



Novel Macromolecular and Biobased Flame Retardants Based on Cellulose Esters and Phosphorylated Sugar Alcohols
Abstract
:1. Introduction
2. Materials and Method
2.1. Materials
2.2. Methods
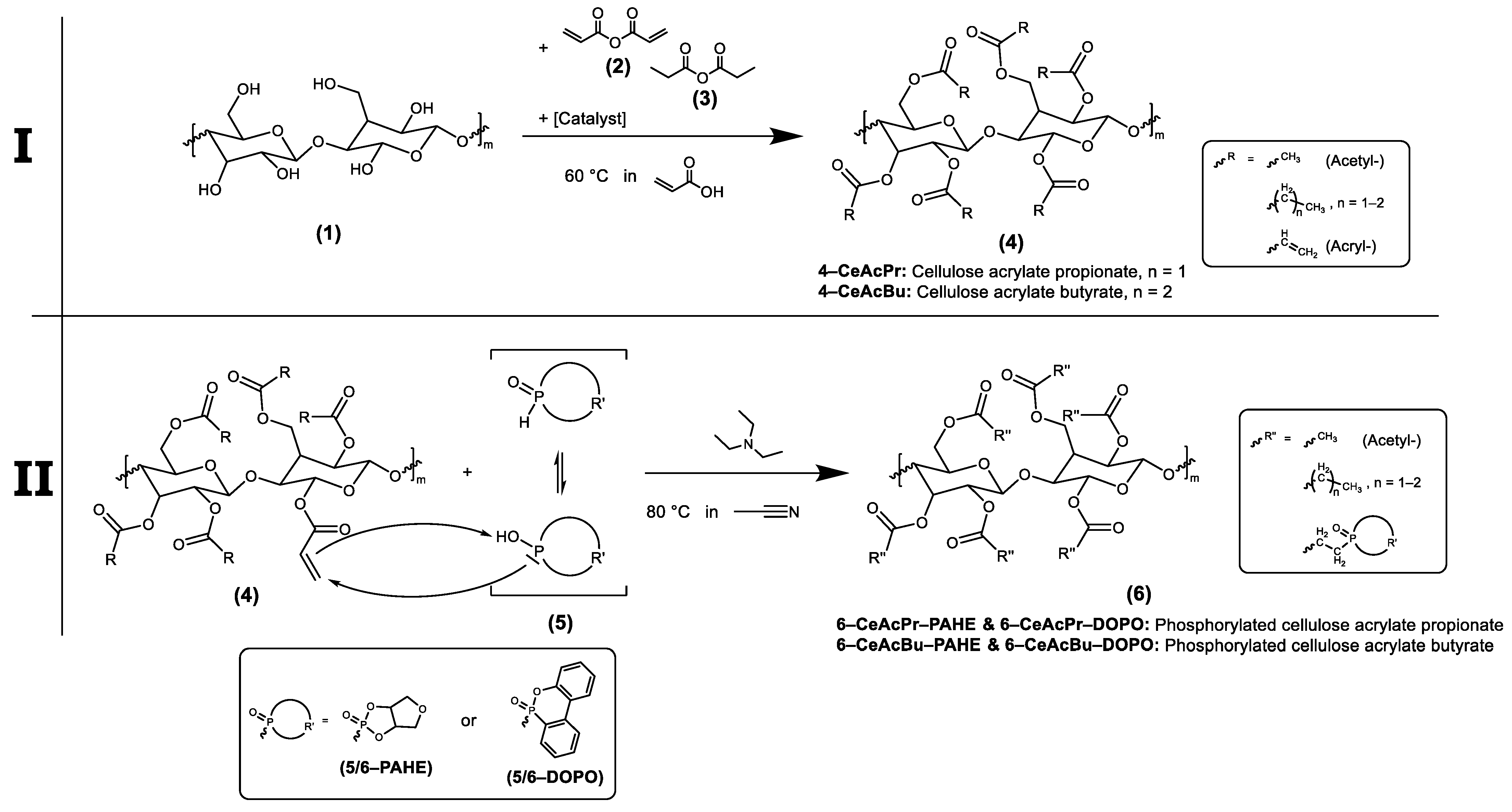
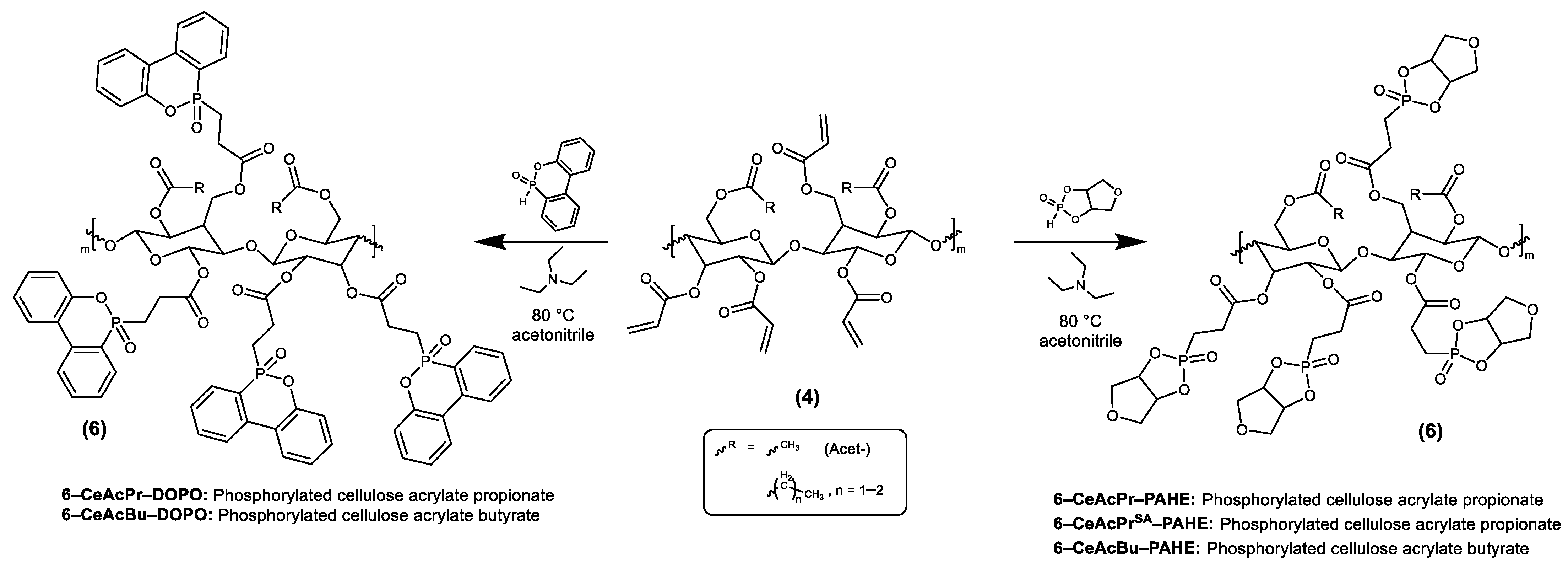
2.3. Synthesis of Flame Retardants
2.3.1. Synthesis of the Acrylic Anhydride Solution (AA–AH) (2)
2.3.2. Synthesis of Tetrahydrofuro[3,4-d][1,3,2]dioxaphosphole 2-oxide (5-PAHE) (Phosphorylated Anhydro Erythritol, PAHE)
2.3.3. Synthesis of Cellulose Acrylate Butyrate (4-CeAcBu)
2.3.4. Synthesis of Phosphorylated Cellulose Acrylate Butyrate with PAHE (6-CeAcBu-PAHE)
2.3.5. Synthesis of Phosphorylated Cellulose Acrylate Butyrate with DOPO (6-CeAcBu-DOPO)
2.3.6. Synthesis of Cellulose Acrylate Propionate (4-CeAcPr)
2.3.7. Synthesis of Cellulose Acrylate Propionate (4-CeAcPrSA)
2.3.8. Synthesis of Phosphorylated Cellulose Acrylate Propionate with PAHE (6-CeAcPr-PAHE)
2.3.9. Synthesis of Phosphorylated Cellulose Acrylate Propionate with PAHE (6-CeAcPrSA-PAHE)
2.3.10. Synthesis of Phosphorylated Cellulose Acrylate Propionate with DOPO (6-CeAcPr-DOPO)
2.4. Preparation of Polymer Compounds and UL 94 V Test Specimens
2.5. Flame Test: UL 94 Vertical/DIN EN 60695-11-10
3. Results and Discussion
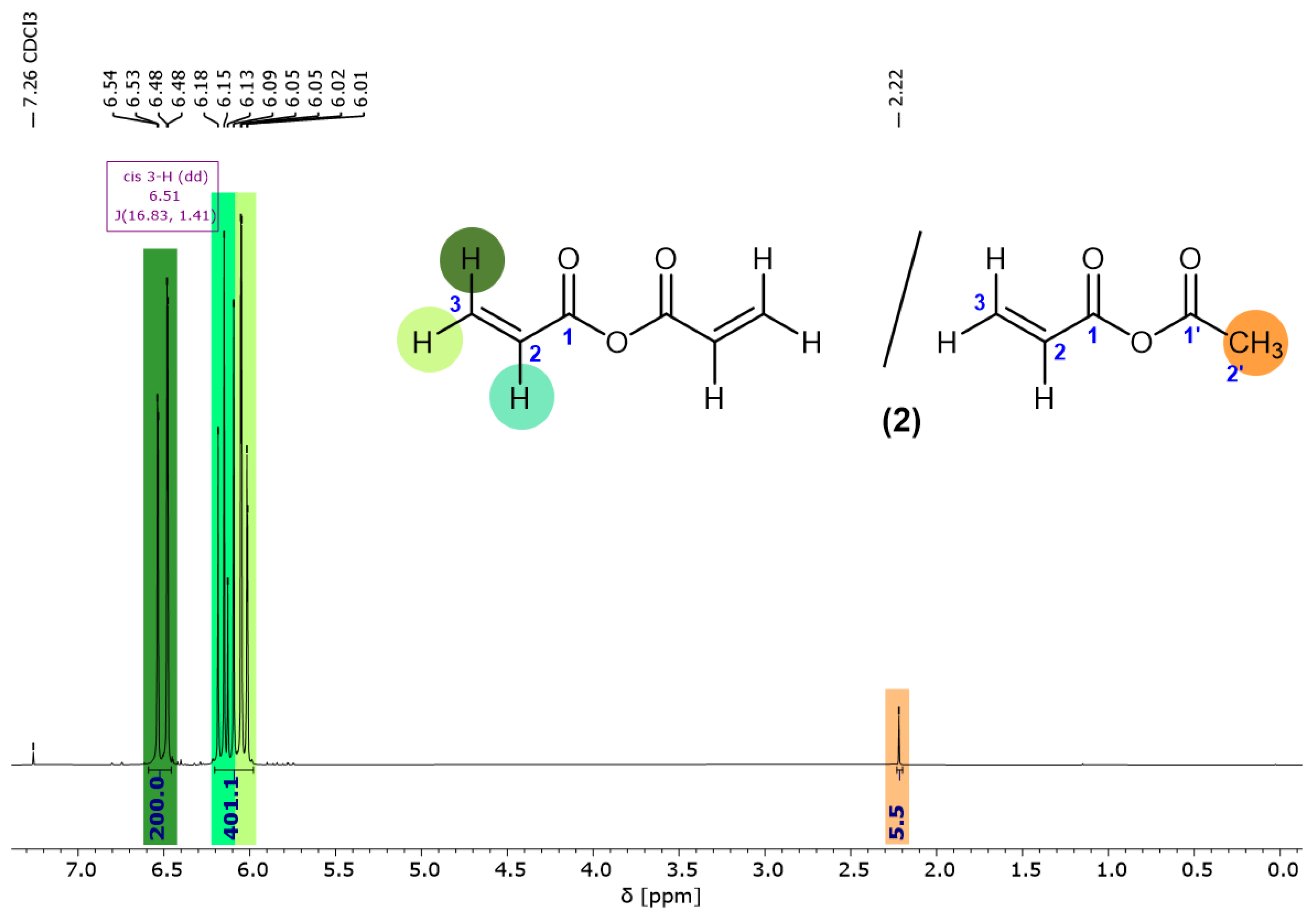
3.1. Characterization of the Acrylic Acid Anhydride Solution (2)
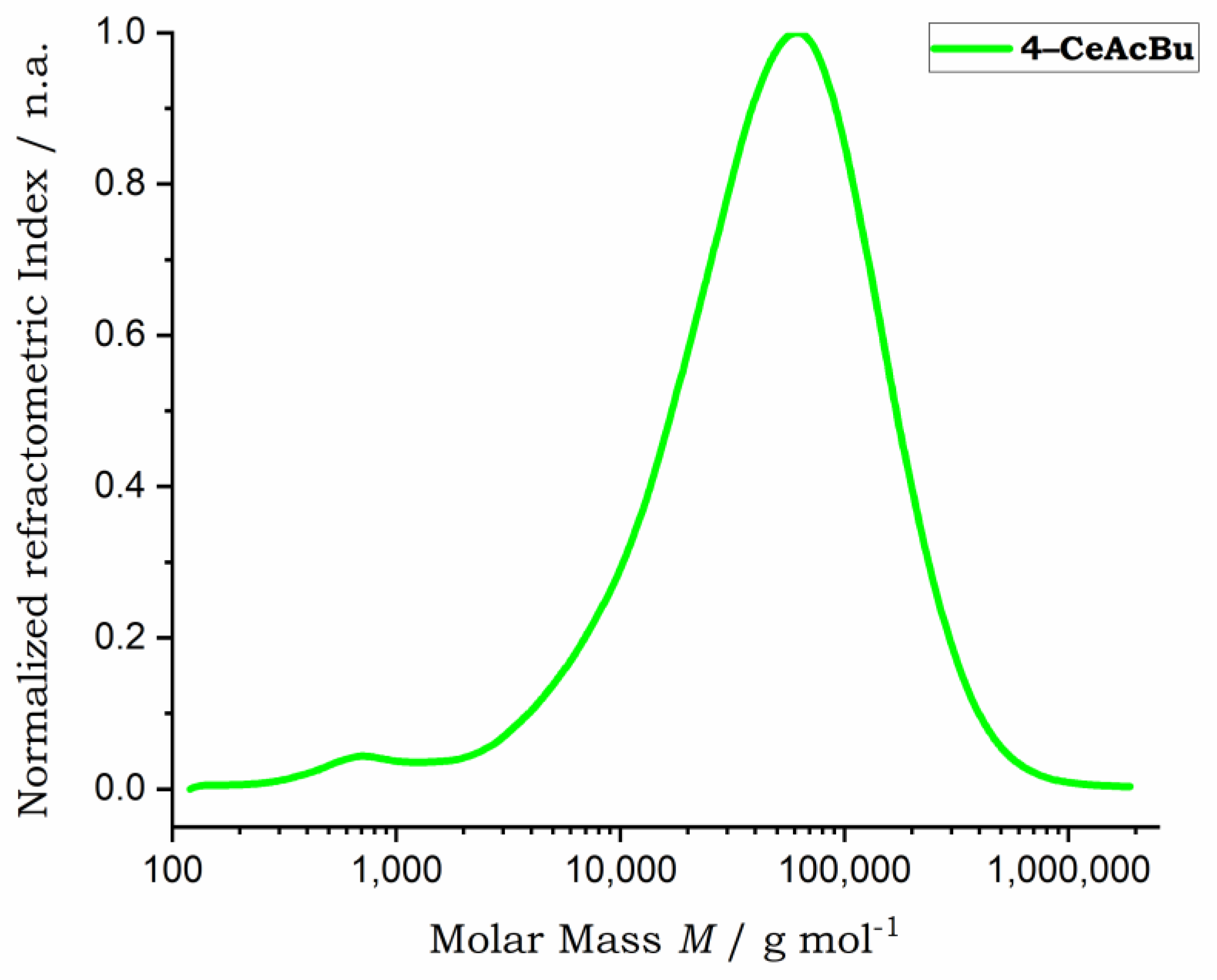
3.2. Characterization of the Cellulose Esters (4)
3.3. Characterization of Tetrahydrofuro[3,4-d][1,3,2]dioxaphosphole 2-oxide (5-PAHE)
3.4. Characterization of the Flame Retardant (Phosphorylated Cellulose Esters, 6)
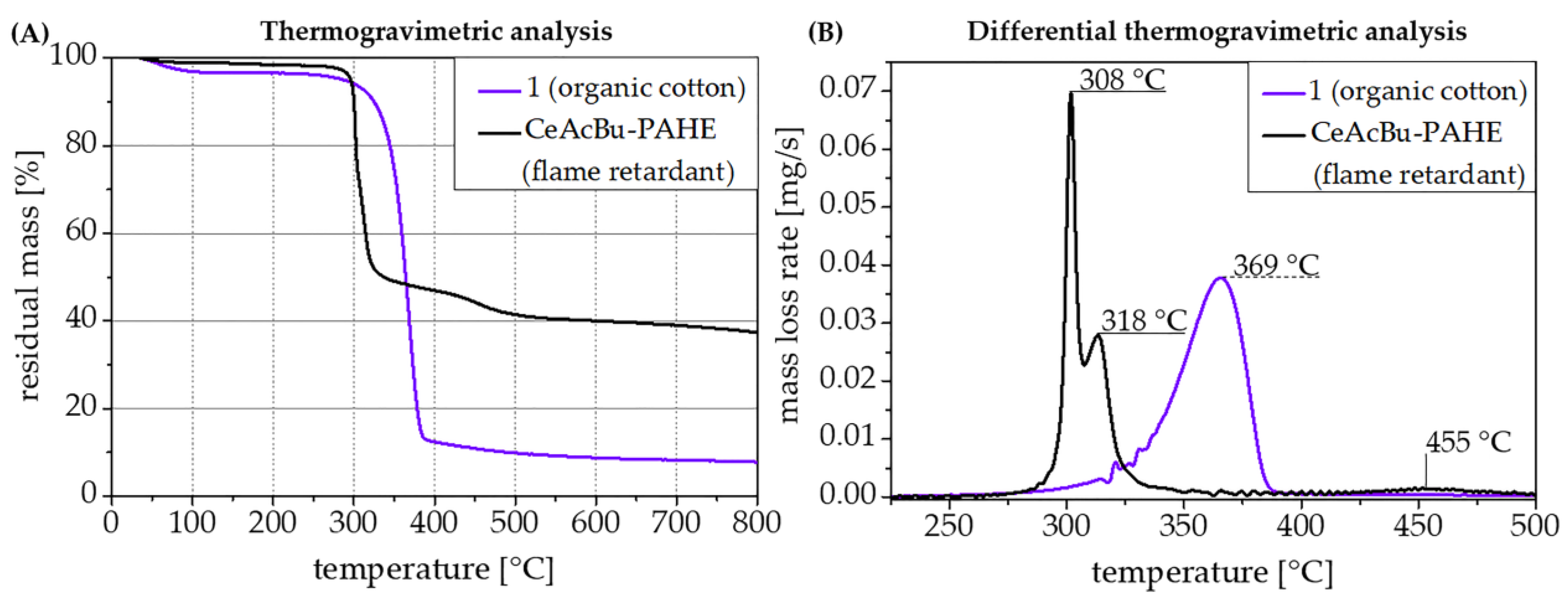
3.5. Thermal Analysis of the Flame Retardants
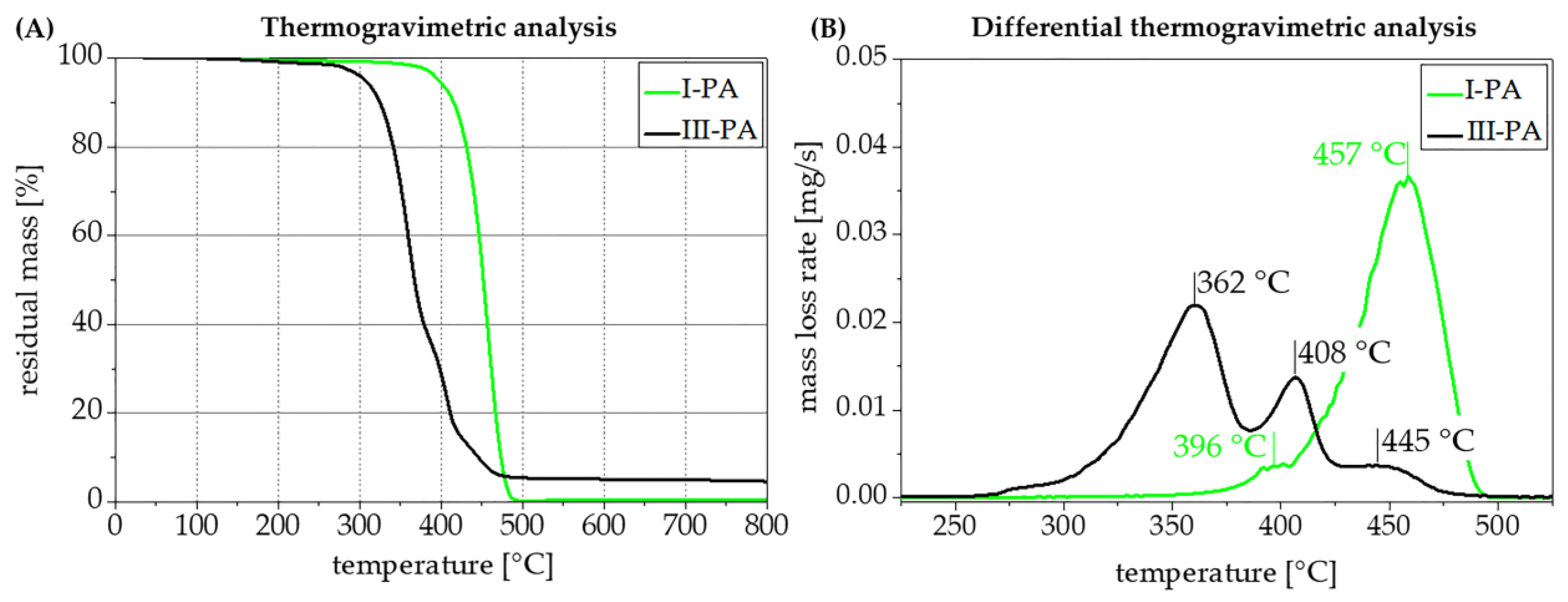
3.6. Processing and Thermal Characterization of Polymer Compounds
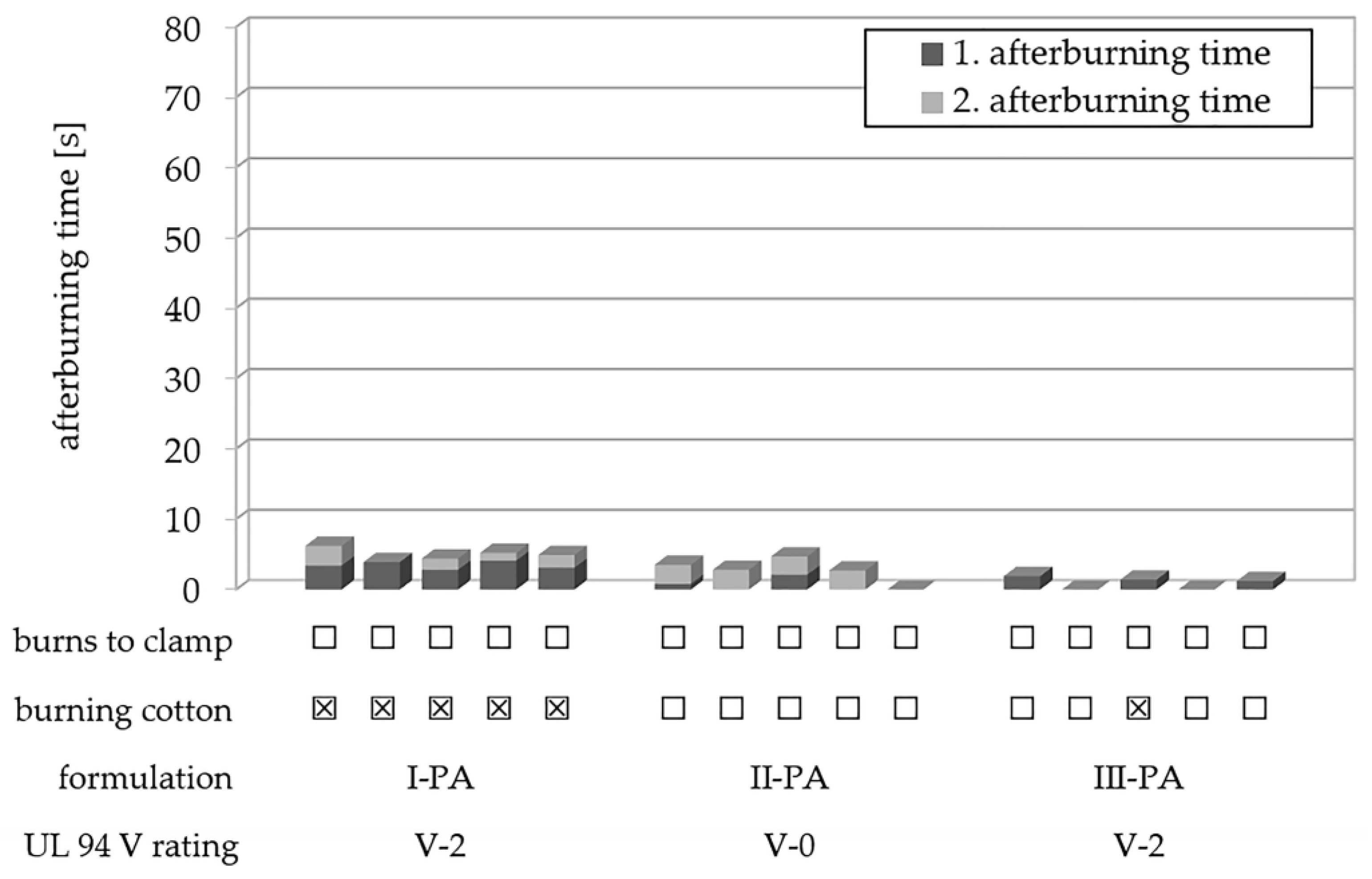
3.7. Flame Retardant Results
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation No. | Specimen No. | DBDE/ ATO | PBDS | 6-CeAcBu-PAHE | UL 94 V Test a | |||
|---|---|---|---|---|---|---|---|---|
| First Burning Time [s] | Second Burning Time [s] | Burning Cotton [yes/no] | UL 94 V Rating | |||||
| I-PP-PE | 1 | - | - | - | 61 | - | Y | NC |
| 2 | - | - | - | 68 | - | Y | ||
| 3 | - | - | - | 60 | - | Y | ||
| 4 | - | - | - | 59 | - | Y | ||
| 5 | - | - | - | 71 | - | Y | ||
| II-PP-PE | 1 | 6.7/3.3 | - | - | 2 | - | Y | V-2 |
| 2 | 6.7/3.3 | - | - | 2 | - | Y | ||
| 3 | 6.7/3.3 | - | - | 1 | 1 | Y | ||
| 4 | 6.7/3.3 | - | - | - | - | Y | ||
| 5 | 6.7/3.3 | - | - | - | 1 | Y | ||
| III-PP-PE | 1 | - | 10 | - | 57 | - | Y | NC |
| 2 | - | 10 | - | 56 | - | Y | ||
| 3 | - | 10 | - | 59 | - | Y | ||
| 4 | - | 10 | - | 52 | - | Y | ||
| 5 | - | 10 | - | 58 | - | Y | ||
| IV-PP-PE | 1 | - | - | 10 | 4 | 1 | Y | NC |
| 2 | - | - | 10 | 4 | 55 | Y | ||
| 3 | - | - | 10 | 6 | 55 | Y | ||
| 4 | - | - | 10 | 7 | 40 | Y | ||
| 5 | - | - | 10 | 4 | 52 | Y | ||
| V-PP-PE | 1 | - | 2 | 8 | 1 | 1 | Y | V-2 |
| 2 | - | 2 | 8 | 1 | 0 | Y | ||
| 3 | - | 2 | 8 | 1 | 0 | Y | ||
| 4 | - | 2 | 8 | 2 | 0 | Y | ||
| 5 | - | 2 | 8 | 1 | 0 | Y | ||
| Formulation No. | Specimen No. | DEPAL | 6-CeAcBu-PAHE | UL 94 V Test a | |||
|---|---|---|---|---|---|---|---|
| First Burning Time [s] | Second Burning Time [s] | Burning Cotton [yes/no] | UL 94 V Rating | ||||
| I-PA | 1 | - | - | 3 | 3 | Y | V-2 |
| 2 | - | - | 4 | - | Y | ||
| 3 | - | - | 3 | 2 | Y | ||
| 4 | - | - | 4 | 1 | Y | ||
| 5 | - | - | 3 | 2 | Y | ||
| II-PA | 1 | 20 | - | 1 | 3 | N | V-0 |
| 2 | 20 | - | 0 | 3 | N | ||
| 3 | 20 | - | 2 | 3 | N | ||
| 4 | 20 | - | 0 | 3 | N | ||
| 5 | 20 | - | 0 | 0 | N | ||
| III-PA | 1 | - | 20 | 2 | 0 | N | V-2 |
| 2 | - | 20 | 0 | 0 | N | ||
| 3 | - | 20 | 1 | 0 | Y | ||
| 4 | - | 20 | 0 | 0 | N | ||
| 5 | - | 20 | 1 | 0 | N | ||
References
- Weltweite und Europäische Kunststoffproduktion in den Jahren von 1950 bis 2021. 2023. Available online: https://de.statista.com/statistik/daten/studie/167099/umfrage/weltproduktion-von-kunststoff-seit-1950/ (accessed on 12 December 2022).
- Polyolefin. 2023. Available online: https://www.kunststoff-deutschland.com/html/polyolefine.html (accessed on 12 December 2022).
- Gebke, S.; Thümmler, K.; Sonnier, R.; Tech, S.; Wagenführ, A.; Fischer, S. Suitability and Modification of Different Renewable Materials as Feedstock for Sustainable Flame Retardants. Molecules 2020, 25, 335. [Google Scholar] [CrossRef]
- Sag, J.; Kukla, P.; Goedderz, D.; Roch, H.; Kabasci, S.; Döring, M.; Schönberger, F. Synthesis of Novel Polymeric Acrylate-Based Flame Retardants Containing Two Phosphorus Groups in Different Chemical Environments and Their Influence on the Flammability of Poly (Lactic Acid). Polymers 2020, 12, 778. [Google Scholar]
- Al-Mosawi, A.I. Flame retardants, their beginning, types, and environmental impact: A review. J. Silic. Based Compos. Mater. 2022, 74, 2. [Google Scholar]
- Costes, L.; Laoutid, F.; Brohez, S.; Dubois, P. A novel reactive phosphorous flame retardant for cotton fabrics with durable flame retardancy and high whiteness due to self-buffering. Mater. Sci. Eng. R Rep. 2017, 117, 1. [Google Scholar] [CrossRef]
- Schönberger, F.; Cielsielski, M.; Sag, J.; Goedderz, D.; Kabasci, S.; Maga, D. Brände mit biobasierten Additiven verhindern. Kunststoffe 2021, 78. [Google Scholar]
- Shah, A.U.R.; Prabhakar, M.N.; Song, J.-I. Current advances in the fire retardancy of natural fiber and bio-based composites—A review. Int. J. Precis. Eng. Manuf. Green Technol. 2017, 4, 247. [Google Scholar] [CrossRef]
- Mensah, R.A.; Shanmugam, V.; Narayanan, S.; Renner, J.S.; Babu, K.; Neisiany, R.E.; Försth, M.; Sas, G.; Das, O. A review of sustainable and environment-friendly flame retardants used in plastics. Polym. Test. 2022, 108, 107511. [Google Scholar] [CrossRef]
- Visakh, P.M.; Semkin, A.O.; Rezaev, I.A.; Fateev, A.V. Review on soft polyurethane flame retardant. Constr. Build. Mater. 2019, 227, 116673. [Google Scholar] [CrossRef]
- Kaynak, C.; Isitman, N.A. Synergistic fire retardancy of colemanite, a natural hydrated calcium borate, in high-impact polystyrene containing brominated epoxy and antimony oxide. Polym. Degrad. Stab. 2011, 96, 798. [Google Scholar]
- Read, N.J.; Heighway-Bury, E.G. Flameproofing of Textile Fabrics with particular reference to the Function of Antimony Compounds. J. Soc. Dye. Colour. 1958, 74, 823. [Google Scholar] [CrossRef]
- Xiong, P.; Yan, X.; Zhu, Q.; Qu, G.; Shi, J.; Liao, C.; Jiang, G. A Review of Environmental Occurrence, Fate, and Toxicity of Novel Brominated Flame Retardants. Environ. Sci. Technol. 2019, 53, 13551. [Google Scholar]
- Ai, L.; Liu, J.; Chen, S.; Xu, Z.; Liu, P.J. Synthesis of melamine phenyl hypophosphite and its synergistic flame retardance with SiO2 on polypropylene. Therm. Anal. Calorim. 2022, 147, 6207. [Google Scholar]
- Morgan, A.B.; Gilman, J.W. An overview of flame retardancy of polymeric materials: Application, technology, and future directions. Fire Mater. 2013, 37, 259. [Google Scholar]
- Sienkiewicz, A.; Czub, P. Flame retardancy of biobased composites—Research development. Materials 2020, 13, 5253. [Google Scholar] [CrossRef]
- Sonnier, R.; Taguet, A.; Ferry, L.; Lopez-Cuesta, J.-M. Towards Bio-Based Flame Retardant Polymers; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Wang, M.; Yin, G.-Z.; Yang, Y.; Fu, W.; Palencia, J.L.D.; Zhao, J.; Wang, N.; Jiang, Y.; Wang, D.-Y. Bio-based flame retardants to polymers: A review. Adv. Ind. Eng. Polym. Res. 2023, 6, 132. [Google Scholar]
- Michael Ciesielski, Elias Chalwatzis, Robin Nezami, Benedikt Sperlich, WO 2021/048335. 2020.
- Hahladakis, J.N.; Velis, C.A.; Weber, R.; Iacovidou, E.; Purnell, P.J. An overview of chemical additives present in plastics: Migration, release, fate and environmental impact during their use, disposal and recycling. Hazard. Mater. 2018, 344, 179. [Google Scholar]
- Jha, N.K.; Misra, A.C.; Bajaj, P.J. Flame-Retardant Additives for Polypropylene. Macromol. Sci. Part C 1984, 24, 69. [Google Scholar] [CrossRef]
- Lewin, M. (Ed.) Handbook of Fiber Chemistry. Chapter 3: Polypropylene Fibers; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Tirri, T.; Aubert, M.; Aziz, H.; Brusentsev, Y.; Pawelec, W.; Wilén, C.-E. Sulfenamides in synergistic combination with halogen free flame retardants in polypropylene. Polym. Degrad. Stab. 2019, 164, 75. [Google Scholar] [CrossRef]
- Weil, E.D.; Levchik, S.V. Flame retardants in commercial use or development for polyolefins. J. Fire Sci. 2008, 26, 5. [Google Scholar] [CrossRef]
- Sabine Fuchs, Uwe Keppeler, Tim Albert, WO 2015/177347. 2015.
- E. M.-Z. Rudolf Pfaendner, DE 10 2013 005 307 A1. 2013.
- Aubert, M.; Nicolas, R.C.; Pawelec, W.; Wilén, C.-E.; Roth, M.; Pfaendner, R. Azoalkanes—Novel flame retardants and their structure–property relationship. Polym. Adv. Technol. 2011, 22, 1529. [Google Scholar] [CrossRef]
- Pinfa. What are Pin FRs–Pinfa. 2018. Available online: https://www.pinfa.eu/flame-retardants/what-are-pin-frs/ (accessed on 10 May 2023).
- Heinze, U.; Klemm, D.; Unger, E.; Pieschel, F. New Starch Phosphate Carbamides of High Swelling Ability: Synthesis and Characterization. Starch-Stärke 2003, 55, 55. [Google Scholar]
- Gebke, S.; Thümmler, K.; Sonnier, R.; Tech, S.; Wagenführ, A.; Fischer, S. Flame retardancy of wood fiber materials using phosphorus-modified wheat starch. Molecules 2020, 25, 335. [Google Scholar] [PubMed]
- Ernst, C. Duden, Basiswissen Schule: Chemie; Dudenverlag: Berlin, Germany, 2007. [Google Scholar]
- Seddiqi, H.; Oliaei, E.; Honarkar, H.; Jin, J.; Geonzon, L.C.; Bacabac, R.G.; Klein-Nulend, J. Cellulose and its derivatives: Towards biomedical applications. Cellulose 2021, 28, 1893. [Google Scholar]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941. [Google Scholar] [PubMed]
- Sun, L.; Wang, H.; Li, W.; Zhang, J.; Zhang, Z.; Lu, Z.; Zhu, P.; Dong, C. Preparation, characterization and testing of flame retardant cotton cellulose material: Flame retardancy, thermal stability and flame-retardant mechanism. Cellulose 2021, 28, 3789. [Google Scholar]
- Chen, Z.; Xiao, P.; Zhang, J.; Tian, W.; Jia, R.; Nawaz, H.; Jin, K.; Zhang, J. A facile strategy to fabricate cellulose-based, flame-retardant, transparent and anti-dripping protective coatings. Chem. Eng. J. 2020, 379, 122270. [Google Scholar]
- Samyn, F.; Bourbigot, S. Thermal decomposition of flame retarded formulations PA6/aluminum phosphinate/melamine polyphosphate/organomodified clay: Interactions between the constituents? Polym. Degrad. Stab. 2012, 97, 2217. [Google Scholar]
- Teixeira, D.; Giovanela, M.; Gonella, L.B.; Crespo, J.S. Influence of flow restriction on the microstructure and mechanical properties of long glass fiber-reinforced polyamide 6.6 composites for automotive applications. Mater. Des. 2013, 47, 287. [Google Scholar]
- Kisner, A.; Rainert, K.T.; Ferrari, F.; Nau, C.T.; Barcellos, I.O.; Pezzin, S.H.; Andreaus, J. Chemical functionalization of polyamide 6.6 fabrics. React. Funct. Polym. 2013, 73, 1349. [Google Scholar]
- Braun, U.; Bahr, H.; Schartel, B. Fire retardancy effect of aluminium phosphinate and melamine polyphosphate in glass fibre reinforced polyamide 6. e-Polymers 2010, 10, 041. [Google Scholar]
- Lin, M.-S.; Chen, A.-J. Preparation and characterization of water soluble and crosslinkable cellulose acrylate. Polymer 1993, 34, 389. [Google Scholar]
- Satgé, C.; Verneuil, B.; Branland, P.; Granet, R.; Krausz, P.; Rozier, J.; Petit, C. Rapid homogeneous esterification of cellulose induced by microwave irradiation. Carbohydr. Polym. 2002, 49, 373. [Google Scholar]
- Yin, C.; Chen, W.; Zhang, J.; Zhang, M.; Zhang, J. A facile and efficient method to fabricate high-resolution immobilized cellulose-based chiral stationary phases via thiol-ene click chemistry. Sep. Purif. Technol. 2019, 210, 175. [Google Scholar]
- Lapienis, G.; Penczek, S.J. Poly (Alkylene phosphate) s bearing tetrahydrofuran rings (1,4-anhydroerythritol) in the main chain. Polym. Sci. Part A Polym. Chem. 1990, 28, 1743. [Google Scholar]
- Otey, F.; Mehltretter, C.J. A Simple Preparation of 1, 4-Anhydroerythritol. Org. Chem. 1960, 26, 1673. [Google Scholar]
- Silverstein, R.M.; Webster, F.X. Spectrometric Identification of Organic Compounds, 6th ed.; Wiley: Hoboken, NJ, USA, 1998. [Google Scholar]
- EN 60695-11-10; Fire Hazard Testing—Part 11–10: Test Flames—50 W Horizontal and Vertical Flame Test Methods. International Electrotechnical Commission: Geneva, Switzerland, 2013.
- Ho, T.-L. Hard soft acids bases (HSAB) principle and organic chemistry. Chem. Rev. 1975, 75, 1. [Google Scholar]
- Zheng, Y.; Song, J.; Cheng, B.; Fang, X.; Yuan, Y. Preparation and flame retardancy of 3-(hydroxyphenylphosphinyl)-propanoic acid esters of cellulose and their fibers. Cellulose 2015, 22, 229. [Google Scholar]
- Gibert, J.-P.; Cuesta, J.-M.L.; Bergeret, A.; Crespy, A. Study of the degradation of fire-retarded PP/PE copolymers using DTA/TGA coupled with FTIR. Polym. Degrad. Stab. 2000, 67, 437. [Google Scholar]
- Gold, G. UL-94-Test for Flammability of Plastic Materials for Parts in Devices and Appliances; Laird Technologies: Tech Notes: London, UK, 2021. [Google Scholar]


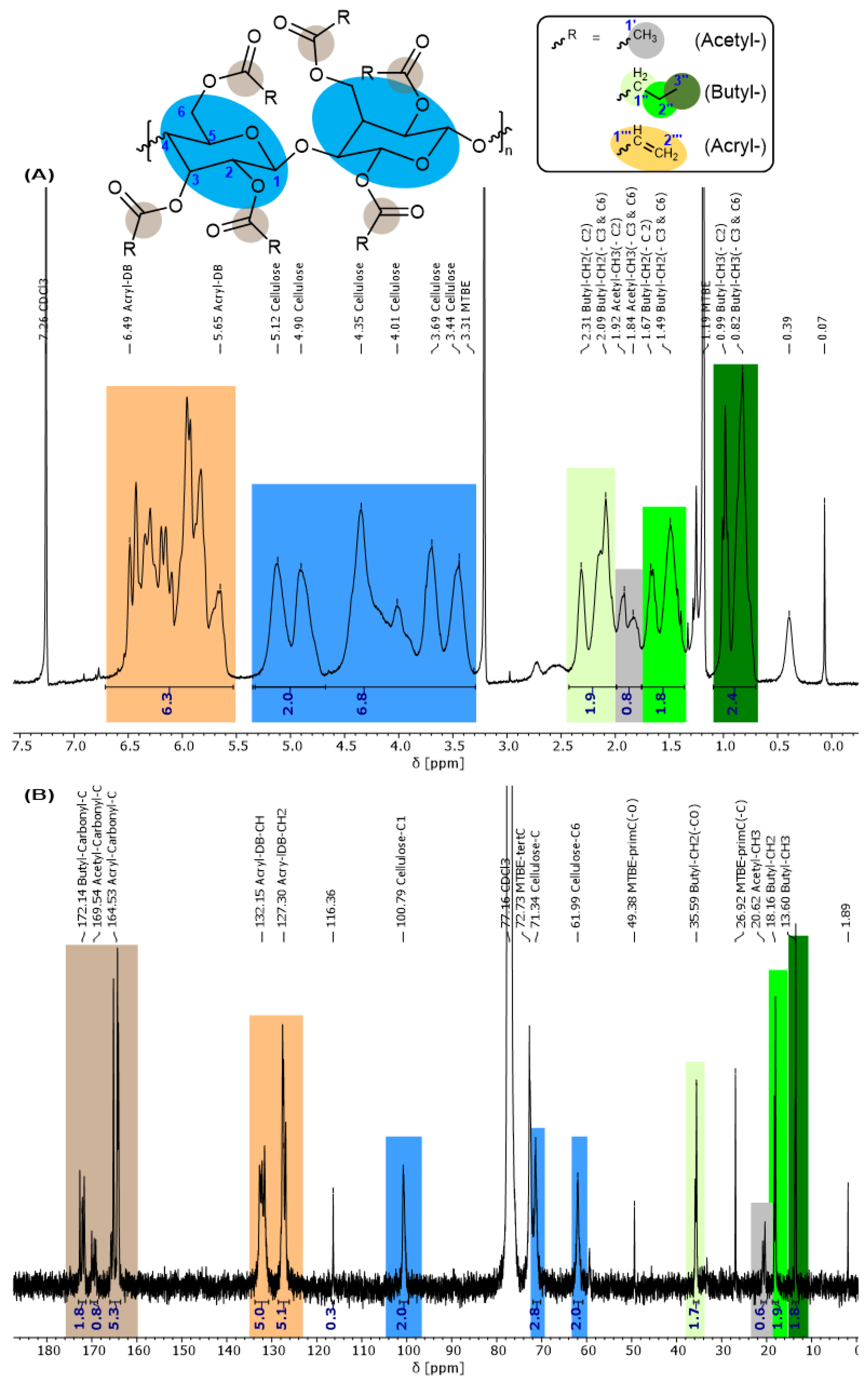

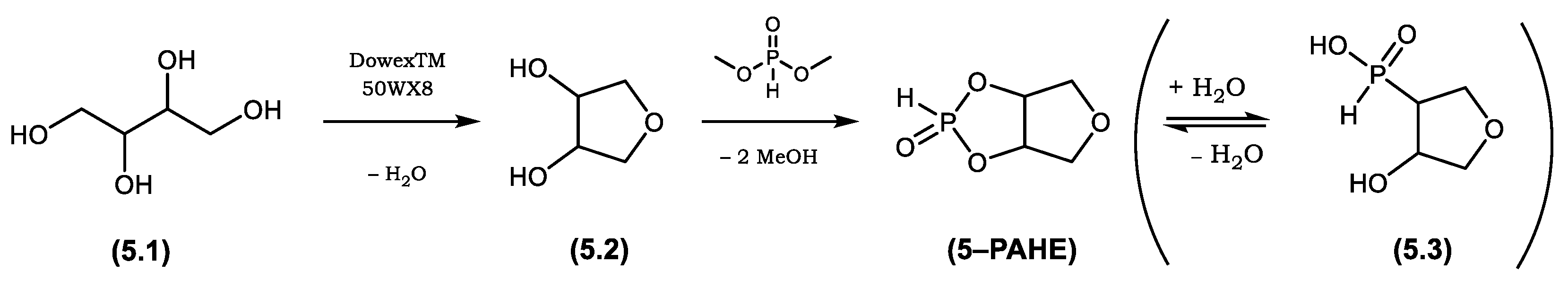
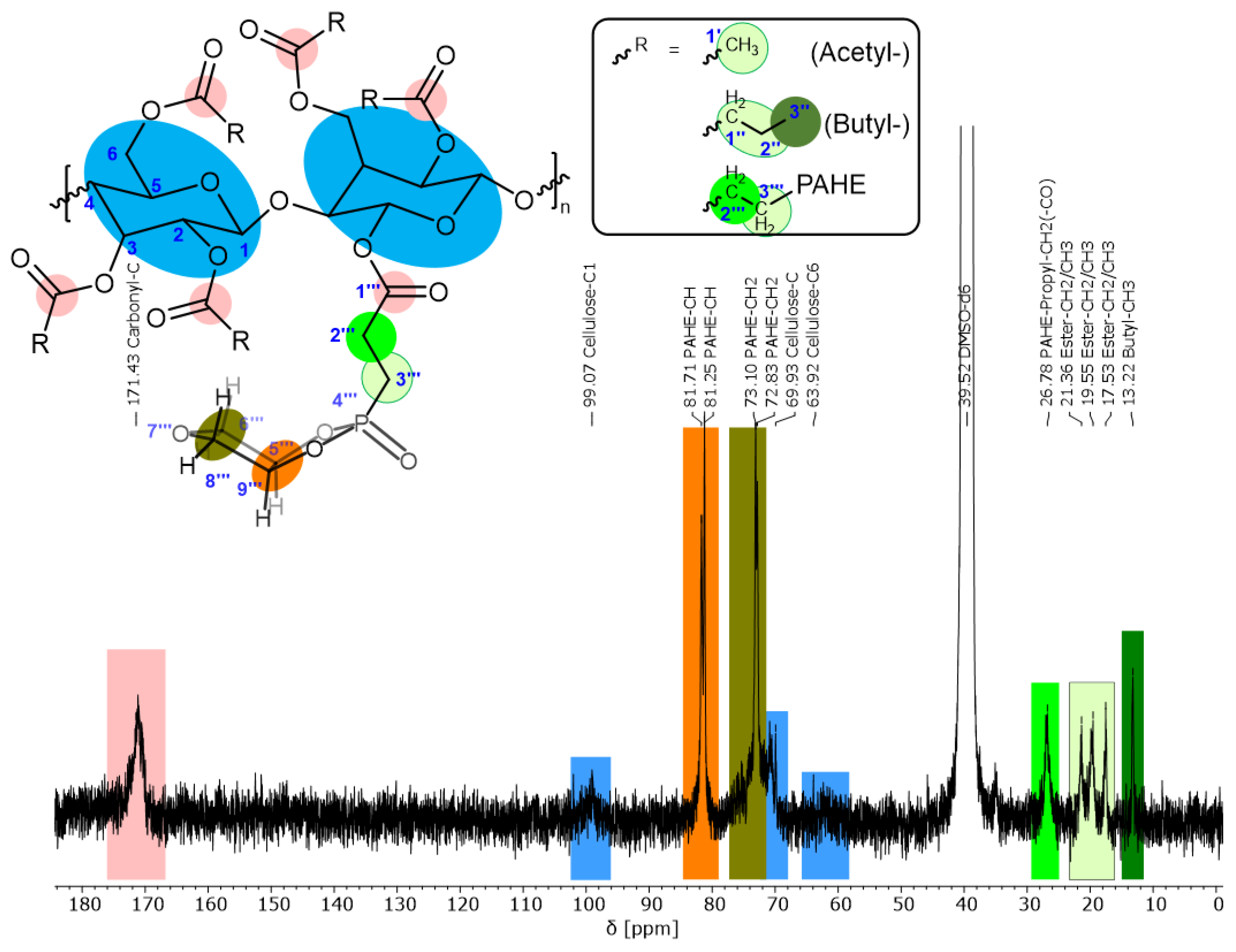
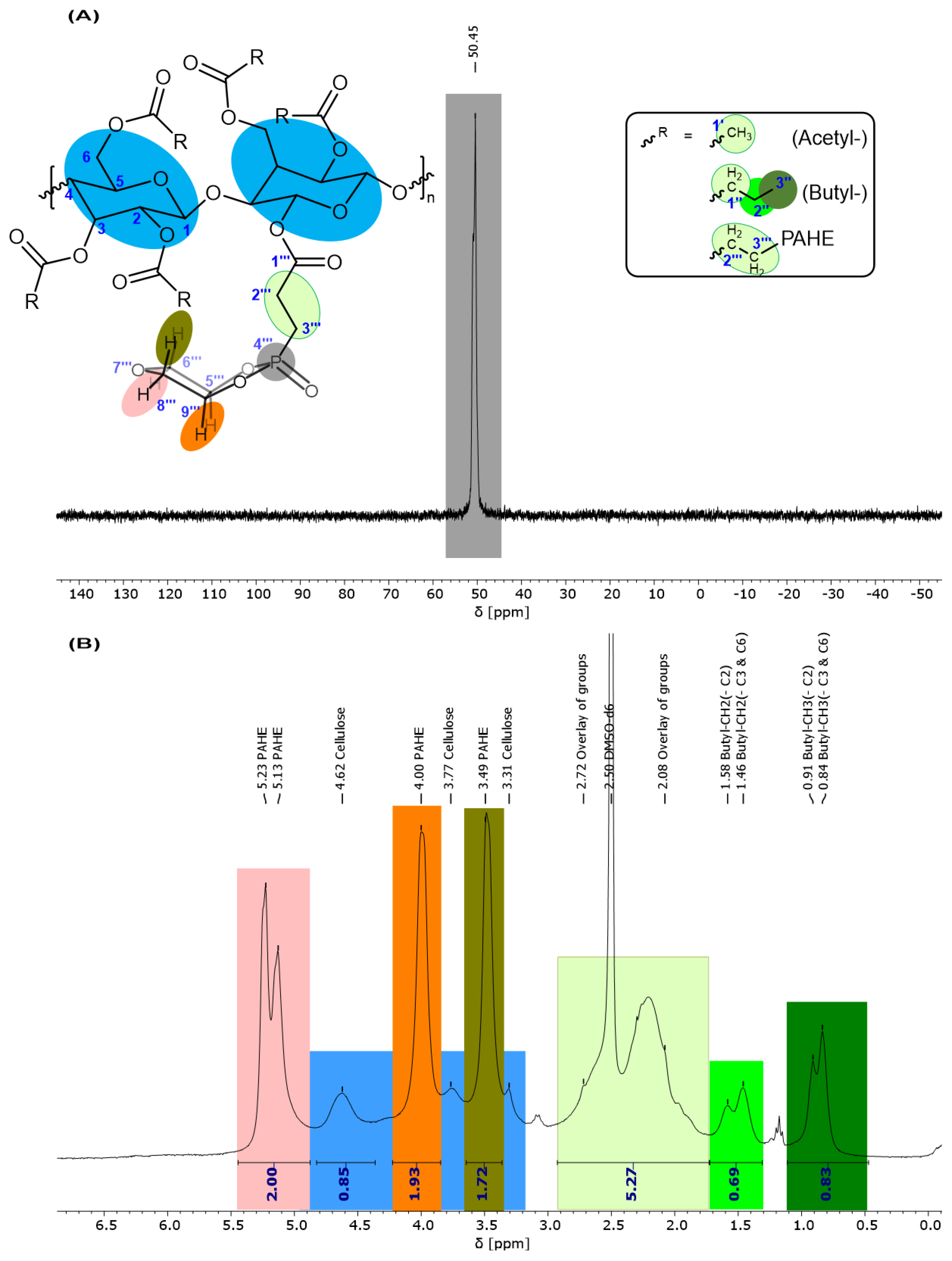




| Flame Retardant | Cellulose Type | Cellulose Modification | Catalyst for Esterification | Functional Group |
|---|---|---|---|---|
| 6-CeAcPrSA-PAHE | cotton | propionate ester | H2SO4 | PAHE |
| 6-CeAcBu-PAHE | cotton | butyrate ester | ZnCl2 | PAHE |
| 6-CeAcBu-DOPO | cotton | butyrate ester | ZnCl2 | DOPO |
| Flame Retardants | 2% Mass Loss [°C] | 5% Mass Loss [°C] | Residual Mass at 800 °C [%] |
|---|---|---|---|
| 6-CeAcPrSA-PAHE | 267 | 277 | 32.6 |
| 6-CeAcBu-PAHE | 288 | 297 | 36.7 |
| 6-CeAcBu-DOPO | 295 | 312 | 18.7 |
| Formulation No. | Composition | 6-CeAcBu-PAHE | PBDS | DBDE | ATO |
|---|---|---|---|---|---|
| I-PP-PE | PP-co-PE | - | - | - | |
| II-PP-PE | PP-co-PE +DBDE + ATO | - | - | 6.7 | 3.3 |
| III-PP-PE | PP-co-PE + PBDS | - | 10 | - | |
| IV-PP-PE | PP-co-PE + 6-CeAcBu-PAHE | 10 | - | - | |
| V-PP-PE | PP-co-PE + 6-CeAcBu-PAHE + PBDS | 8 | 2 | - |
| Formulation No. | Composition | 6.1B | DEPAL |
|---|---|---|---|
| I-PA | PA | - | - |
| II-PA | PA + DEPAL | - | 20 |
| III-PA | PA + 6-CeAcBu-PAHE | 20 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplan, M.; Ciesielski, M.; Fuchs, S.; Getterle, C.; Schönberger, F.; Pfaendner, R. Novel Macromolecular and Biobased Flame Retardants Based on Cellulose Esters and Phosphorylated Sugar Alcohols. Polymers 2023, 15, 3195. https://doi.org/10.3390/polym15153195
Kaplan M, Ciesielski M, Fuchs S, Getterle C, Schönberger F, Pfaendner R. Novel Macromolecular and Biobased Flame Retardants Based on Cellulose Esters and Phosphorylated Sugar Alcohols. Polymers. 2023; 15(15):3195. https://doi.org/10.3390/polym15153195
Chicago/Turabian StyleKaplan, Matay, Michael Ciesielski, Sabine Fuchs, Christoffer Getterle, Frank Schönberger, and Rudolf Pfaendner. 2023. "Novel Macromolecular and Biobased Flame Retardants Based on Cellulose Esters and Phosphorylated Sugar Alcohols" Polymers 15, no. 15: 3195. https://doi.org/10.3390/polym15153195
APA StyleKaplan, M., Ciesielski, M., Fuchs, S., Getterle, C., Schönberger, F., & Pfaendner, R. (2023). Novel Macromolecular and Biobased Flame Retardants Based on Cellulose Esters and Phosphorylated Sugar Alcohols. Polymers, 15(15), 3195. https://doi.org/10.3390/polym15153195