, 139–152. [Google Scholar] [CrossRef]
- Tunca, U.; Ozyurek, Z.; Erdogan, T.; Hizal, G. Novel miktofunctional initiator for the preparation of an ABCtype miktoarm star polymer via a combination of controlled polymerization techniques. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 4228–4236. [Google Scholar] [CrossRef]
- Joel, C.; Bennie, R.B.; Raj, A.N.P.; David, S.T.; Abraham, S.D. Removal of toxic metal ions by macroacyclic ligands via liquid–liquid extraction technique. Sep. Sci. Technol. 2021, 56, 1231–1240. [Google Scholar] [CrossRef]
- Hajipour, A.R.; Habibi, S.; Ruoho, A.E. Synthesis and complexation study of calix[4]arene diamine derivative incorporated in a polymeric backbone with chiral monomers. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 107–117. [Google Scholar] [CrossRef]
- Vujasinovic, I.; Veljkovic, J.; Mlinaric–Majerski, K. New Tin Templates for the Synthesis of Macrocyclic Polythiaether–Polythiaester Ligands. J. Org. Chem. 2004, 69, 8550–8553. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Time (h) | Conv c (%) | Mn,th d | Mn,GPC e | Đ e | f f |
|---|---|---|---|---|---|---|
| PM-1 a | 0.5 | 21.4 | 2696 | 6500 | 1.28 | 0.41 |
| PM-2 a | 1.0 | 55.3 | 6086 | 17,440 | 1.31 | 0.35 |
| PM-3 a | 2.0 | 65.1 | 7066 | 20,300 | 1.75 | 0.35 |
| PS-1 b | 0.5 | 22.8 | 2927 | 6780 | 1.30 | 0.43 |
| PS-2 b | 1.0 | 25.7 | 3228 | 8700 | 1.34 | 0.37 |
| PS-3 b | 2.0 | 80.3 | 8907 | 25,270 | 1.49 | 0.35 |
| Entry | Temp (°C) | Mn,th × 104 | f Mn,GPC × 104 | Đ f |
|---|---|---|---|---|
| ABA–1 a | 110 | 3.41 | 4.60 | 1.52 |
| ABA–2 b | 100 | 3.00 | 3.12 | 1.34 |
| ABA–3 c | 110 | 1.85 | 2.23 | 1.41 |
| ABA–4 d | 100 | 1.81 | 2.16 | 1.31 |
| ABA–5 e | 100 | 7.19 | 13.15 | 1.21 |
| ABA–6 e | 100 | 7.65 | 8.28 | 1.40 |
| Extractability (%) | ||||||
|---|---|---|---|---|---|---|
| Metal ion | Hg2+ | Ag+ | Cd2+ | Pb2+ | Cu2+ | Zn2+ |
| PXTPCLOH | 58.62 | 47.30 | 42.86 | 12.63 | 32.04 | 21.32 |
| PXTPS | 43.03 | 37.56 | 29.35 | 8.79 | 17.25 | 14.64 |
| 8H–DAB [87] | – | 42.36 | 35.22 | 35.32 | – | |
| Calix[4]arenediamine derivative(7a’) [88] | 46.61 | – | 33.06 | – | 14.22 | – |
| Calix[4]arenediamine derivative(7b) [88] | 19.07 | – | 8.26 | – | 9.91 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mısır, M.; Savaskan Yılmaz, S.; Bilgin, A. Synthesis and Characterization of ABA-Type Triblock Copolymers Using Novel Bifunctional PS, PMMA, and PCL Macroinitiators Bearing p-xylene-bis(2-mercaptoethyloxy) Core. Polymers 2023, 15, 3813. https://doi.org/10.3390/polym15183813
Mısır M, Savaskan Yılmaz S, Bilgin A. Synthesis and Characterization of ABA-Type Triblock Copolymers Using Novel Bifunctional PS, PMMA, and PCL Macroinitiators Bearing p-xylene-bis(2-mercaptoethyloxy) Core. Polymers. 2023; 15(18):3813. https://doi.org/10.3390/polym15183813
Chicago/Turabian StyleMısır, Murat, Sevil Savaskan Yılmaz, and Ahmet Bilgin. 2023. "Synthesis and Characterization of ABA-Type Triblock Copolymers Using Novel Bifunctional PS, PMMA, and PCL Macroinitiators Bearing p-xylene-bis(2-mercaptoethyloxy) Core" Polymers 15, no. 18: 3813. https://doi.org/10.3390/polym15183813
APA StyleMısır, M., Savaskan Yılmaz, S., & Bilgin, A. (2023). Synthesis and Characterization of ABA-Type Triblock Copolymers Using Novel Bifunctional PS, PMMA, and PCL Macroinitiators Bearing p-xylene-bis(2-mercaptoethyloxy) Core. Polymers, 15(18), 3813. https://doi.org/10.3390/polym15183813