
Thermal and Calorimetric Investigations of Some Phosphorus-Modified Chain Growth Polymers 2: Polystyrene

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. A Typical Procedure for Bulk Polymerization
2.3. Characterization
3. Results and Discussion
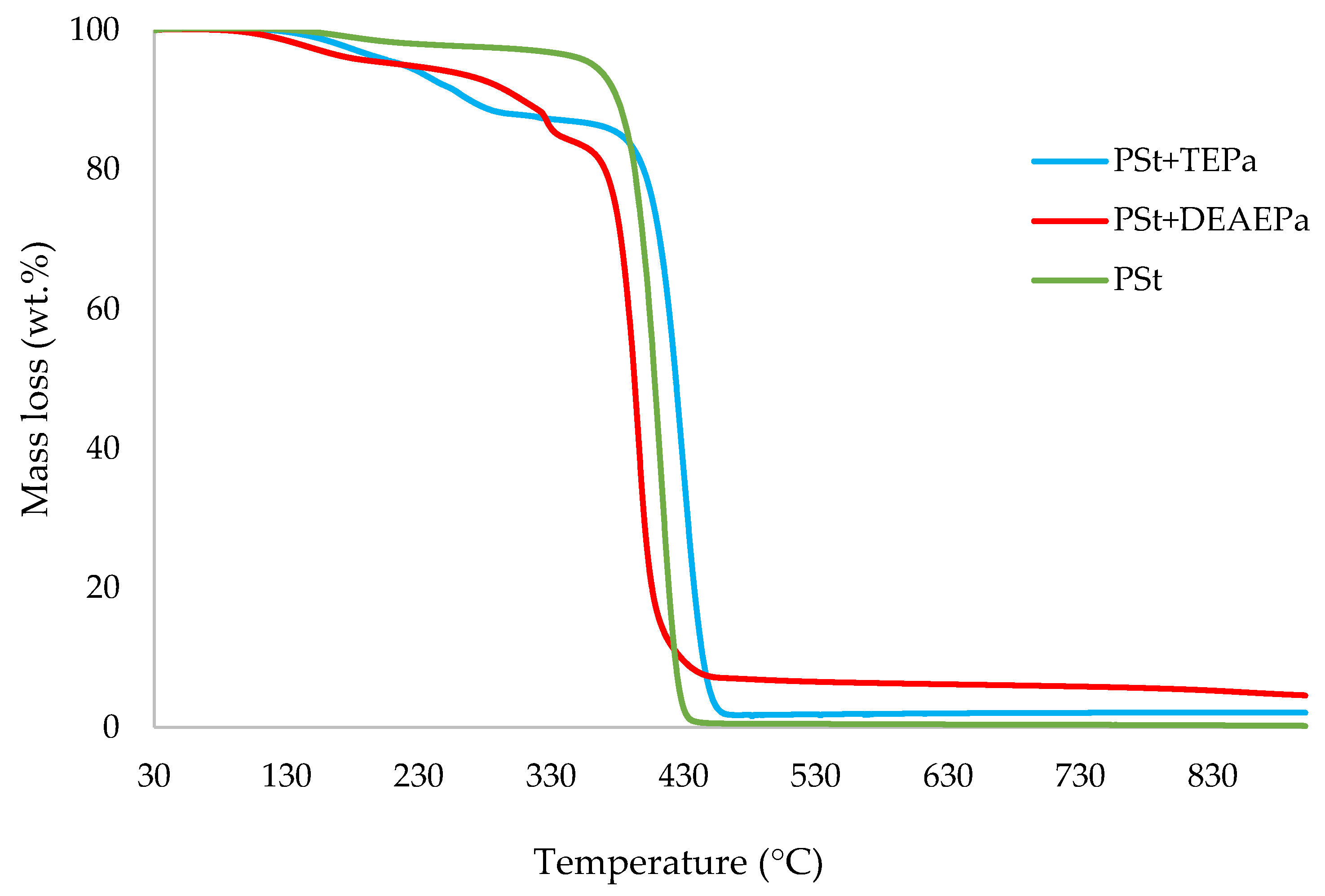
3.1. Thermogravimetric Analysis (TGA)
3.2. Kinetic Analysis of the TGA Thermograms
3.3. Differential Scanning Calorimetry
3.4. Pyrolysis Combustion Flow Calorimetry (PCFC)
3.5. ‘Bomb’ Calorimetry
3.6. Some Generalizations among the Test Parameters
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynwood, C. Polystyrene: Synthesis, Characteristics, and Applications; Nova Publishers: Hauppauge, NY, USA, 2014. [Google Scholar]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Atia, A.A.; Donia, A.M.; ELwakeel, K.Z. Adsorption behaviour of non-transition metal ions on a synthetic chelating resin bearing iminoacetate functions. Sep. Purif. Technol. 2005, 43, 43–48. [Google Scholar] [CrossRef]
- Atia, A.A.; Donia, A.M.; Elwakeel, K.Z. Selective separation of mercury (II) using a synthetic resin containing amine and mercaptan as chelating groups. React. Funct. Polym. 2005, 65, 267–275. [Google Scholar] [CrossRef]
- Donia, A.M.; Atia, A.A.; Elwakeel, K.Z. Gold (III) recovery using synthetic chelating resins with amine, thio and amine/mercaptan functionalities. Sep. Purif. Technol. 2005, 42, 111–116. [Google Scholar] [CrossRef]
- Elwakeel, K.Z.; Guibal, E. Potential use of magnetic glycidyl methacrylate resin as a mercury sorbent: From basic study to the application to wastewater treatment. J. Environ. Chem. Eng. 2016, 4, 3632–3645. [Google Scholar] [CrossRef]
- Sayadi, A.A.; Tapia, J.V.; Neitzert, T.R.; Clifton, G.C. Effects of expanded polystyrene (EPS) particles on fire resistance, thermal conductivity and compressive strength of foamed concrete. Constr. Build. Mater. 2016, 112, 716–724. [Google Scholar] [CrossRef]
- Price, D.; Cunliffe, L.K.; Bullett, K.; Hull, T.R.; Milnes, G.J.; Ebdon, J.R.; Hunt, B.J.; Joseph, P. Thermal behaviour of covalently bonded phosphate and phosphonate flame retardant polystyrene systems. Polym. Degrad. Stab. 2007, 92, 1101–1114. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Melt-flow behaviours of thermoplastic materials under fire conditions: Recent experimental studies and some theoretical approaches. Materials 2015, 8, 8793–8803. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Raj, B. Reactions and Mechanisms in Thermal Analysis of Advanced Materials; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Konkel, C.S.; Price, D.; Pyrah, K.; Hull, T.R.; Milnes, G.J.; Hill, S.B.; Lindsay, C.I. Thermal degradation and flame retardance in copolymers of methyl methacrylate with diethyl (methacryloyloxymethyl) phosphonate. Polym. Degrad. Stab. 2000, 70, 425–436. [Google Scholar] [CrossRef]
- McNeill, I.; Zulfiqar, M.; Kousar, T. A detailed investigation of the products of the thermal degradation of polystyrene. Polym. Degrad. Stab. 1990, 28, 131–151. [Google Scholar] [CrossRef]
- Jankowski, P.; Kedzierski, M. Synthesis of polystyrene of reduced flammability by suspension polymerization in the presence of halogen-free additives. Polimery 2011, 56, 20–26. [Google Scholar] [CrossRef]
- Cao, J.-P.; Zhao, X.; Zhao, J.; Zha, J.-W.; Hu, G.-H.; Dang, Z.-M. Improved thermal conductivity and flame retardancy in polystyrene/poly (vinylidene fluoride) blends by controlling selective localization and surface modification of SiC nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 6915–6924. [Google Scholar] [CrossRef] [PubMed]
- Edenharter, A.; Feicht, P.; Diar-Bakerly, B.; Beyer, G.; Breu, J. Superior flame retardant by combining high aspect ratio layered double hydroxide and graphene oxide. Polymer 2016, 91, 41–49. [Google Scholar] [CrossRef]
- Kausar, A.; Haider, S.; Muhammad, B. Nanocomposite based on polystyrene/polyamide blend and bentonite: Morphology, thermal, and nonflammability properties. Nanomater. Nanotechnol. 2017, 7, 1847980417702785. [Google Scholar] [CrossRef]
- Baby, A.; Tretsiakova-McNally, S.; Arun, M.; Joseph, P.; Zhang, J. Reactive and additive modifications of styrenic polymers with phosphorus-containing compounds and their effects on fire retardance. Molecules 2020, 25, 3779. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Shi, Y.; Zhu, S.; Fu, L.; Feng, Y.; Lv, Y.; Yang, F.; Liu, M.; Shui, W. Induced assembly of polystyrene composites for simultaneously improving flame retardant and electromagnetic shielding properties. Polym. Adv. Technol. 2021, 32, 4251–4262. [Google Scholar] [CrossRef]
- Gao, C.; Shi, Y.; Chen, Y.; Zhu, S.; Feng, Y.; Lv, Y.; Yang, F.; Liu, M.; Shui, W. Constructing segregated polystyrene composites for excellent fire resistance and electromagnetic wave shielding. J. Colloid Interface Sci. 2022, 606, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Negrell-Guirao, C.; Iftene, F.; Boutevin, B.; Chougrani, K. Recent progress on phosphonate vinyl monomers and polymers therefore obtained by radical (co) polymerization. Polym. Chem. 2012, 3, 265–274. [Google Scholar] [CrossRef]
- Benin, V.; Cui, X.; Morgan, A.B.; Seiwert, K. Synthesis and flammability testing of epoxy functionalized phosphorous-based flame retardants. J. Appl. Polym. Sci. 2015, 132, 1–10. [Google Scholar] [CrossRef]
- Wang, K.; Morgan, A.B.; Benin, V. Preparation and studies of new phosphorus-containing diols as potential flame retardants. Fire Mater. 2017, 41, 973–982. [Google Scholar] [CrossRef]
- Costes, L.; Laoutid, F.; Brohez, S.; Dubois, P. Bio-based flame retardants: When nature meets fire protection. Mater. Sci. Eng. R Rep. 2017, 117, 1–25. [Google Scholar] [CrossRef]
- Hobbs, C.E. Recent advances in bio-based flame retardant additives for synthetic polymeric materials. Polymers 2019, 11, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, R.A.; Shanmugam, V.; Narayanan, S.; Renner, J.S.; Babu, K.; Neisiany, R.E.; Försth, M.; Sas, G.; Das, O. A review of sustainable and environment-friendly flame retardants used in plastics. Polym. Test. 2022, 108, 107511. [Google Scholar] [CrossRef]
- Wilkie, C.A.; Morgan, A.B. Fire Retardancy of Polymeric Materials; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Ebdon, J.R.; Price, D.; Hunt, B.J.; Joseph, P.; Gao, F.; Milnes, G.J.; Cunliffe, L.K. Flame retardance in some polystyrenes and poly (methyl methacrylate) s with covalently bound phosphorus-containing groups: Initial screening experiments and some laser pyrolysis mechanistic studies. Polym. Degrad. Stab. 2000, 69, 267–277. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-Mcnally, S. Reactive modifications of some chain-and step-growth polymers with phosphorus-containing compounds: Effects on flame retardance—A review. Polym. Adv. Technol. 2011, 22, 395–406. [Google Scholar] [CrossRef]
- Salmeia, K.A.; Gaan, S. An overview of some recent advances in DOPO-derivatives: Chemistry and flame retardant applications. Polym. Degrad. Stab. 2015, 113, 119–134. [Google Scholar] [CrossRef]
- Cullis, C.F.; Hirschler, M.M. The Combustion of Organic Polymers; Oxford University Press: London, UK, 1981; Volume 5. [Google Scholar]
- Joseph, P.; Ebdon, J.R. Recent developments in flame-retarding thermoplastics and thermosets. Fire Retard. Mater. 2000, 1, 220–263. [Google Scholar]
- Ebdon, J.; Bunt, B.; Joseph, P.; Konkel, C. Flame-retarding thermoplastics: Additive versus reactive approach. In Speciality Polymer Additives: Principles and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 231–257. [Google Scholar]
- Price, D.; Pyrah, K.; Hull, T.R.; Milnes, G.J.; Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Konkel, C.S. Flame retarding poly (methyl methacrylate) with phosphorus-containing compounds: Comparison of an additive with a reactive approach. Polym. Degrad. Stab. 2001, 74, 441–447. [Google Scholar] [CrossRef]
- Arun, M.; Bigger, S.; Guerrieri, M.; Joseph, P. Thermal and calorimetric investigations of some phosphorus-modified chain growth polymers 1: Polymethyl methacrylate. Polymers 2022, 14, 1447. [Google Scholar] [CrossRef]
- Lyon, R.E.; Walters, R.N. Pyrolysis combustion flow calorimetry. J. Anal. Appl. Pyrolysis 2004, 71, 27–46. [Google Scholar] [CrossRef]
- Cogen, J.M.; Lin, T.S.; Lyon, R.E. Correlations between pyrolysis combustion flow calorimetry and conventional flammability tests with halogen-free flame retardant polyolefin compounds. Fire Mater. Int. J. 2009, 33, 33–50. [Google Scholar] [CrossRef]
- Sonnier, R.; Vahabi, H.; Ferry, L.; Lopez-Cuesta, J.-M. Pyrolysis-combustion flow calorimetry: A powerful tool to evaluate the flame retardancy of polymers. Fire Polym. VI New Adv. Flame Retard. Chem. Sci. 2012, 1118, 361–390. [Google Scholar]
- Tretsiakova-McNally, S.; Joseph, P. Pyrolysis combustion flow calorimetry studies on some reactively modified polymers. Polymers 2015, 7, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Bigger, S.W.; Cran, M.J.; Bohn, M.A. Novel theoretical and computer-assisted modeling of isothermal and non-isothermal depolymerization kinetics. Polym. Test. 2015, 44, 1–7. [Google Scholar] [CrossRef]
- Bigger, S.W.; Cran, M.J.; Tawakkal, I.S. Two novel algorithms for the thermogravimetric assessment of polymer degradation under non-isothermal conditions. Polym. Test. 2015, 43, 139–146. [Google Scholar] [CrossRef]
- Thomas, A.; Moinuddin, K.; Tretsiakova-McNally, S.; Joseph, P. A kinetic analysis of the thermal degradation behaviours of some bio-based substrates. Polymers 2020, 12, 1830. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Combustion behaviours of chemically modified polyacrylonitrile polymers containing phosphorylamino groups. Polym. Degrad. Stab. 2012, 97, 2531–2535. [Google Scholar] [CrossRef]
- Faravelli, T.; Pinciroli, M.; Pisano, F.; Bozzano, G.; Dente, M.; Ranzi, E. Thermal degradation of polystyrene. J. Anal. Appl. Pyrolysis 2001, 60, 103–121. [Google Scholar] [CrossRef]
- Joseph, P.; Arun, M.; Bigger, S.; Guerrieri, M.; Pospiech, D.; Harnisch, C. Mode of action of condensed-and gaseous-phase fire retardation in some phosphorus-modified polymethyl methacrylate-and polystyrene-based bulk polymers. Polymers 2021, 13, 3402. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Styrene (mL) | Additive/Reactive | Formula Weight | Additive/Reactive Weight (g/mL) | BPO/Dicumyl Peroxide (mg) |
|---|---|---|---|---|---|
| 1 | 50.00 | --- | --- | --- | 50.0/25.0 |
| 2 | 36.57 | TPP | 262 | 6.76 g | 44.0/22.0 |
| 3 | 36.10 | TPPO | 278 | 7.17 g | 43.0/21.5 |
| 4 | 37.90 | DOPO | 216 | 5.57 g | 43.0/21.5 |
| 5 | 40.09 | DEHPi | 138 | 3.56 mL | 44.0/22.0 |
| 6 | 39.29 | TEPi | 166 | 4.42 mL | 44.0/22.0 |
| 7 | 38.84 | TEPa | 182 | 4.40 mL | 44.0/22.0 |
| 8 | 39.00 | DEPP | 180 | 4.65 g | 44.0/22.0 |
| 9 | 37.53 | DEBP | 228 | 5.88 g | 44.0/22.0 |
| 10 | 23.32 | DE-1-AEP | 236 | 3.81 g | 26.0/13.0 |
| 11 | 23.00 | ADPMAE | 265 | 4.27 g | 26.0/13.0 |
| 12 | 23.00 | DEAEPa | 252 | 4.07 g | 26.0/13.0 |
| 13 | 23.00 | DEpVBP | 254 | 4.10 g | 26.0/13.0 |
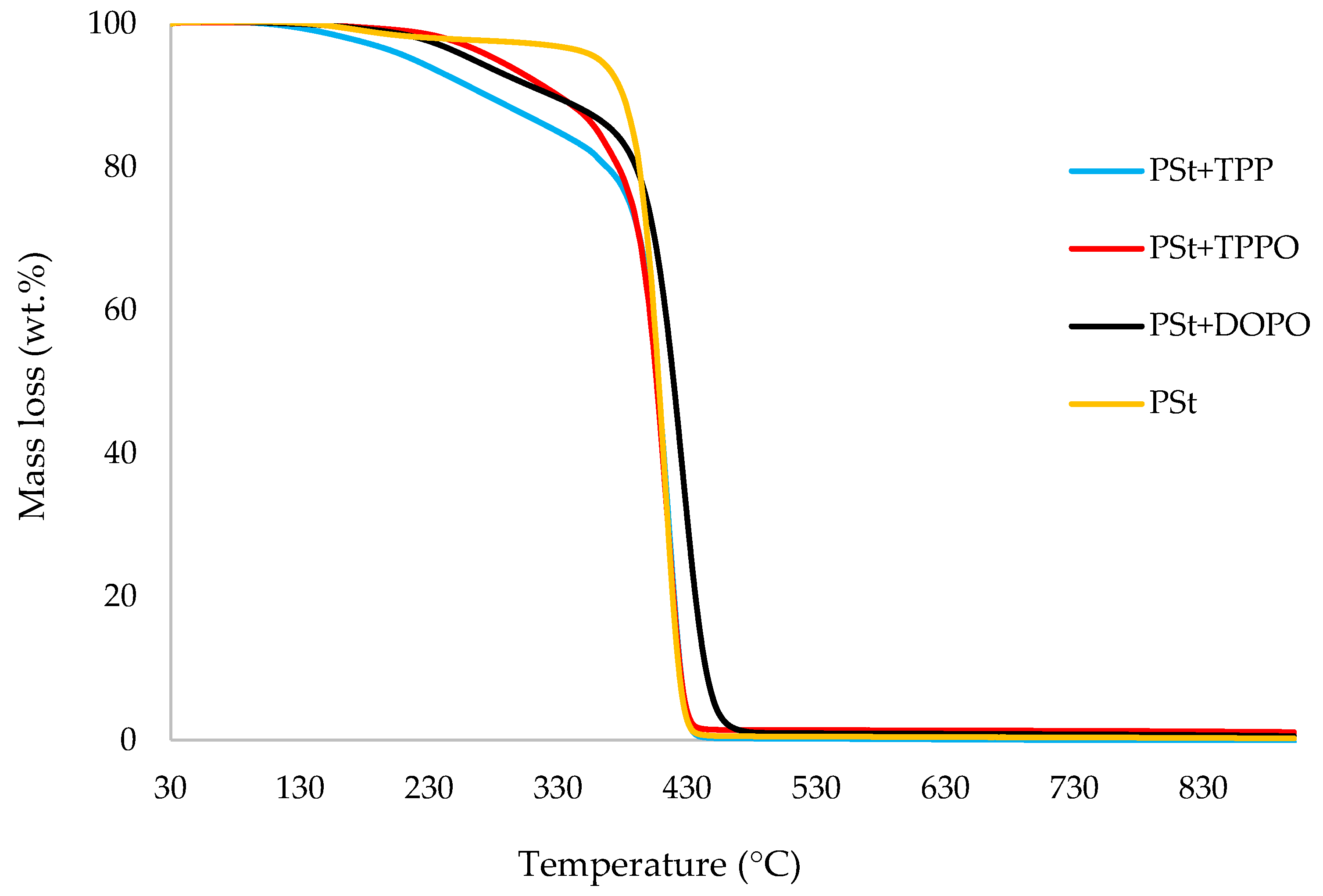
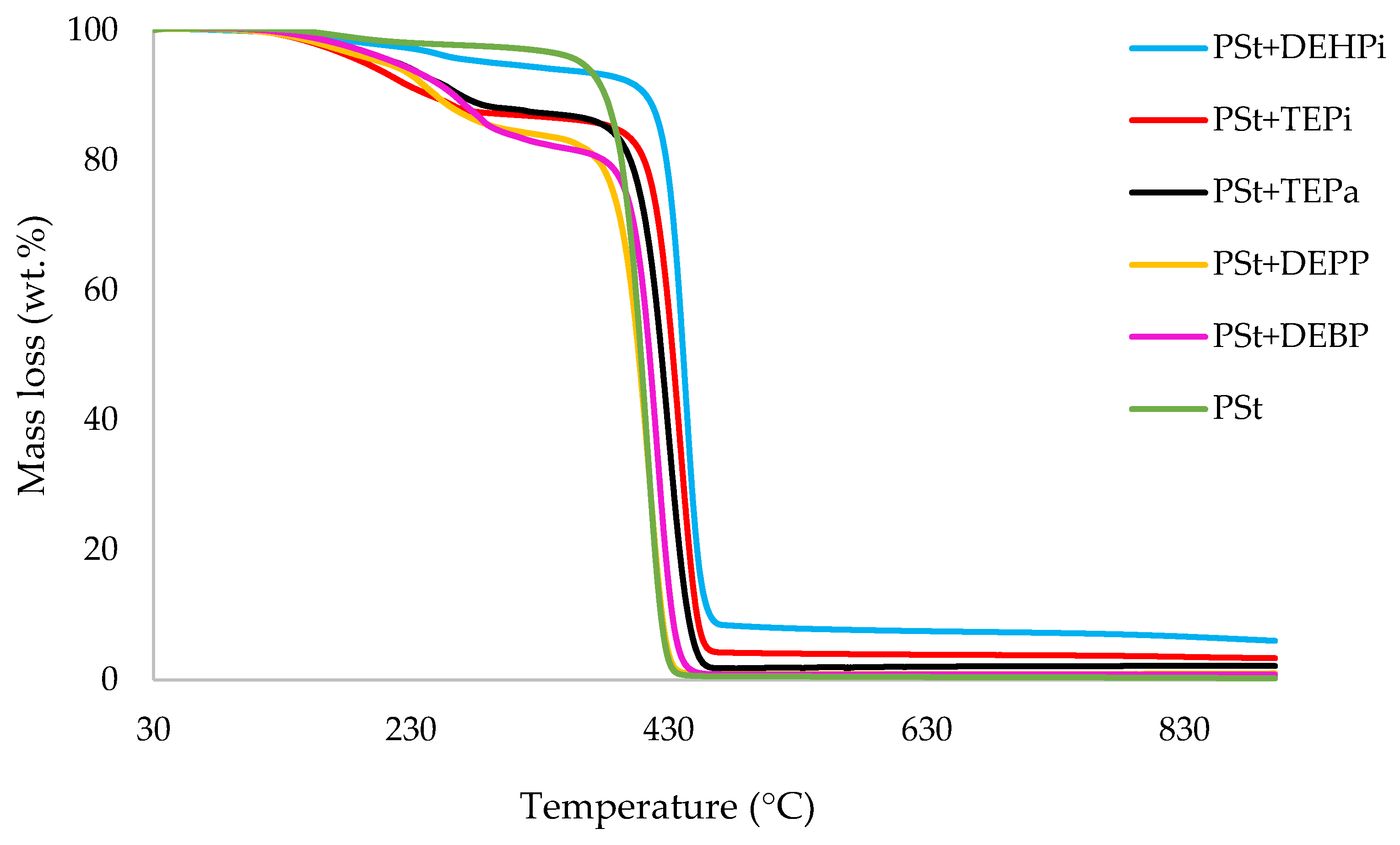
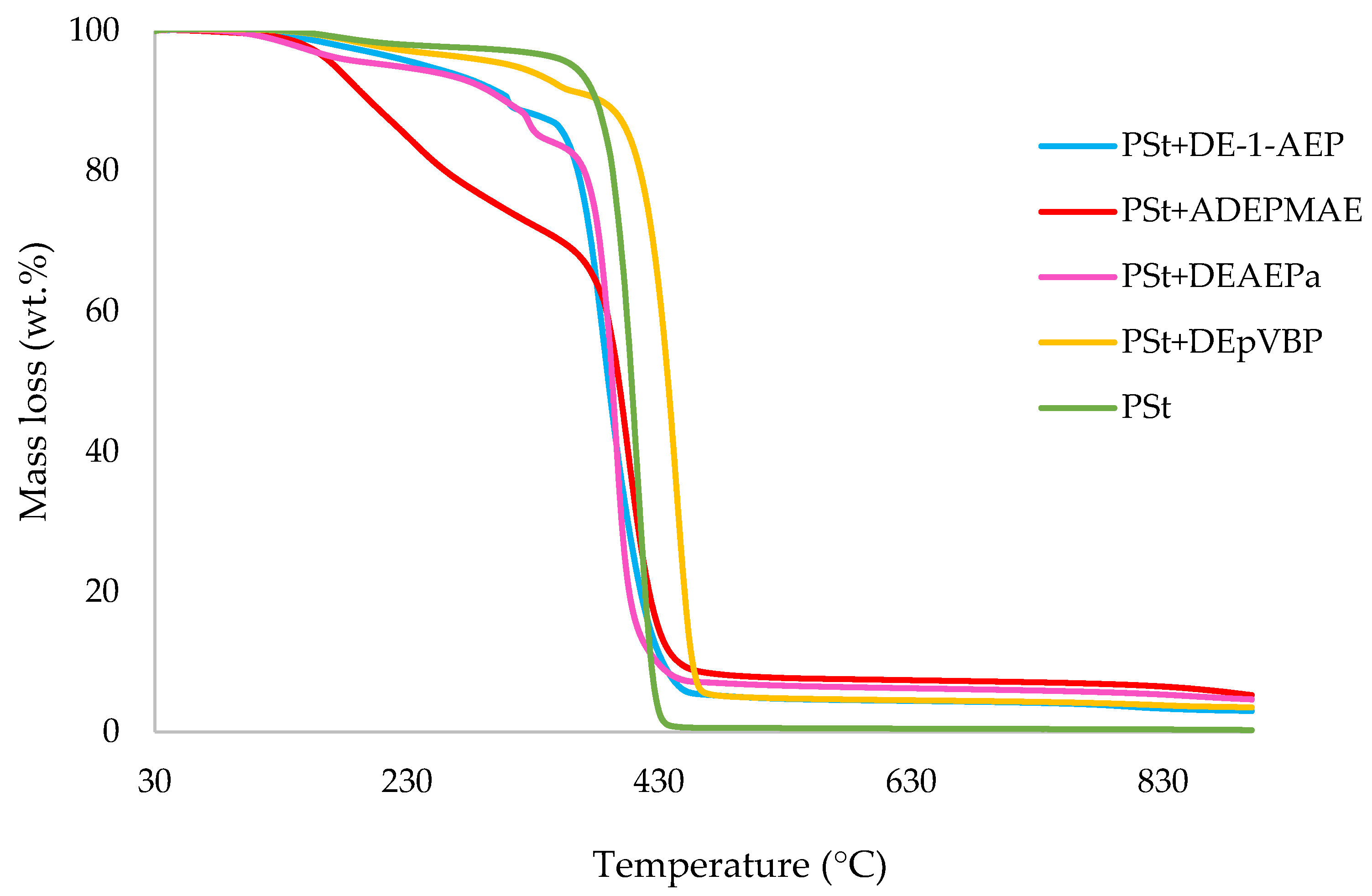
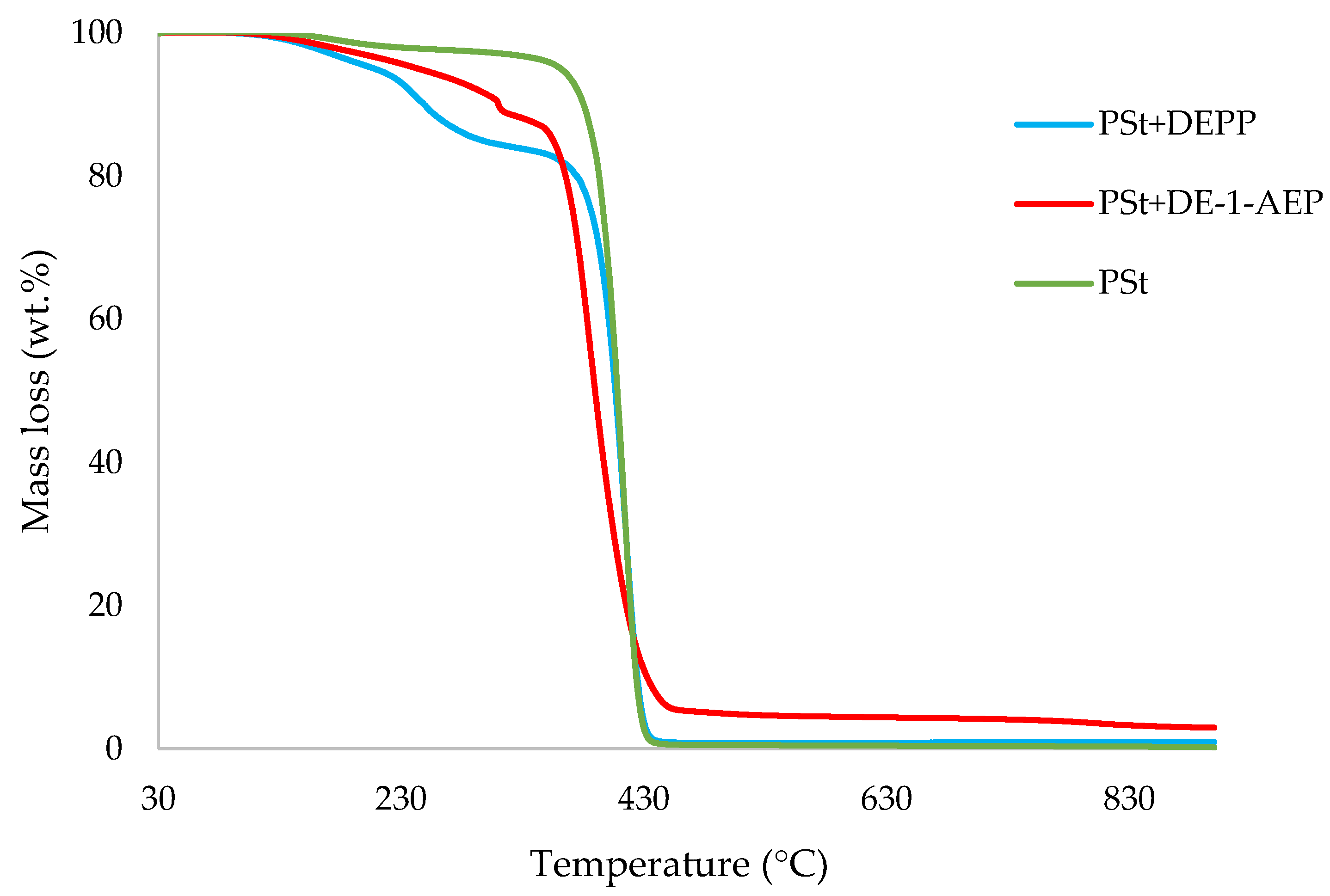
| Sl. No. | Sample | Induction Temp. (°C) | Temp. at 50 wt.% (°C) | Residue at 500 °C (wt.%) | Final Residue at 800 °C (wt.%) |
|---|---|---|---|---|---|
| 1 | Polystyrene | 129 | 408 | 0.5 | 0.4 |
| 2 | PSt+TPP | 105 | 408 | 0.2 | 0.0 |
| 3 | PSt+TPPO | 123 | 407 | 1.4 | 1.3 |
| 4 | PSt+DOPO | 118 | 420 | 1.0 | 0.8 |
| 5 | PSt+DEHPi | 73.0 | 441 | 8.1 | 6.9 |
| 6 | PSt+TEPi | 75.0 | 433 | 4.1 | 3.6 |
| 7 | PSt+TEPa | 119 | 424 | 1.8 | 2.1 |
| 8 | PSt+DEPP | 96.0 | 407 | 0.9 | 0.9 |
| 9 | PSt+DEBP | 100 | 415 | 0.9 | 0.8 |
| 10 | PSt+DE-1-AEP | 109 | 390 | 4.9 | 3.6 |
| 11 | PSt+ADEPMAE | 72.0 | 398 | 7.9 | 6.7 |
| 12 | PSt+DEAEPa | 74.0 | 393 | 6.8 | 5.5 |
| 13 | PSt+DEpVBP | 121 | 437 | 4.9 | 4.0 |
| Sl. No. | Sample | Apparent Activation Energy (Ea, kJ mol−1) | A (min−1) | R2 Values | α-Value Range |
|---|---|---|---|---|---|
| 1 | PSt | 270 | 2.68 × 1020 | 0.998 | 0.1 to 0.9 |
| 2 | PSt+TPP | 118 | 2.69 × 108 | 0.9125 | 0.2 to 0.8 |
| 3 | PSt+TPPO | 162 | 9.52 × 1011 | 0.9706 | 0.2 to 0.8 |
| 4 | PSt+DOPO | 165 | 8.94 × 1011 | 0.9939 | 0.2 to 0.8 |
| 5 | PSt+DEHPi | 305 | 1.23 × 1022 | 0.9943 | 0.1 to 0.9 |
| 6 | PSt+TEPi | 229 | 3.88 × 1016 | 0.9908 | 0.2 to 0.8 |
| 7 | PSt+TEPa | 210 | 2.24 × 1015 | 0.9935 | 0.2 to 0.8 |
| 8 | PSt+DEPP | 157 | 3.82 × 1011 | 0.9652 | 0.2 to 0.8 |
| 9 | PSt+DEBP | 137 | 6.79 × 109 | 0.9218 | 0.2 to 0.8 |
| 10 | PSt+DE-1-AEP | 97 | 1.00 × 107 | 0.9815 | 0.1 to 0.9 |
| 11 | PSt+ADEPMAE | 130 | 3.82 × 109 | 0.9978 | 0.4 to 0.9 |
| 12 | PSt+DEAEPa | 216 | 4.26 × 1016 | 0.9871 | 0.2 to 0.8 |
| 13 | PSt+DEpVBP | 223 | 1.08 × 1016 | 0.9923 | 0.2 to 0.8 |
| Sl. No. | Samples | Heats of Pyrolysis, ∆Hpyro (mJ mg−1) | Tg (°C) (±5 °C) |
|---|---|---|---|
| 1 | PSt | 810 | 96 |
| 2 | PSt+TPP | 650 | 55 |
| 3 | PSt+TPPO | 720 | 76 |
| 4 | PSt+DOPO | 730 | 61 |
| 5 | PSt+DEHPi | 540 | 70 |
| 6 | PSt+TEPi | 560 | 65 |
| 7 | PSt+TEPa | 680 | 62 |
| 8 | PSt+DEPP | 690 | 74 |
| 9 | PSt+DEBP | 660 | 62 |
| 10 | PSt+DE-1-AEP | 560 | 63 |
| 11 | PSt+ADEPMAE | 470 | 70 |
| 12 | PSt+DEAEPa | 324 | 65 |
| 13 | PSt+DEpVBP | 520 | 75 |
| Sl. No. | Samples | Temp to pHRR (°C) | pHRR (W g−1) | THR (kJ g−1) | HRC (J g−1 K−1) | Char Yield (wt.%) | EHC (kJ g−1) |
|---|---|---|---|---|---|---|---|
| 1 | PSt | 434 | 840 | 37.1 | 852 | 4.4 | 38.8 |
| 2 | PSt+TPP | 434 | 682 | 35.9 | 686 | 6.5 | 38.4 |
| 3 | PSt+TPPO | 434 | 717 | 36.7 | 729 | 0 | 36.7 |
| 4 | PSt+DOPO | 446 | 618 | 36.3 | 621 | 0 | 36.3 |
| 5 | PSt+DEHPi | 462 | 778 | 32.9 | 778 | 5.9 | 35 |
| 6 | PSt+TEPi | 463 | 771 | 31.9 | 776 | 5.6 | 33.8 |
| 7 | PSt+TEPa | 444 | 743 | 34.2 | 772 | 0 | 34.2 |
| 8 | PSt+DEPP | 438 | 815 | 34.2 | 813 | 0 | 34.2 |
| 9 | PSt+DEBP | 440 | 792 | 34.5 | 794 | 0 | 34.5 |
| 10 | PSt+DE-1-AEP | 409 | 501 | 33 | 499 | 7.4 | 35.6 |
| 11 | PSt+ADEPMAE | 431 | 425 | 23.8 | 424 | 5.7 | 25.2 |
| 12 | PSt+DEAEPa | 416 | 757 | 31.9 | 755 | 6.4 | 34.1 |
| 13 | PSt+DEpVBP | 468 | 655 | 34.6 | 653 | 2.9 | 35.6 |
| Sl. No. | Sample | ΔHcomb (kJ g−1) |
|---|---|---|
| 1 | PSt | 41.50 |
| 2 | PSt+TPP | 41.02 |
| 3 | PSt+TPPO | 40.30 |
| 4 | PSt+DOPO | 39.50 |
| 5 | PSt+DEHPi | 38.70 |
| 6 | PSt+TEPi | 38.89 |
| 7 | PSt+TEPa | 38.99 |
| 8 | PSt+DEPP | 39.55 |
| 9 | PSt+DEBP | 39.13 |
| 10 | PSt+DE-1-AEP | 38.23 |
| 11 | PSt+ADEPMAE | 33.03 |
| 12 | PSt+DEpVBP | 39.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arun, M.; Bigger, S.; Guerrieri, M.; Joseph, P.; Tretsiakova-McNally, S. Thermal and Calorimetric Investigations of Some Phosphorus-Modified Chain Growth Polymers 2: Polystyrene. Polymers 2022, 14, 1520. https://doi.org/10.3390/polym14081520
Arun M, Bigger S, Guerrieri M, Joseph P, Tretsiakova-McNally S. Thermal and Calorimetric Investigations of Some Phosphorus-Modified Chain Growth Polymers 2: Polystyrene. Polymers. 2022; 14(8):1520. https://doi.org/10.3390/polym14081520
Chicago/Turabian StyleArun, Malavika, Stephen Bigger, Maurice Guerrieri, Paul Joseph, and Svetlana Tretsiakova-McNally. 2022. "Thermal and Calorimetric Investigations of Some Phosphorus-Modified Chain Growth Polymers 2: Polystyrene" Polymers 14, no. 8: 1520. https://doi.org/10.3390/polym14081520
APA StyleArun, M., Bigger, S., Guerrieri, M., Joseph, P., & Tretsiakova-McNally, S. (2022). Thermal and Calorimetric Investigations of Some Phosphorus-Modified Chain Growth Polymers 2: Polystyrene. Polymers, 14(8), 1520. https://doi.org/10.3390/polym14081520