

New Light-Green Thermally Activated Delayed Fluorescence Polymer Based on Dimethylacridine-Triphenyltriazine Light-Emitting Unit and Tetraphenylsilane Moiety as Non-Conjugated Backbone

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Equipment
2.3. Thin Film Preparation for AFM and FE-SEM Analysis
2.4. Synthesis of p-TPS-DMAC-TRZ

3. Results and Discussion
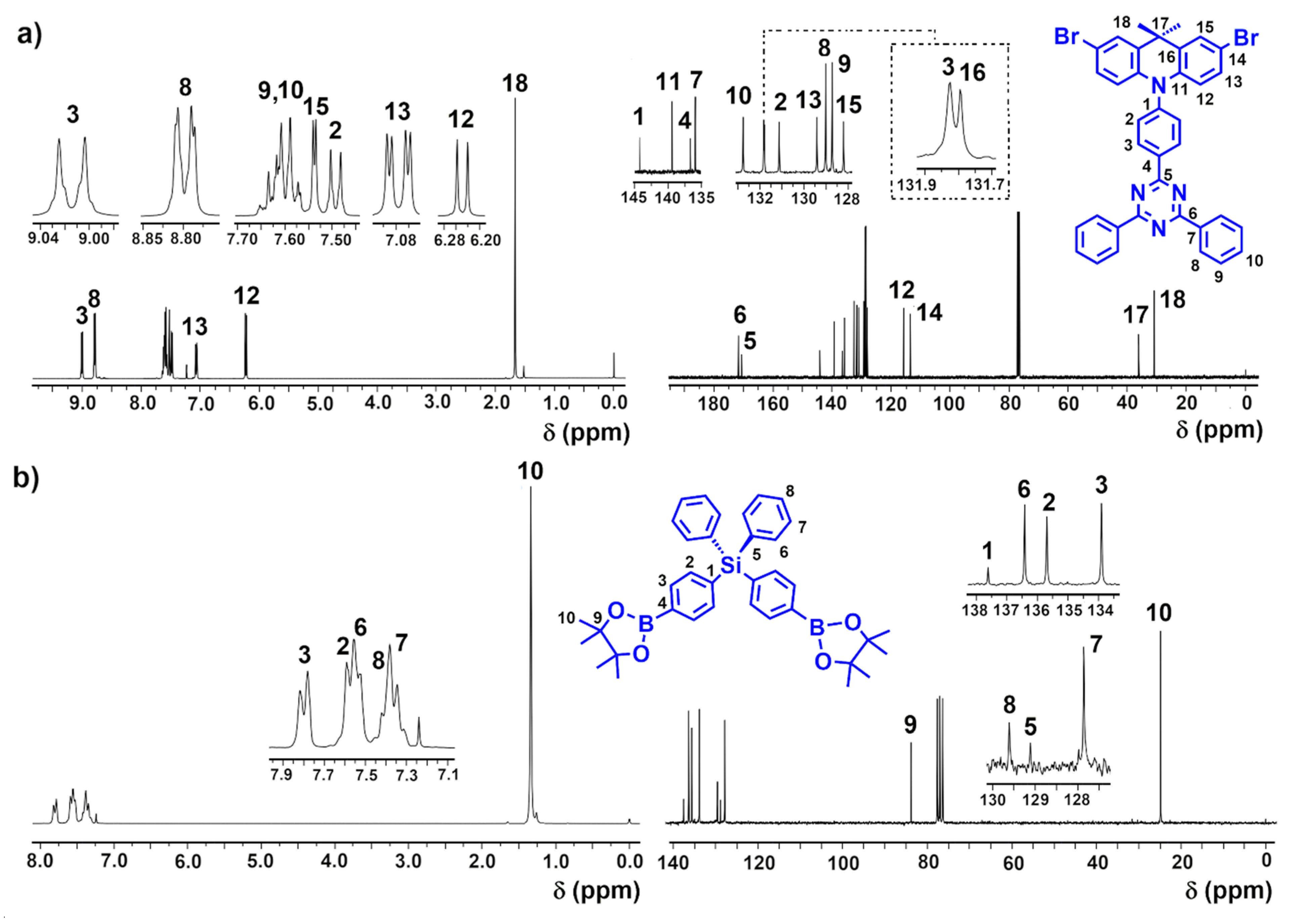
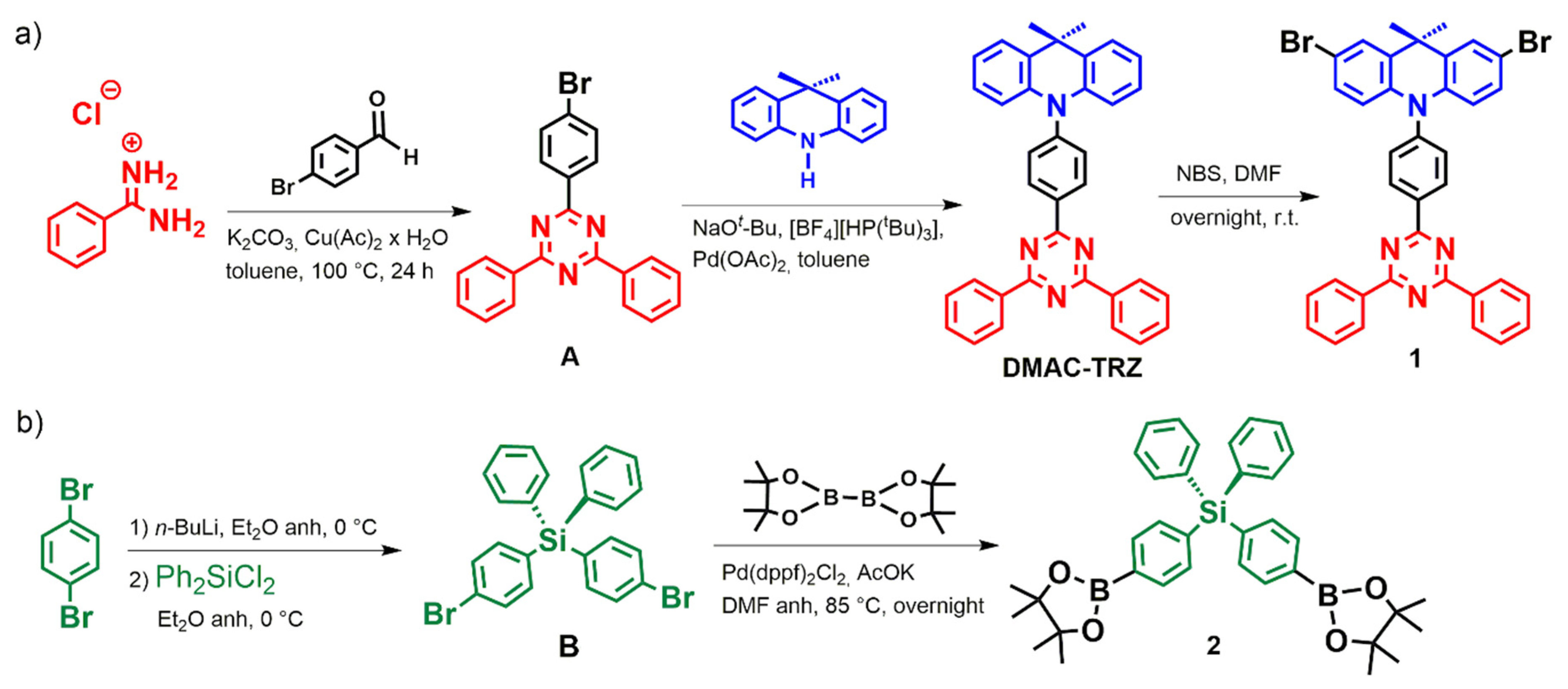
3.1. Synthesis and Characterization of Monomers
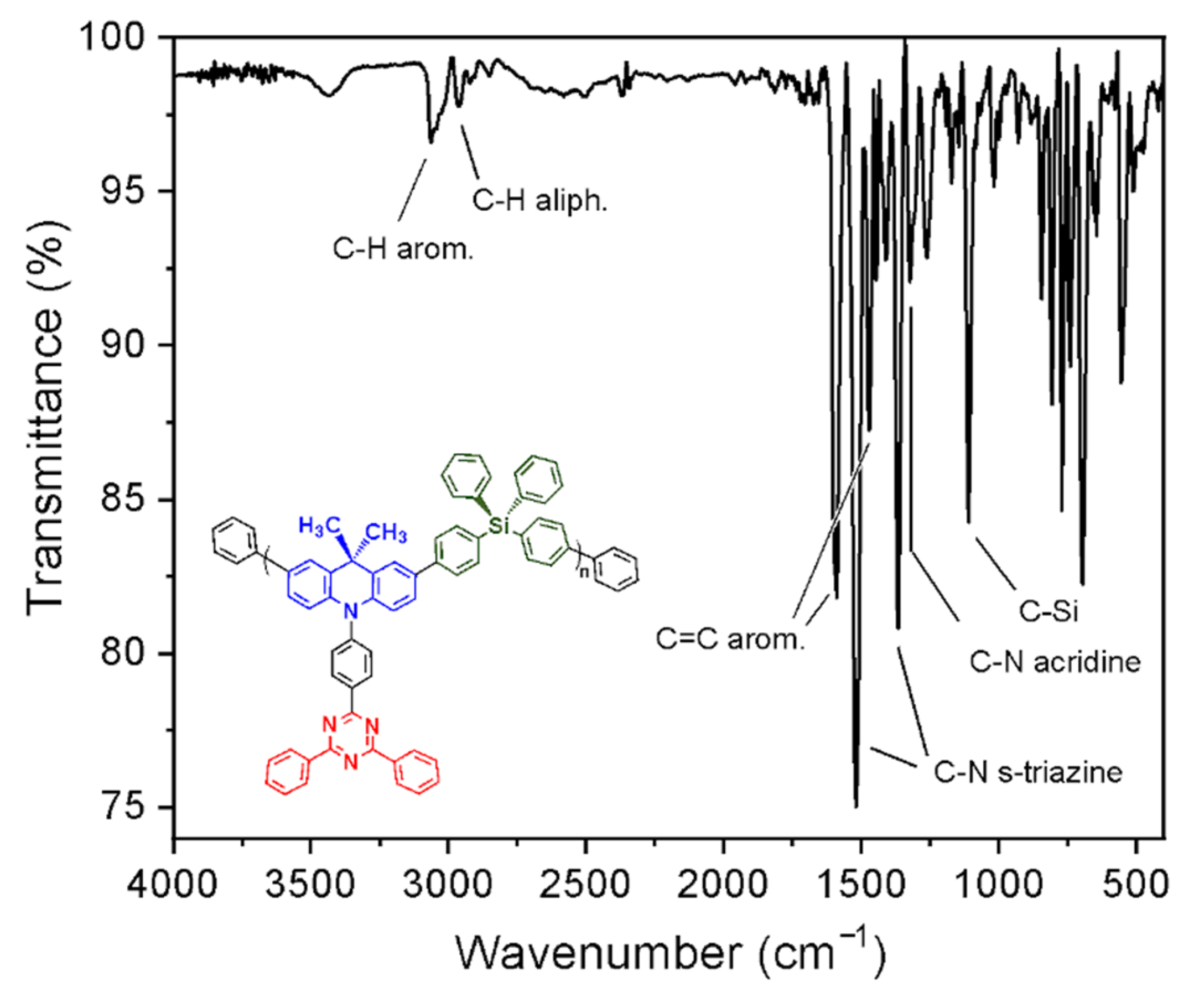
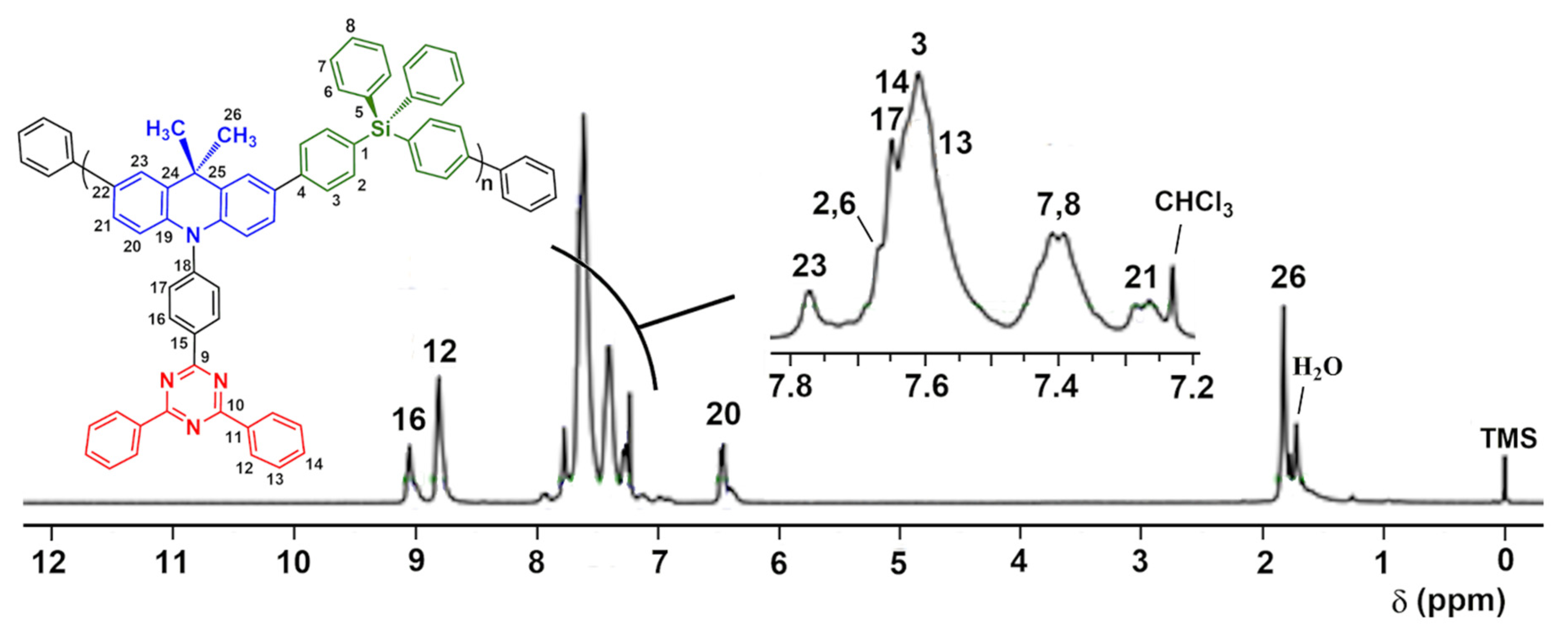
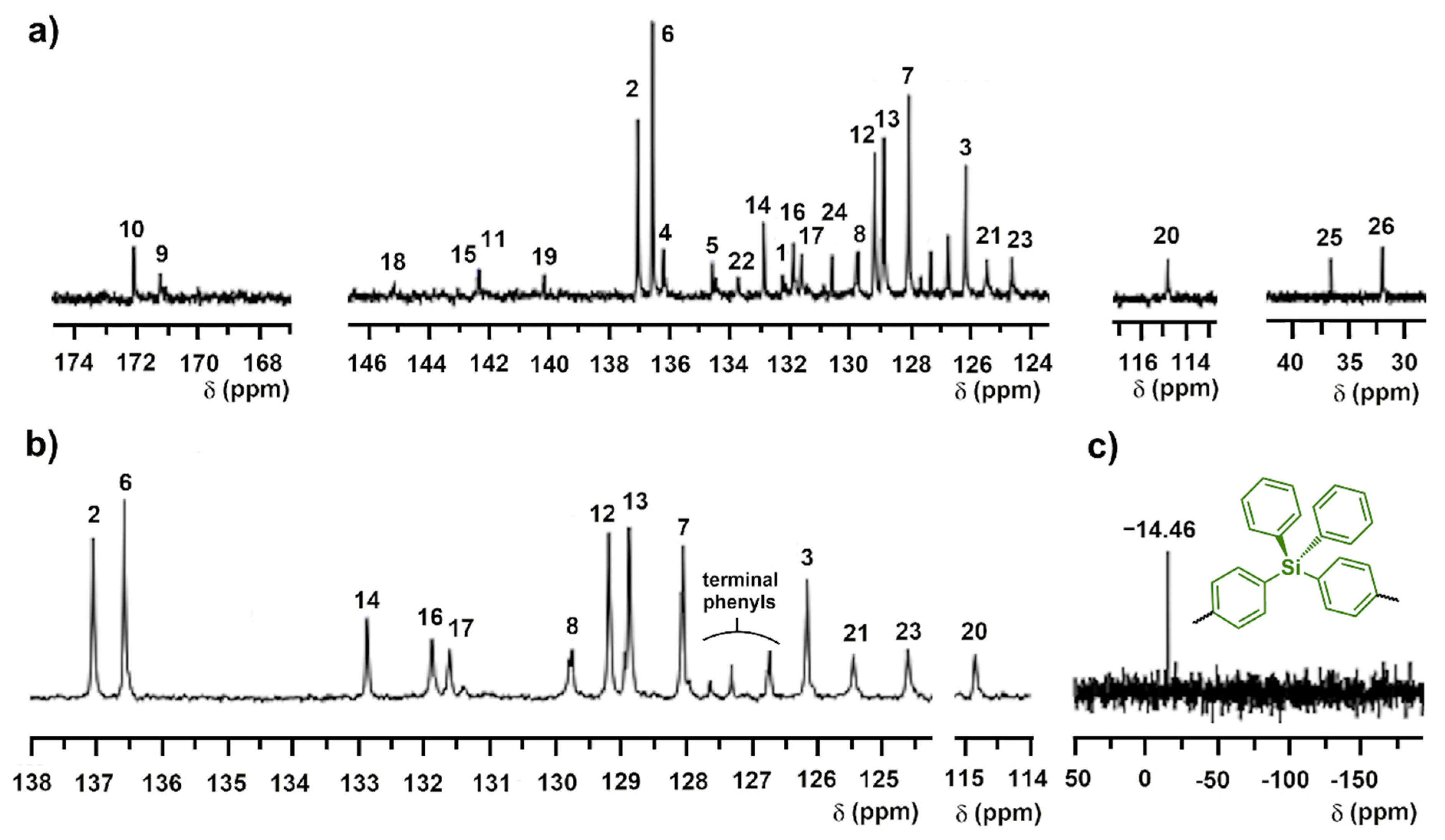
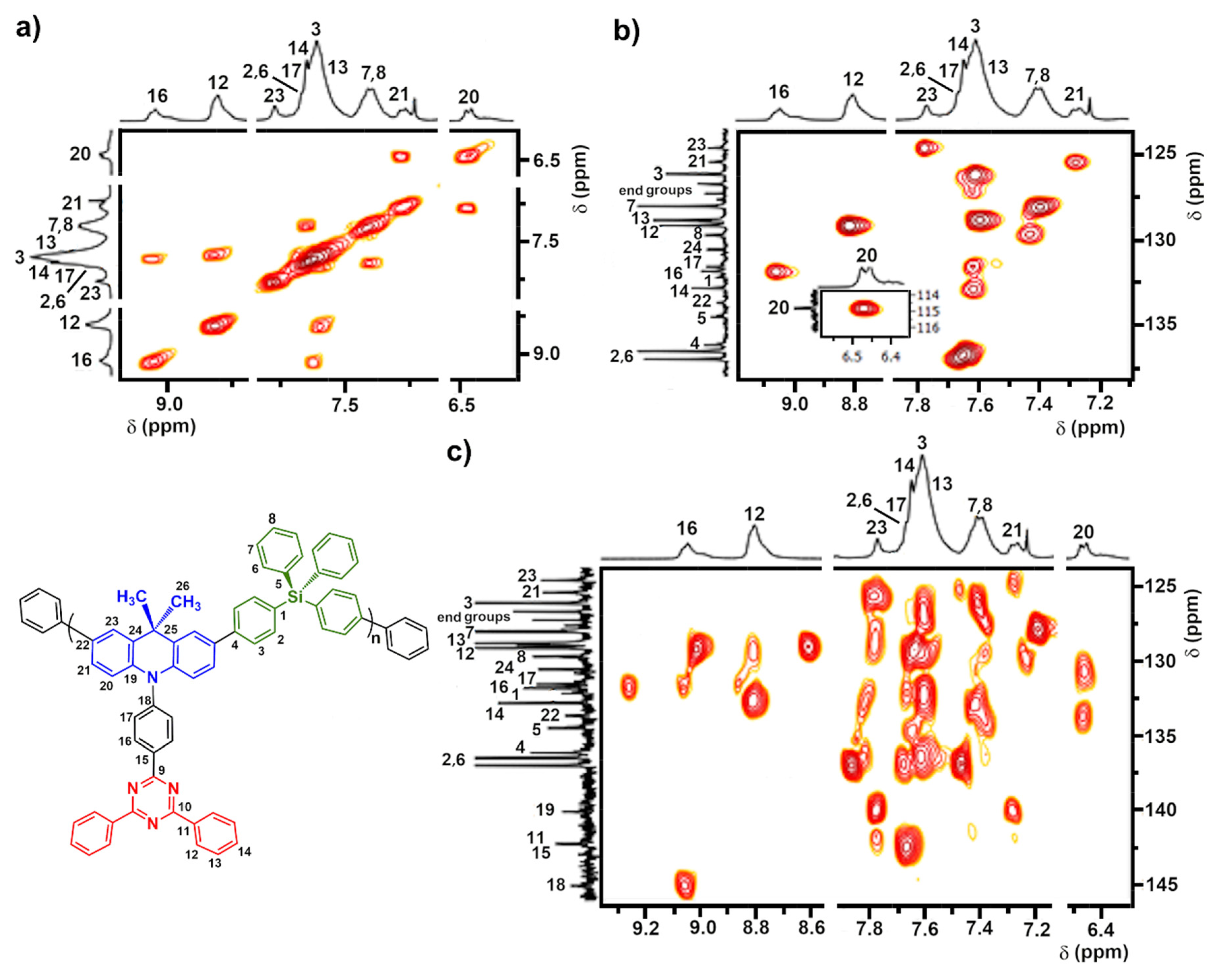
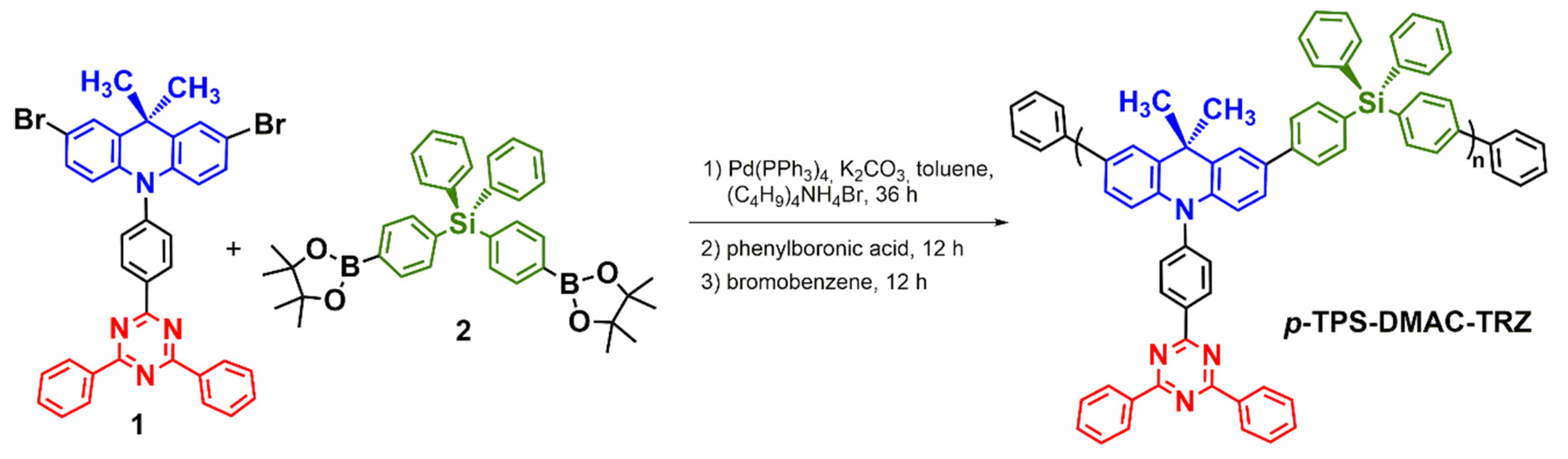
3.2. Synthesis and Structural Characterization of p-TPS-DMAC-TRZ
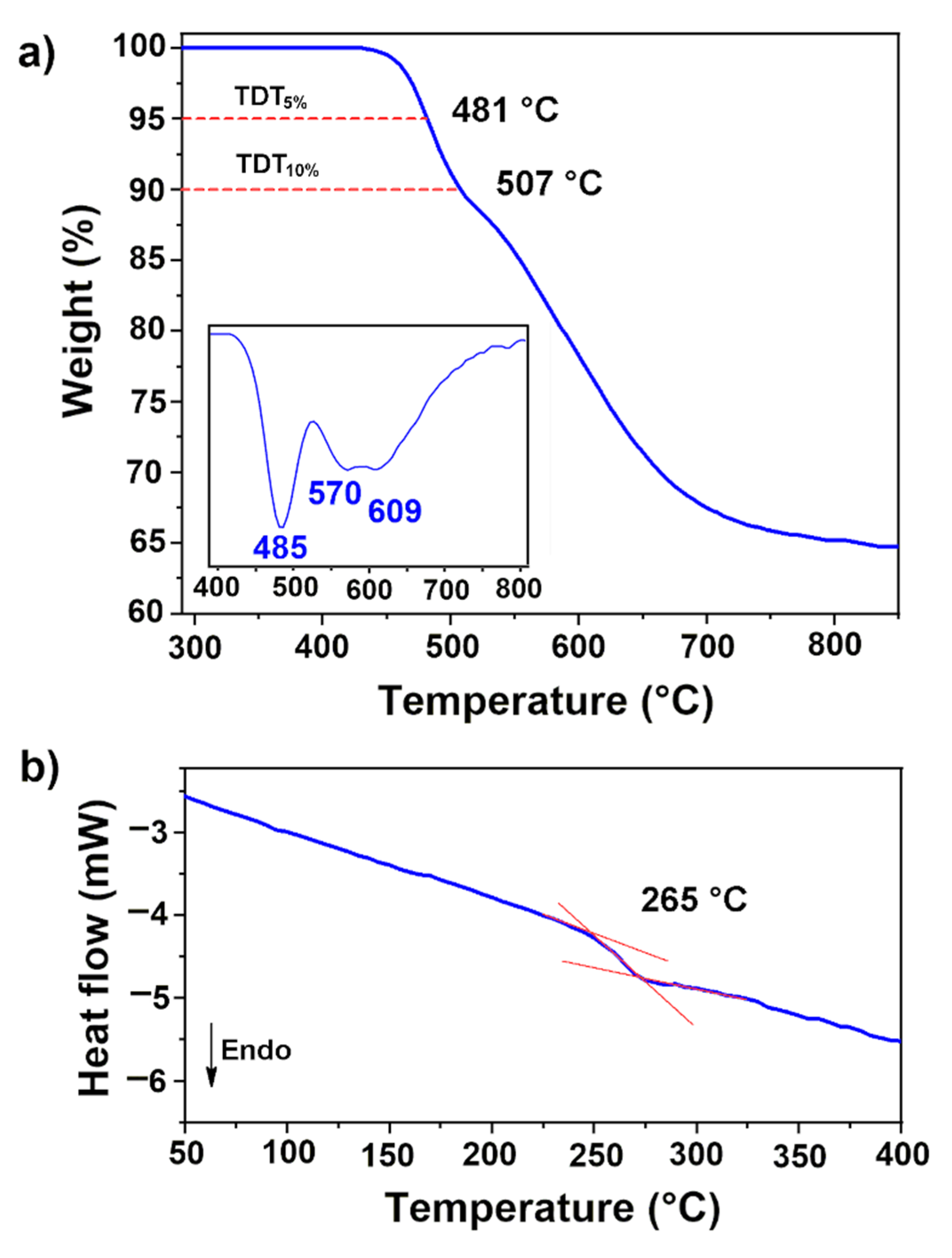
3.3. Solubility, Molecular Weights and Thermal Properties
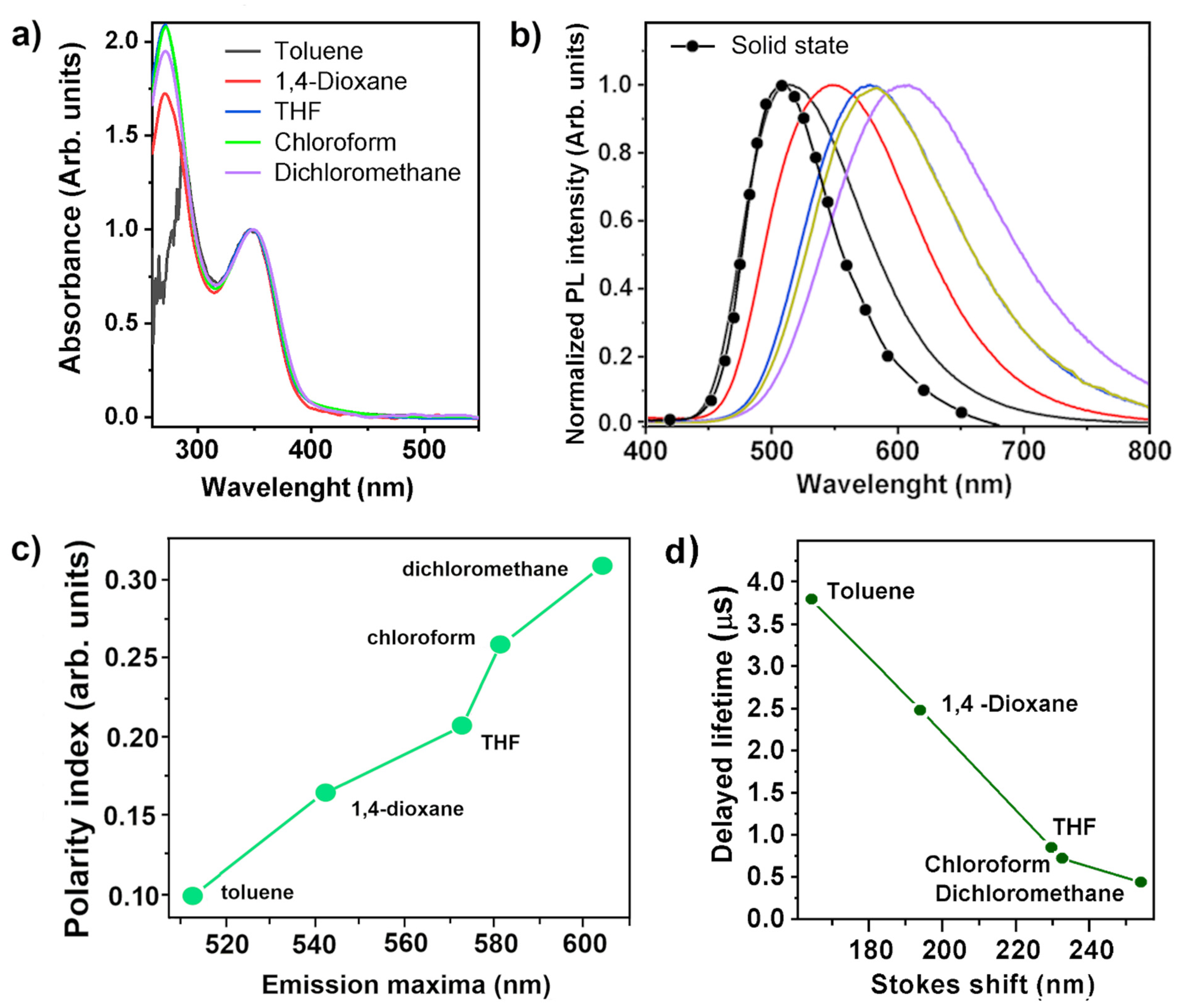
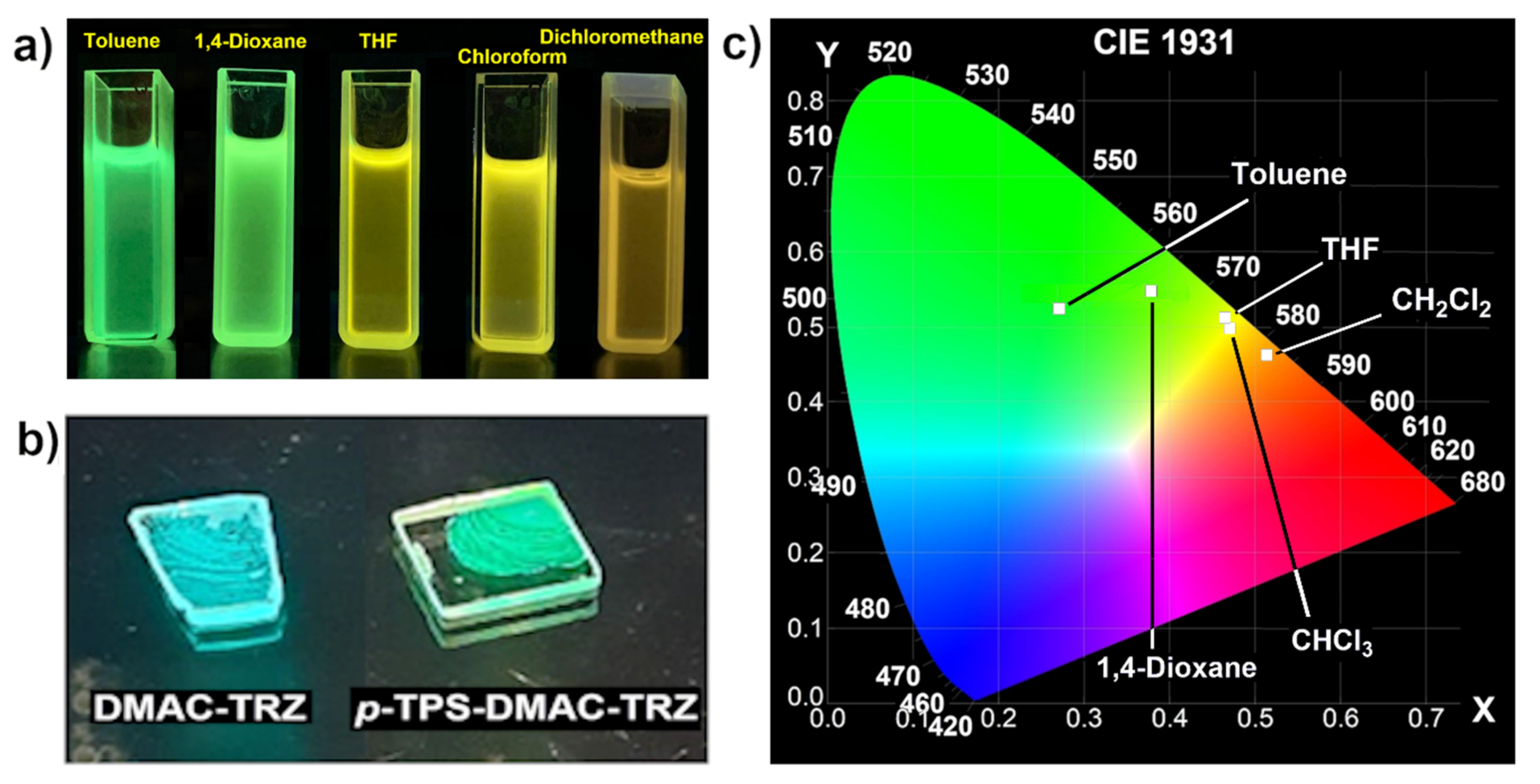
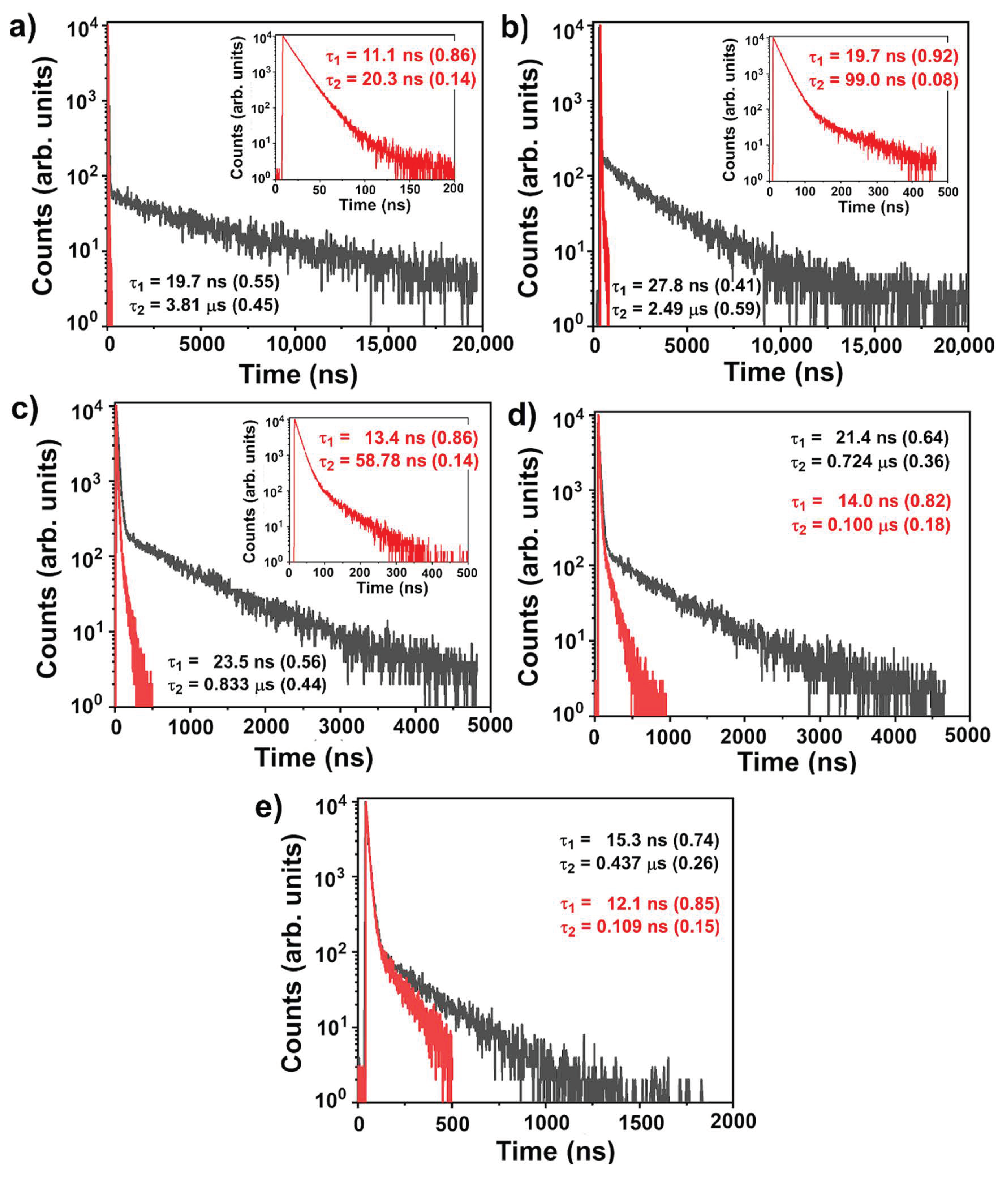
3.4. Photophysical Properties
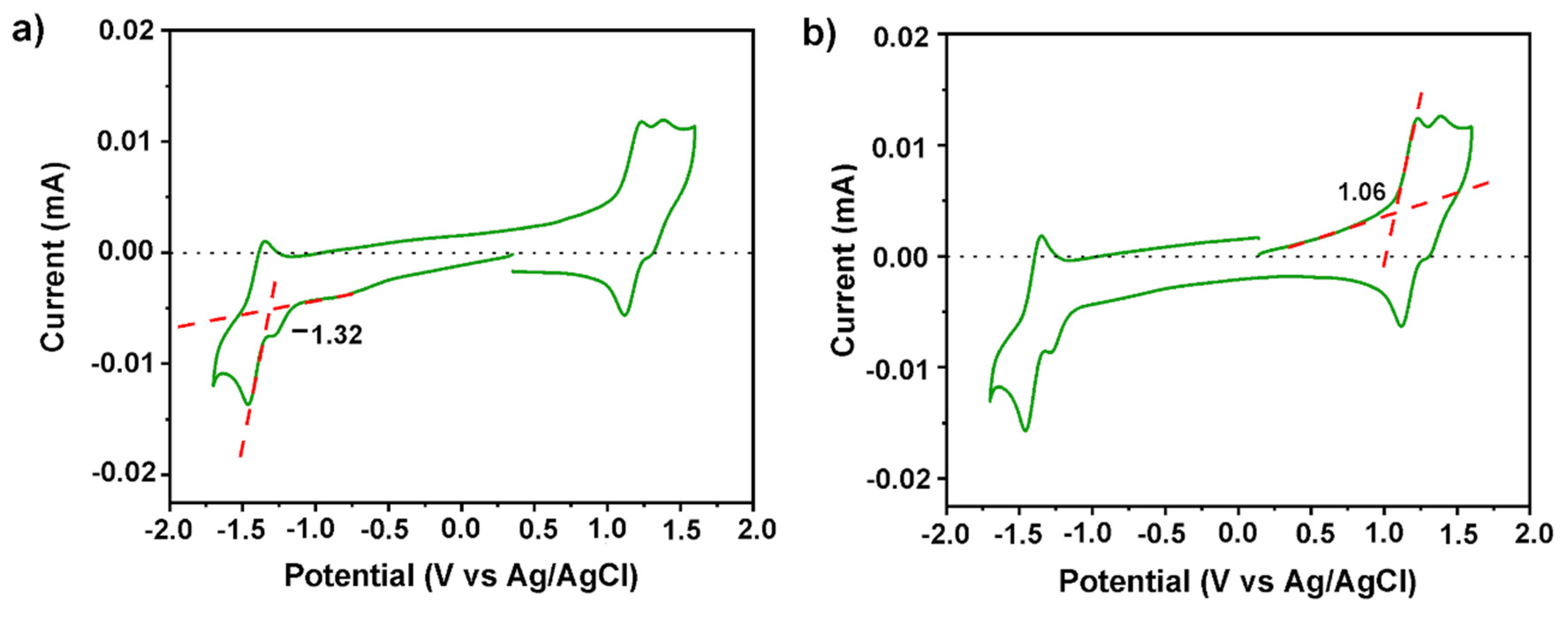
3.5. Electrochemical Properties
3.6. Morphological Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, Y.; Gong, S.; Xie, G.; Yang, C. Design Strategy for Solution-Processable Thermally Activated Delayed Fluorescence Emitters and Their Applications in Organic Light-Emitting Diodes. Adv. Opt. Mater. 2018, 6, 1800568. [Google Scholar] [CrossRef]
- Zhang, D.; Huang, T.; Duan, L. Emerging Self-Emissive Technologies for Flexible Displays. Adv. Mater. 2020, 32, 1902391. [Google Scholar] [CrossRef] [PubMed]
- Reineke, S.; Thomschke, M.; Lüssem, B.; Leo, K. White Organic Light-Emitting Diodes: Status and Perspective. Rev. Mod. Phys. 2013, 85, 1245–1293. [Google Scholar] [CrossRef]
- Anand, V.; Mishra, R.; Barot, Y. Recent Advances in the Development of Pure Organic White Light Emitters. Dye. Pigment. 2021, 191, 109390. [Google Scholar] [CrossRef]
- Attili, S.K.; Lesar, A.; McNeill, A.; Camacho-Lopez, M.; Moseley, H.; Ibbotson, S.; Samuel, I.D.W.; Ferguson, J. An Open Pilot Study of Ambulatory Photodynamic Therapy Using a Wearable Low-Irradiance Organic Light-Emitting Diode Light Source in the Treatment of Nonmelanoma Skin Cancer. Br. J. Dermatol. 2009, 161, 170–173. [Google Scholar] [CrossRef]
- Murawski, C.; Gather, M.C. Emerging Biomedical Applications of Organic Light-Emitting Diodes. Adv. Opt. Mater. 2021, 9, 2100269. [Google Scholar] [CrossRef]
- Shi, Y.Z.; Wu, H.; Wang, K.; Yu, J.; Ou, X.M.; Zhang, X.H. Recent Progress in Thermally Activated Delayed Fluorescence Emitters for Nondoped Organic Light-Emitting Diodes. Chem. Sci. 2022, 13, 3625–3651. [Google Scholar] [CrossRef]
- Zhang, Y.; Biswas, R. High Light Outcoupling Efficiency from Periodically Corrugated OLEDs. ACS Omega 2021, 6, 9291–9301. [Google Scholar] [CrossRef]
- Corrêa Santos, D.; Vieira Marques, M.d.F. Blue Light Polymeric Emitters for the Development of OLED Devices. J. Mater. Sci. Mater. Electron. 2022, 33, 12529–12565. [Google Scholar] [CrossRef]
- Adachi, C. Third-Generation Organic Electroluminescence Materials. Jpn. J. Appl. Phys. 2014, 53, 60101. [Google Scholar] [CrossRef]
- Sarma, M.; Wong, K.T. Exciplex: An Intermolecular Charge-Transfer Approach for TADF. ACS Appl. Mater. Interfaces 2018, 10, 19279–19304. [Google Scholar] [CrossRef] [PubMed]
- Achelle, S.; Hodée, M.; Massue, J.; Fihey, A.; Katan, C. Diazine-Based Thermally Activated Delayed Fluorescence Chromophores. Dye. Pigment. 2022, 200, 110157. [Google Scholar] [CrossRef]
- Wei, X.; Hu, T.; Li, Z.; Liu, Y.; Hu, X.; Gao, H.; Liu, G.; Wang, P.; Yi, Y.; Wang, Y. Rational Strategy of Exciplex-Type Thermally Activated Delayed Fluorescent (TADF) Emitters: Stacking of Donor and Acceptor Units of the Intramolecular TADF Molecule. Chem. Eng. J. 2022, 433, 133546. [Google Scholar] [CrossRef]
- Endo, A.; Sato, K.; Yoshimura, K.; Kai, T.; Kawada, A.; Miyazaki, H.; Adachi, C. Efficient Up-Conversion of Triplet Excitons into a Singlet State and Its Application for Organic Light Emitting Diodes. Appl. Phys. Lett. 2011, 98, 83302. [Google Scholar] [CrossRef]
- Nakanotani, H.; Tsuchiya, Y.; Adachi, C. Thermally-Activated Delayed Fluorescence for Light-Emitting Devices. Chem. Lett. 2021, 50, 938–948. [Google Scholar] [CrossRef]
- Im, Y.; Kim, M.; Cho, Y.J.; Seo, J.A.; Yook, K.S.; Lee, J.Y. Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater. 2017, 29, 1946–1963. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent Advances in Organic Thermally Activated Delayed Fluorescence Materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M.R. All-Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Nat. Rev. Mater. 2018, 3, 18020. [Google Scholar] [CrossRef]
- Xie, Y.; Li, Z. Thermally Activated Delayed Fluorescent Polymers. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 575–584. [Google Scholar] [CrossRef]
- Yin, X.; He, Y.; Wang, X.; Wu, Z.; Pang, E.; Xu, J.; Wang, J. Recent Advances in Thermally Activated Delayed Fluorescent Polymer—Molecular Designing Strategies. Front. Chem. 2020, 8, 725. [Google Scholar] [CrossRef]
- Zhang, B.; Cheng, Y. Recent Advances in Conjugated TADF Polymer Featuring in Backbone-Donor/Pendant-Acceptor Structure: Material and Device Perspectives. Chem. Rec. 2019, 19, 1624–1643. [Google Scholar] [CrossRef]
- Godumala, M.; Choi, S.; Kim, H.J.; Lee, C.; Park, S.; Moon, J.S.; Si Woo, K.; Kwon, J.H.; Cho, M.J.; Choi, D.H. Novel Dendritic Large Molecules as Solution-Processable Thermally Activated Delayed Fluorescent Emitters for Simple Structured Non-Doped Organic Light Emitting Diodes. J. Mater. Chem. C 2018, 6, 1160–1170. [Google Scholar] [CrossRef]
- Zong, W.; Qiu, W.; Yuan, P.; Wang, F.; Liu, Y.; Xu, S.; Su, S.J.; Cao, S. Thermally Activated Delayed Fluorescence Polymers for High-Efficiency Solution-Processed Non-Doped OLEDs: Convenient Synthesis by Binding TADF Units and Host Units to the Pre-Synthesized Polycarbazole-Based Backbone via Click Reaction. Polymer 2022, 240, 124468. [Google Scholar] [CrossRef]
- Yook, K.S.; Lee, J.Y. Organic Materials for Deep Blue Phosphorescent Organic Light-Emitting Diodes. Adv. Mater. 2012, 24, 3169–3190. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, W.; Fan, Z.; Li, J.; Chen, X.; Luo, G.; Zhang, X. Synthesis and Characterization of High Triplet Energy Polyfluorene Bearing M-Tetraphenylsilane Segment as a Polymer Host for Green Phosphorescent Polymer Light Emitting Diodes. Synth. Met. 2015, 204, 70–75. [Google Scholar] [CrossRef]
- Xu, F.; Kim, J.H.; Kim, H.U.; Jang, J.H.; Yook, K.S.; Lee, J.Y.; Hwang, D.H. Synthesis of High-Triplet-Energy Host Polymer for Blue and White Electrophosphorescent Light-Emitting Diodes. Macromolecules 2014, 47, 7397–7406. [Google Scholar] [CrossRef]
- Sun, D.; Zhou, X.; Li, H.; Sun, X.; Zheng, Y.; Ren, Z.; Ma, D.; Bryce, M.R.; Yan, S. A Versatile Hybrid Polyphenylsilane Host for Highly Efficient Solution-Processed Blue and Deep Blue Electrophosphorescence. J. Mater. Chem. C 2014, 2, 8277–8284. [Google Scholar] [CrossRef]
- Yeh, H.C.; Chien, C.H.; Shih, P.I.; Yuan, M.C.; Shu, C.F. Polymers Derived from 3,6-Fluorene and Tetraphenylsilane Derivatives: Solution-Processable Host Materials for Green Phosphorescent OLEDs. Macromolecules 2008, 41, 3801–3807. [Google Scholar] [CrossRef]
- Sun, D.; Ren, Z.; Bryce, M.R.; Yan, S. Arylsilanes and Siloxanes as Optoelectronic Materials for Organic Light-Emitting Diodes (OLEDs). J. Mater. Chem. C 2015, 3, 9496–9508. [Google Scholar] [CrossRef]
- Choi, S.; Godumala, M.; Lee, J.H.; Kim, G.H.; Moon, J.S.; Kim, J.Y.; Yoon, D.W.; Yang, J.H.; Kim, J.; Cho, M.J.; et al. Optimized Structure of Silane-Core Containing Host Materials for Highly Efficient Blue TADF OLEDs. J. Mater. Chem. C 2017, 5, 6570–6577. [Google Scholar] [CrossRef]
- Choi, S.; Yoon, J.W.; Godumala, M.; Kim, H.J.; Park, S.H.; Kim, S.K.; Lee, H.; Kwon, J.H.; Cho, M.J.; Choi, D.H. 2D-σ-2A Type Cruciform Host Material with Silane Core for Highly Efficient Solution-Processable Green Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes. Dye. Pigment. 2019, 167, 120–126. [Google Scholar] [CrossRef]
- Yun, J.H.; Lee, K.H.; Chung, W.J.; Lee, J.Y.; Lee, Y.; Lyu, J.J. Thermally Activated Delayed Fluorescence Type Exciplex Host for Long Lifetime in Deep Blue Phosphorescent Organic Light-Emitting Diodes. Chem. Eng. J. 2021, 417, 128086. [Google Scholar] [CrossRef]
- Tsai, W.L.; Huang, M.H.; Lee, W.K.; Hsu, Y.J.; Pan, K.C.; Huang, Y.H.; Ting, H.C.; Sarma, M.; Ho, Y.Y.; Hu, H.C.; et al. A Versatile Thermally Activated Delayed Fluorescence Emitter for Both Highly Efficient Doped and Non-Doped Organic Light Emitting Devices. Chem. Commun. 2015, 51, 13662–13665. [Google Scholar] [CrossRef]
- Bergen, A.; Bohne, C.; Fuentealba, D.; Ihmels, H.; Pace, T.C.S.; Waidelich, M.; Yihwa, C.; Willem Bats, J. Studies of the Solvatochromic Emission Properties of N-Aroylurea Derivatives I: Influence of the Substitution Pattern. Photochem. Photobiol. Sci. 2012, 11, 752–767. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Yang, L.; Lu, X.; Zou, H.; Yang, W.; Zhang, Y.; Li, Z.; Ma, M. Synthesis, Spectra, and Theoretical Investigations of 1,3,5-Triazines Compounds as Ultraviolet Rays Absorber Based on Time-Dependent Density Functional Calculations and Three-Dimensional Quantitative Structure-Property Relationship. J. Fluoresc. 2018, 28, 707–723. [Google Scholar] [CrossRef]
- Kim, K.H.; Baek, J.Y.; Cheon, C.W.; Moon, C.K.; Sim, B.; Choi, M.Y.; Kim, J.J.; Kim, Y.H. Highly Efficient Non-Doped Deep Blue Fluorescent Emitters with Horizontal Emitting Dipoles Using Interconnecting Units between Chromophores. Chem. Commun. 2016, 52, 10956–10959. [Google Scholar] [CrossRef]
- Tanaka, H.; Shizu, K.; Miyazaki, H.; Adachi, C. Efficient Green Thermally Activated Delayed Fluorescence (TADF) from a Phenoxazine–Triphenyltriazine (PXZ–TRZ) Derivative. Chem. Commun. 2012, 48, 11392–11394. [Google Scholar] [CrossRef]
- Dorel, R.; Grugel, C.P.; Haydl, A.M. The Buchwald–Hartwig Amination After 25 Years. Angew. Chem. Int. Ed. 2019, 58, 17118–17129. [Google Scholar] [CrossRef]
- Heravi, M.M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. Buchwald-Hartwig Reaction: An Overview. J. Organomet. Chem. 2018, 861, 17–104. [Google Scholar] [CrossRef]
- Tundidor-Camba, A.; González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Tagle, L.H.; Hauyón, R.A.; Sobarzo, P.A.; González, A.; Ortiz, P.A.; Maya, E.M.; Terraza, C.A. Silylated Oligomeric Poly(Ether-Azomethine)s from Monomers Containing Biphenyl Moieties: Synthesis and Characterization. RSC Adv. 2018, 8, 1296–1312. [Google Scholar] [CrossRef]
- Guo, X.; Gao, B.; Cui, X.; Wang, J.; Dong, W.; Duan, Q.; Fei, T.; Su, Z. PL Sensor for Sensitive and Selective Detection of 2,4,6-Trinitrophenol Based on Carbazole and Tetraphenylsilane Polymer. Dye. Pigment. 2021, 191, 109379. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, C.; Godumala, M.; Choi, S.; Park, S.Y.; Cho, M.J.; Park, S.; Choi, D.H. Solution-Processed Thermally Activated Delayed Fluorescence Organic Light-Emitting Diodes Using a New Polymeric Emitter Containing Non-Conjugated Cyclohexane Units. Polym. Chem. 2018, 9, 1318–1326. [Google Scholar] [CrossRef]
- Lee, S.Y.; Yasuda, T.; Komiyama, H.; Lee, J.; Adachi, C. Thermally Activated Delayed Fluorescence Polymers for Efficient Solution-Processed Organic Light-Emitting Diodes. Adv. Mater. 2016, 28, 4019–4024. [Google Scholar] [CrossRef]
- Xie, G.; Luo, J.; Huang, M.; Chen, T.; Wu, K.; Gong, S.; Yang, C. Inheriting the Characteristics of TADF Small Molecule by Side-Chain Engineering Strategy to Enable Bluish-Green Polymers with High PLQYs up to 74% and External Quantum Efficiency over 16% in Light-Emitting Diodes. Adv. Mater. 2017, 29, 1604223. [Google Scholar] [CrossRef] [PubMed]
- Hauyon, R.A.; Garrido-Gatica, G.; Sobarzo, P.A.; González-Henríquez, C.M.; Tagle, L.H.; Rodríguez-González, F.E.; Jessop, I.A.; Recabarren-Gajardo, G.; Tundidor-Camba, A.; Terraza, C.A. New Cardo Silylated Poly(Azomethine)s Containing 9,9′-Diphenylfluorene Units as Materials with Brønsted Acid-Dependent Fluorescence. Polym. Int. 2020, 69, 239–247. [Google Scholar] [CrossRef]
- Sobarzo, P.A.; Mariman, A.P.; Sánchez, C.O.; Hauyon, R.A.; Rodríguez-González, F.E.; Medina, J.; Jessop, I.A.; Recabarren-Gajardo, G.; Tundidor-Camba, A.; Terraza, C.A. Comparison between Poly(Azomethine)s and Poly(p-Phenylvinylene)s Containing a Di-R-Diphenylsilane (R = Methyl or Phenyl) Moiety. Optical, Electronic and Thermal Properties. Eur. Polym. J. 2021, 159, 110714. [Google Scholar] [CrossRef]
- Lin, K.H.; Paterson, L.; May, F.; Andrienko, D. Glass Transition Temperature Prediction of Disordered Molecular Solids. npj Comput. Mater. 2021, 7, 179. [Google Scholar] [CrossRef]
- Zhao, Y.; Fu, C.; Fu, L.; Liu, Y.; Lu, Z.; Pu, X. Data-Driven Machine Learning Models for Quick Prediction of Thermal Stability Properties of OLED Materials. Mater. Today Chem. 2021, 22, 100625. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Chen, X.; Xiao, X.; Zhao, J. Application of Time-Resolved Electron Paramagnetic Resonance Spectroscopy in the Mechanistic Study of Thermally Activated Delayed Fluorescence (TADF) Materials. J. Mater. Chem. C 2021, 10, 4546–4557. [Google Scholar] [CrossRef]
- Chen, T.; Chen, Z.; Ni, F.; Xie, G.; Yang, C. Sky-Blue Thermally Activated Delayed Fluorescence Polymers by Using a Conjugation-Confined Poly (Aryl Ether) Main Chain. Polym. Chem. 2021, 12, 2490–2497. [Google Scholar] [CrossRef]
- Dos Santos, P.L.; Ward, J.S.; Bryce, M.R.; Monkman, A.P. Using Guest-Host Interactions to Optimize the Efficiency of TADF OLEDs. J. Phys. Chem. Lett. 2016, 7, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, V.; Brooks, J.; Tamayo, A.; Alexander, A.M.; Djurovich, P.I.; D’Andrade, B.W.; Adachi, C.; Forrest, S.R.; Thompson, M.E. High Efficiency Single Dopant White Electrophosphorescent Light Emitting Diodesy. New J. Chem. 2002, 26, 1171–1178. [Google Scholar] [CrossRef]
- Shayani-jam, H. Electrochemical Study of Adsorption and Electrooxidation of 4,4′-Biphenol on the Glassy Carbon Electrode: Determination of the Orientation of Adsorbed Molecules. Mon. Chem. 2019, 150, 183–192. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Bai, Q.; Sun, X.; Zhao, L.; Liu, H.; Gao, Y.; Zhang, H.; Yang, B.; Lu, P. Molecular Understanding of Diphenylether-, 9,9-Biphenylfluorene- and Tetraphenylsilane-Centered Wide Bandgap Host Materials for Highly Efficient Blue Phosphorescent OLEDs. Dye. Pigment. 2019, 160, 898–908. [Google Scholar] [CrossRef]
- Pan, L.; Hu, B.; Zhu, X.; Chen, X.; Shang, J.; Tan, H.; Xue, W.; Zhu, Y.; Liu, G.; Li, R.W. Role of Oxadiazole Moiety in Different D-A Polyazothines and Related Resistive Switching Properties. J. Mater. Chem. C 2013, 1, 4556–4564. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Solution λabsmax/ λemmax (nm) | ||||
| Toluene | 1,4-Dioxane | THF | Chloroform | Dichloromethane |
| 349/513 | 348/542 | 348/578 | 349/581 | 350/604 |
| In film (contribution) | ||||
| λem,max (nm) | Εgopt (eV) a | τ77 K (ns) | τ298 K (ns) | PLQY |
| 508 | 3.11 | 24.6 (0.80), | 23.6 ns (0.36), | 0.29 |
| 180 (0.20) | 0.290 μs (0.14), | 0.62 b | ||
| 2.06 μs (0.50) | ||||
| PL Lifetimes in N2 Solution of p-TPS-DMAC-TRZ (Contribution) Prompt (ns) Delayed (μs) | ||||
| Toluene | 1,4-Dioxane | THF | Chloroform | Dichloromethane |
| 19.7 (0.55) 3.81 (0.45) | 27.8 (0.41) 2.49 (0.59) | 23.5 (0.56) 0.833 (0.44) | 21.4 (0.64) 0.724 (0.36) | 15.26 (0.74) 0.437 (0.26) |
| PL Lifetimes (ns) in Air Solution of p-TPS-DMAC-TRZ (Contribution) | ||||
| Toluene | 1,4-Dioxane | THF | Chloroform | Dichloromethane |
| 11.1 (0.86) 20.3 (0.14) | 19.9 (0.92) 99.0 (0.08) | 13.4 (0.86) 58.8 (0.14) | 13.4 (0.82) 100.0 (0.18) | 12.1 (0.85) 109.0 (0.15) |
| PLQY c in Solution of p-TPS-DMAC-TRZ N2/Air | ||||
| Toluene | 1,4-Dioxane | THF | Chloroform | Dichloromethane |
| 0.45/0.25 | 0.66/0.42 | 0.29/0.15 | 0.20/0.14 | 0.16/0.13 |
| Prompt and Delayed PLQY of p-TPS-DMAC-TRZ in N2 Purged Solution | ||||
| Toluene | 1,4-Dioxane | THF | Chloroform | Dichloromethane |
| 0.25/0.20 | 0.27/0.39 | 0.16/0.13 | 0.13/0.07 | 0.12/0.04 |
| Ered (V) | Eox (V) | EHOMO a (eV) | ELUMO b (eV) | Egelec c (eV) |
|---|---|---|---|---|
| −1.32 | 1.06 | −5.14 | −2.76 | 2.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauyon, R.A.; Fuentealba, D.; Pizarro, N.; Ortega-Alfaro, M.C.; Ugalde-Saldívar, V.M.; Sobarzo, P.A.; Medina, J.; García, L.; Jessop, I.A.; González-Henríquez, C.M.; et al. New Light-Green Thermally Activated Delayed Fluorescence Polymer Based on Dimethylacridine-Triphenyltriazine Light-Emitting Unit and Tetraphenylsilane Moiety as Non-Conjugated Backbone. Polymers 2023, 15, 67. https://doi.org/10.3390/polym15010067
Hauyon RA, Fuentealba D, Pizarro N, Ortega-Alfaro MC, Ugalde-Saldívar VM, Sobarzo PA, Medina J, García L, Jessop IA, González-Henríquez CM, et al. New Light-Green Thermally Activated Delayed Fluorescence Polymer Based on Dimethylacridine-Triphenyltriazine Light-Emitting Unit and Tetraphenylsilane Moiety as Non-Conjugated Backbone. Polymers. 2023; 15(1):67. https://doi.org/10.3390/polym15010067
Chicago/Turabian StyleHauyon, René A., Denis Fuentealba, Nancy Pizarro, María C. Ortega-Alfaro, Víctor M. Ugalde-Saldívar, Patricio A. Sobarzo, Jean Medina, Luis García, Ignacio A. Jessop, Carmen M. González-Henríquez, and et al. 2023. "New Light-Green Thermally Activated Delayed Fluorescence Polymer Based on Dimethylacridine-Triphenyltriazine Light-Emitting Unit and Tetraphenylsilane Moiety as Non-Conjugated Backbone" Polymers 15, no. 1: 67. https://doi.org/10.3390/polym15010067
APA StyleHauyon, R. A., Fuentealba, D., Pizarro, N., Ortega-Alfaro, M. C., Ugalde-Saldívar, V. M., Sobarzo, P. A., Medina, J., García, L., Jessop, I. A., González-Henríquez, C. M., Tundidor-Camba, A., & Terraza, C. A. (2023). New Light-Green Thermally Activated Delayed Fluorescence Polymer Based on Dimethylacridine-Triphenyltriazine Light-Emitting Unit and Tetraphenylsilane Moiety as Non-Conjugated Backbone. Polymers, 15(1), 67. https://doi.org/10.3390/polym15010067