
A Computational Procedure for Atomistic Modelling of Polyphosphazenes towards Better Capturing Molecular-Level Structuring and Thermo-Mechanical Properties


Abstract
:
1. Introduction
2. Models and Methodology
2.1. Simulation Details
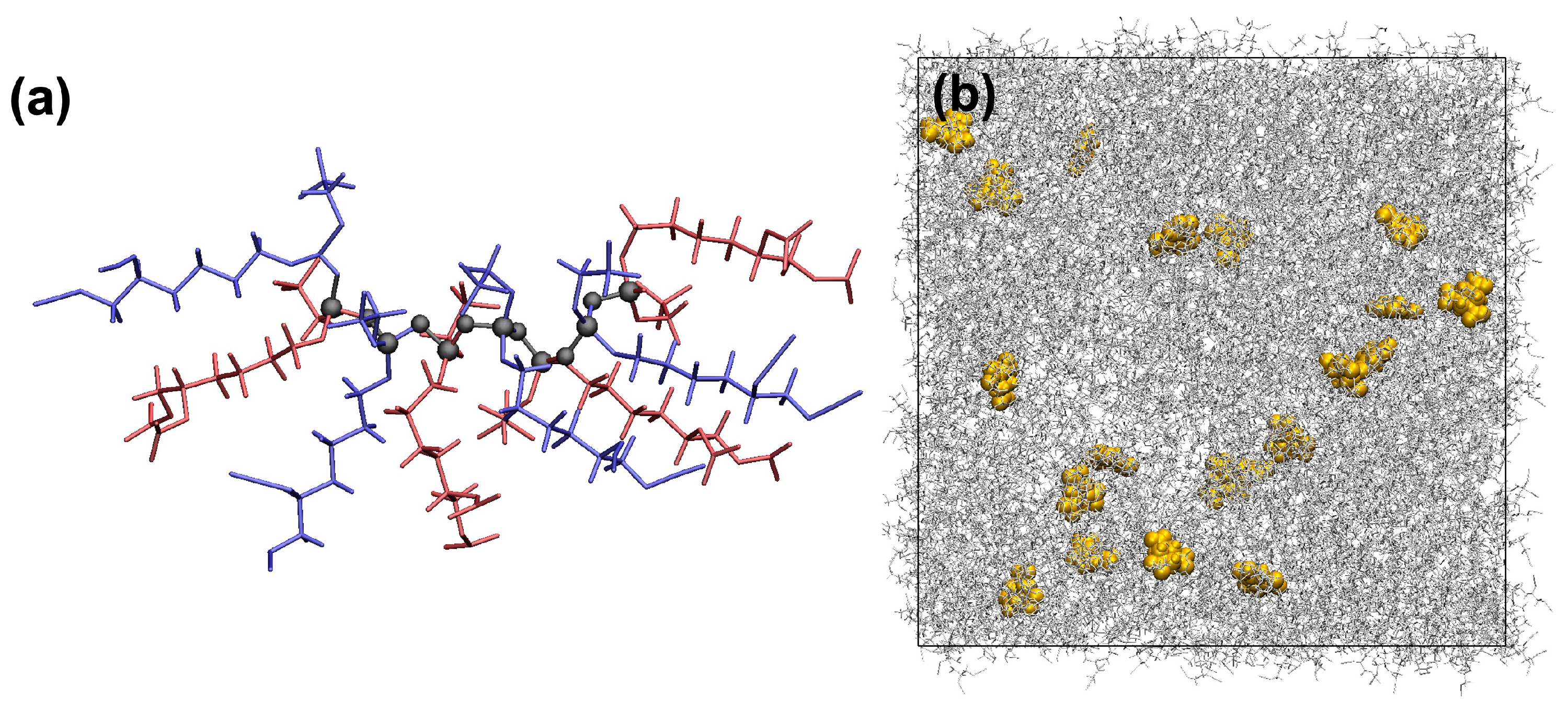
2.2. Preparation of Liquid Samples
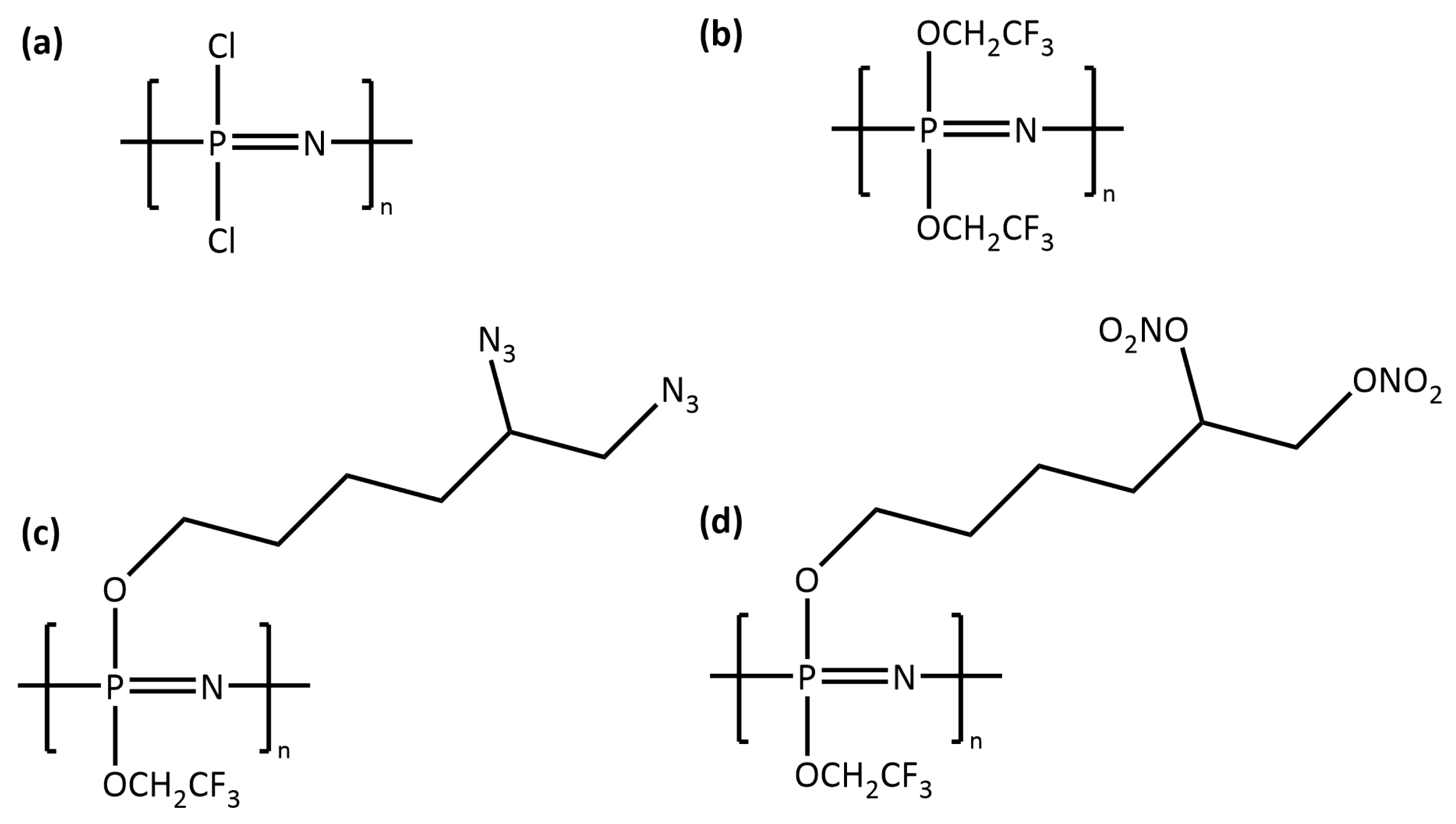
2.3. Polymerisation Step
2.4. Analysis and Prediction of Thermo-Mechanical Properties
3. Results and Discussion
3.1. Liquid Samples
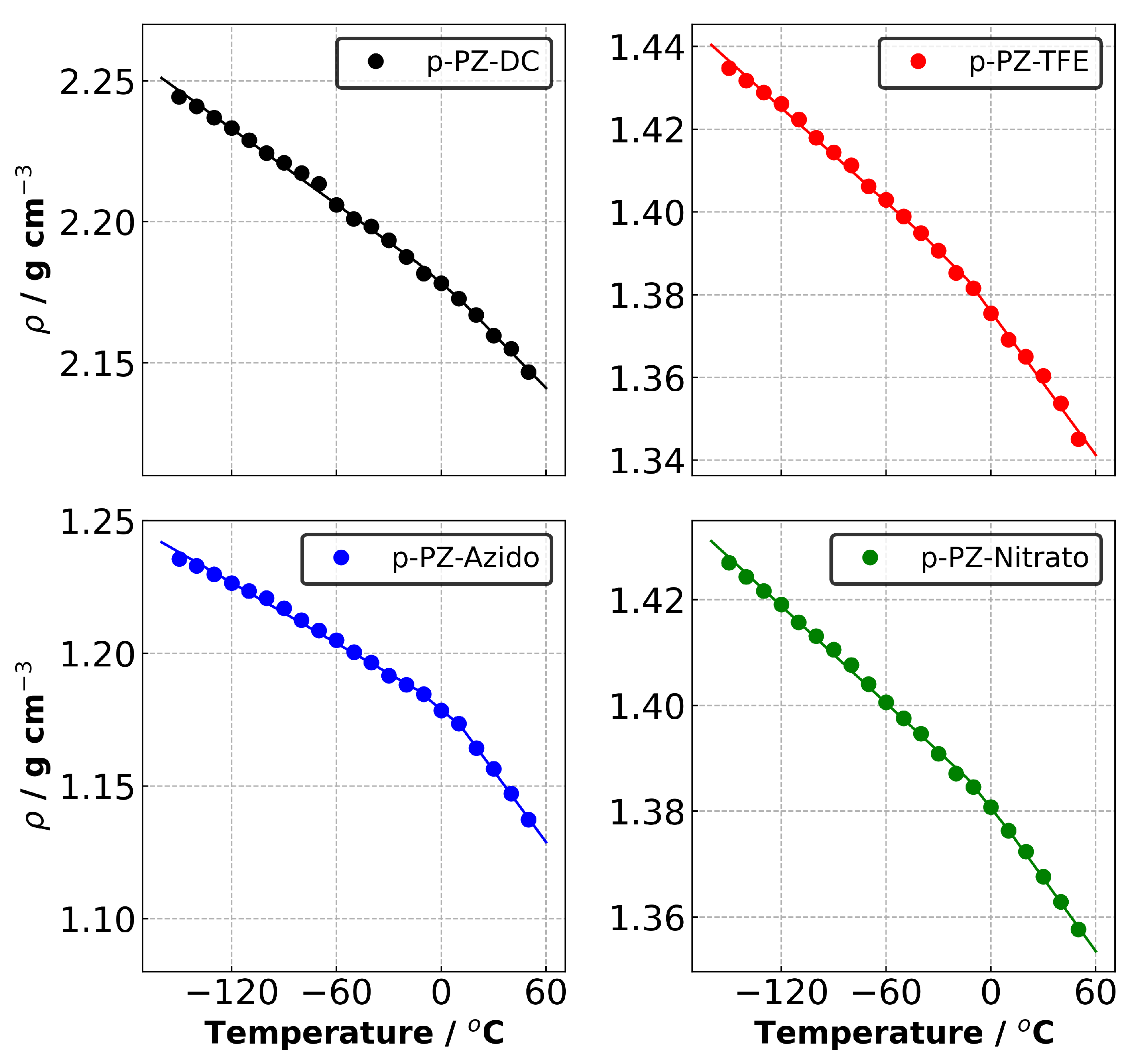
3.2. Thermal Properties
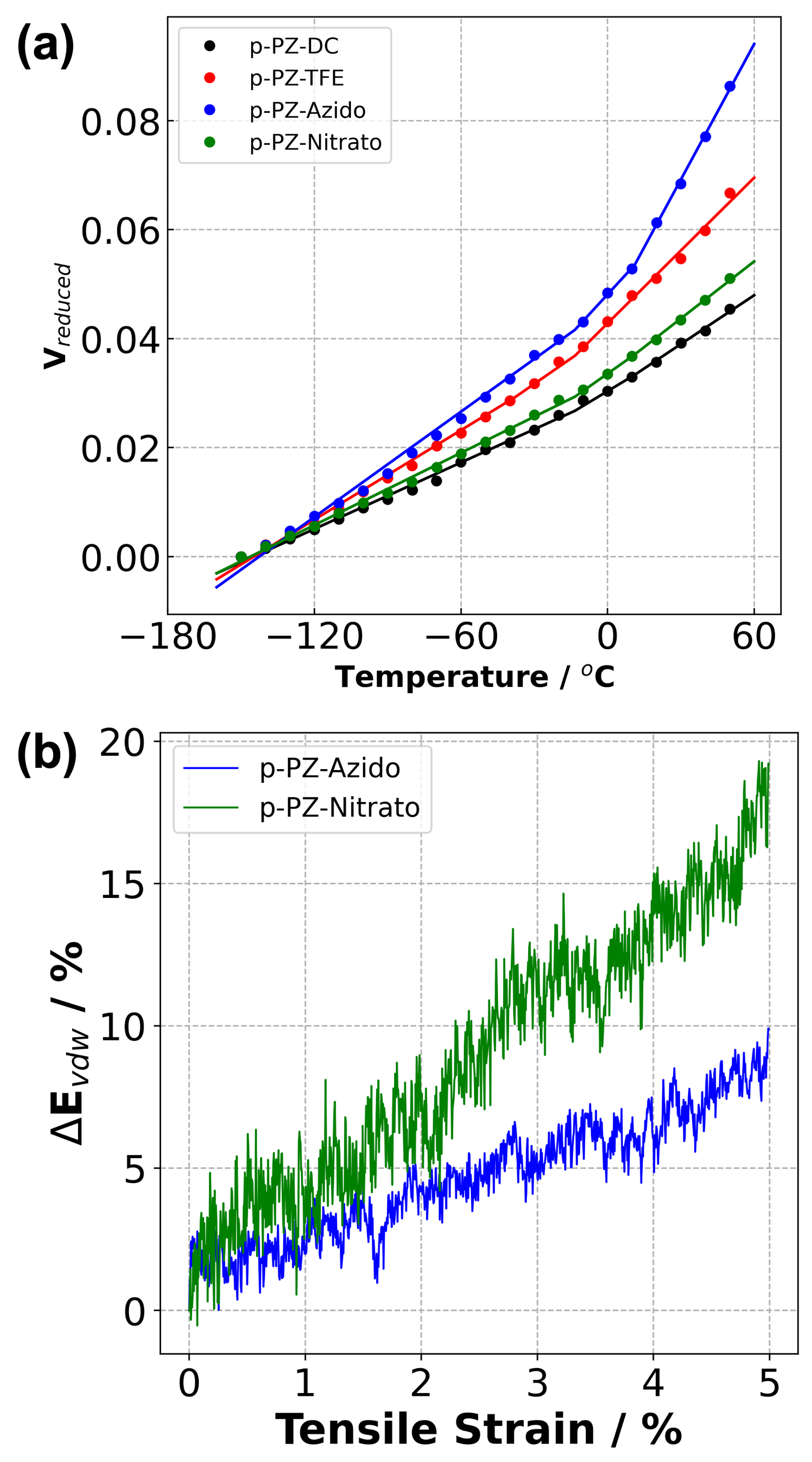
3.3. Mechanical Properties
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PZ | Polyphosphazenes |
| MD | Molecular dynamics |
| PAC | Partial atomic charge |
| SA | Simulated annealing |
| RDF | Radial distribution function |
| CVTE | Coefficient of volumetric thermal expansion |
| T | Glass transition temperature |
References
- Harry, R.; Allcock, R.M.W. Design and synthesis of ion-conductive polyphosphazenes for fuel cell applications: Review. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 2258–2368. [Google Scholar]
- Chen, J.; Li, C.; Wang, J.; Li, L.; Wei, Z. A general strategy to enhance the alkaline stability of anion exchange membranes. J. Mater. Chem. A 2017, 5, 6318–6327. [Google Scholar] [CrossRef]
- Allen, C.W. The use of phosphazenes as fire resistant materials. J. Fire Sci. 1993, 11, 320–328. [Google Scholar] [CrossRef]
- Zhou, X.; Qiu, S.; Mu, X.; Zhou, M.; Cai, W.; Song, L.; Xing, W.; Hu, Y. Polyphosphazenes-based flame retardants: A review. Compos. B Eng. 2020, 202, 108397. [Google Scholar] [CrossRef]
- Zhou, X.; Qiu, S.; He, L.; Wang, X.; Zhu, Y.; Chu, F.; Wang, B.; Song, L.; Hu, Y. Synthesis of star-shaped allyl phosphazene small molecules for enhancing fire safety and toughness of high performance BMI resin. Chem. Eng. J. 2021, 425, 130655. [Google Scholar] [CrossRef]
- Golding, P.; Trussell, S.J.; Colclough, M.E.; Hamid, J. Energetic Polyphosphazenes. U.S. Patent 8,268,959, 18 September 2012. [Google Scholar]
- Ullah, R.S.; Wang, L.; Yu, H.; Abbasi, N.M.; Akram, M.; ul Abdin, Z.; Saleem, M.; Haroon, M.; Khan, R.U. Synthesis of polyphosphazenes with different side groups and various tactics for drug delivery. RSC Adv. 2017, 7, 23363–23391. [Google Scholar] [CrossRef] [Green Version]
- Ogueri, K.S.; Ogueri, K.S.; Allcock, H.R.; Laurencin, C.T. Polyphosphazene polymers: The next generation of biomaterials for regenerative engineering and therapeutic drug delivery. J. Vac. Sci. Technol. 2020, 38, 030801. [Google Scholar] [CrossRef]
- Rothemund, S.; Teasdale, I. Preparation of polyphosphazenes: A tutorial review. Chem. Soc. Rev. 2016, 45, 5200–5215. [Google Scholar] [CrossRef] [Green Version]
- Allcock, H.R.; Kellam, E. The synthesis and applications of novel aryloxy/oligoethyleneoxy substituted polyphosphazenes as solid polymer electrolytes. Solid State Ion. 2003, 156, 401–414. [Google Scholar] [CrossRef]
- Singh, A.; Krogman, N.R.; Sethuraman, S.; Nair, L.S.; Sturgeon, J.L.; Brown, P.W.; Laurencin, C.T.; Allcock, H.R. Effect of Side Group Chemistry on the Properties of Biodegradable l-Alanine Cosubstituted Polyphosphazenes. Biomacromolecules 2006, 7, 914–918. [Google Scholar] [CrossRef]
- Weikel, A.L.; Owens, S.G.; Fushimi, T.; Allcock, H.R. Synthesis and Characterization of Methionine- and Cysteine-Substituted Phosphazenes. Macromolecules 2010, 43, 5205–5210. [Google Scholar] [CrossRef]
- Han, H.; Ma, H.; Yu, J.; Zhu, H.; Wang, Z. Preparation and performance of novel tetraphenylphosphonium-functionalized polyphosphazene membranes for alkaline fuel cells. Eur. Polym. J. 2019, 114, 109–117. [Google Scholar] [CrossRef]
- Clubb, J.W.; Kay, J.A.; Golding, P. Novel Energetic Phosphazene Polymer Binder Systems for Environmentally Friendly Rocket Motor Propellants; WP-2141; Strategic Environmental Research and Development Program: Alexandria, VA, USA, 2015; Available online: https://www.serdp-estcp.org/index.php//Program-Areas/Weapons-Systems-and-Platforms/Energetic-Materials-and-Munitions/Rocket-and-Missile-Propellants/WP-2141 (accessed on 23 February 2022).
- Tarazona, M.; Saiz, E. A conformational model for poly(dichlorophosphazene) derived from molecular dynamics simulations. Polymer 2000, 41, 3337–3347. [Google Scholar] [CrossRef]
- Fried, J.; Ren, P. Molecular simulation of the glass transition of polyphosphazenes. Comp. Theor. Polym. Sci. 1999, 9, 111–116. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Basharat, M.; Wu, S.; Zhang, S.; Zhang, X.; Ma, H.; Liu, W.; Wu, D.; Wu, Z. Effect of side groups on glass transition temperatures of Poly(ethoxy/phenoxy)phosphazenes: Prediction and synthesis. Polymer 2021, 230, 124068. [Google Scholar] [CrossRef]
- Kroger, J.L.; Fried, J.R. Molecular Simulations of Polyphosphazenes for Biomedical Applications. J. Inorg. Organomet. Polym. Mater. 2012, 22, 973–984. [Google Scholar] [CrossRef]
- Laguna, M.; Tarazona, M.P.; Carriedo, G.A.; Alonso, F.J.G.; Fidalgo, J.I.; Saiz, E. Thermal degradation and solution properties of poly(2,2’-dioxybiphenyl phosphazene). Macromolecules 2002, 35, 7505–7515. [Google Scholar] [CrossRef]
- Wang, Y.; Balbuena, P.B. Combined ab Initio Quantum Mechanics and Classical Molecular Dynamics Studies of Polyphosphazene Polymer Electrolytes: Competitive Solvation of Li+ and LiCF3SO3. J. Phys. Chem. B 2004, 108, 15694–15702. [Google Scholar] [CrossRef]
- Sun, H.; Ren, P.; Fried, J. The COMPASS force field: Parameterization and validation for phosphazenes. Comp. Theor. Polym. Sci. 1998, 8, 229–246. [Google Scholar] [CrossRef]
- Caminiti, R.; Gleria, M.; Lipkowitz, K.B.; Lombardo, G.M.; Pappalardo, G.C. Molecular Dynamics Simulations Combined with Large Angle X-ray Scattering Technique for the Determination of the Structure, Conformation, and Conformational Dynamics of Polyphosphazenes in Amorphous Phase: Study of Poly[di(4-methylphenoxy)phosphazene]. J. Am. Chem. Soc. 1997, 119, 2196–2204. [Google Scholar] [CrossRef]
- Fried, J.R. Molecular Simulation of Polyphosphazenes. In Polyphosphazenes in Biomedicine, Engineering, and Pioneering Synthesis; American Chemical Society: Washington, DC, USA, 2018; pp. 241–252. [Google Scholar]
- Allcock, H.R.; Chen, C. Polyphosphazenes: Phosphorus in Inorganic—Organic Polymers. J. Org. Chem. 2020, 85, 14286–14297. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Goddard, W.A. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 1991, 95, 3358–3363. [Google Scholar] [CrossRef]
- Demir, B.; Dumée, L.F. Modelling Amorphous Nanoporous Polymers Doped with an Ionic Liquid via an Adaptable Computational Procedure. Ind. Eng. Chem. Res. 2021, 60, 11893–11904. [Google Scholar] [CrossRef]
- Demir, B. Atomistic Modeling of Dual-Cured Thermosets Based on Glycidyl Methacrylate and Hardeners with Various Architecture and Functionality. ACS Appl. Polym. Mater. 2021, 3, 5030–5038. [Google Scholar] [CrossRef]
- Demir, B. A Bespoke Computational Procedure to Incorporate CO2 as a Renewable Feedstock into Polycarbonates. ACS Appl. Polym. Mater. 2021, 3, 2722–2731. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Constant-pressure equations of motion. Phys. Rev. A 1986, 34, 2499–2500. [Google Scholar] [CrossRef]
- Hockney, R.; Eastwood, J. Computer Simulation Using Particles; Adam Hilger: London, UK, 1989. [Google Scholar]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyckens, D.J.; Servinis, L.; Scheffler, C.; Wolfel, E.; Demir, B.; Walsh, T.R.; Henderson, L.C. Synergistic interfacial effects of ionic liquids as sizing agents and surface modified carbon fibers. J. Mater. Chem. A 2018, 6, 4504–4514. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural Relaxation Made Simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef] [Green Version]
- Demir, B.; Henderson, L.C.; Walsh, T.R. Design Rules for Enhanced Interfacial Shear Response in Functionalized Carbon Fiber Epoxy Composites. ACS Appl. Mater. Inter. 2017, 9, 11846–11857. [Google Scholar] [CrossRef]
- Demir, B.; Walsh, T.R. A Versatile Computational Procedure for Chain-Growth Polymerization Using Molecular Dynamics Simulations. ACS Appl. Polym. Mater. 2019, 1, 3027–3038. [Google Scholar] [CrossRef]
- Demir, B.; Walsh, T.R. A robust and reproducible procedure for cross-linking thermoset polymers using molecular simulation. Soft Matter 2016, 12, 2453–2464. [Google Scholar] [CrossRef]
- Balani, K.; Verma, V.; Agarwal, A.; Narayan, R. Biosurfaces: A Materials Science and Engineering Perspective; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Brown, T.L. Chemistry the Central Science/Theodore L. Brown; Singapore Pearson Education: Harlow, UK, 2015. [Google Scholar]
- Allcock, H.R.; Arcus, R.A. Crystalline Transitions and Related Physical Properties of Poly(dichlorophosphazene). Macromolecules 1979, 12, 1130–1136. [Google Scholar] [CrossRef]
- Hirose, T.; Kamiya, Y.; Mizoguchi, K. Gas transport in poly[bis(trifluoroethoxy)phosphazene]. J. Appl. Polym. Sci. 1989, 38, 809–820. [Google Scholar] [CrossRef]
- Nagai, K.; Freeman, B.D.; Cannon, A.; Allcock, H.R. Gas permeability of poly(bis-trifluoroethoxyphosphazene) and blends with adamantane amino/trifluoroethoxy (50/50) polyphosphazene. J. Membr. Sci. 2000, 172, 167–176. [Google Scholar] [CrossRef]
- Demir, B.; Hamerton, I. An automated in-situ polymerisation procedure for multi-functional cyanate ester resins via ring formation. Polymer 2021, 228, 123938. [Google Scholar] [CrossRef]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-forming Liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kugel, R.L.; Valan, K.J. Phosphonitrilic Compounds. VI. High Molecular Weight Poly(alkoxy- and aryloxyphosphazenes). Inorg. Chem. 1966, 5, 1709–1715. [Google Scholar] [CrossRef]
- Allen, G.; Lewis, C.; Todd, S. Polyphosphazenes: Part 2. Characterization. Polymer 1970, 11, 44–60. [Google Scholar] [CrossRef]
- Connelly, T.M., Jr.; Gillham, J.K. Polyphosphazenes: Thermomechanical transitions. J. Appl. Polym. Sci. 1976, 20, 473–488. [Google Scholar] [CrossRef]
- Maher, A.E. Synthesis and Characterization of Mixed-Substituent Poly(Organophosphazenes). Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2004. [Google Scholar]
- Gleria, M.; De Jaeger, R. Phosphazenes: A Worldwide Insight; Nova Science Publishers: Hauppauge, NY, USA, 2004. [Google Scholar]
- Ma, Y.; Liu, Y.; Yu, T.; Lai, W.; Ge, Z.; Jiang, Z. Structure—Property relationship of nitramino oxetane polymers: A computational study on the effect of pendant chains. RSC Adv. 2019, 9, 3120–3127. [Google Scholar] [CrossRef] [Green Version]
- Seymour, R.B.; Carraher, C.E., Jr. Structure-Property Relationships in Polymers; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Zheng, Y.; Zhang, Z.; Qiu, M.; Wu, J.; Liu, W.; Zhang, S.; Zhang, T.; Wu, Z. Influence of n-Butoxy group on properties of Poly(aryloxyphosphazene) elastomers and evaluation on their performances as thermal and electrical insulations. Compos. Part A Appl. Sci. Manuf. 2021, 149, 106533. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | |
|---|---|
| PZ-DC | 1.565 |
| PZ-TFE | 1.428 |
| PZ-Azido | 1.169 |
| PZ-Nitrato | 1.298 |
| p-PZ-DC | p-PZ-TFE | p-PZ-Azido | p-PZ-Nitrato | |
|---|---|---|---|---|
| Density | 2.167 | 1.365 | 1.164 | 1.372 |
| Change in Density | 0.123 | 0.171 | 0.169 | 0.100 |
| T | −32.6 | −51.8 | −29.8 | −39.5 |
| CVTE (<T) | 2.03 | 2.74 | 3.22 | 2.21 |
| CVTE (>T) | 2.99 | 4.46 | 8.29 | 3.49 |
| Young’s Modulus | 4.19 | 1.09 | 1.56 | 3.58 |
| Poisson’s Ratio | 0.31 | 0.41 | 0.47 | 0.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Demir, B. A Computational Procedure for Atomistic Modelling of Polyphosphazenes towards Better Capturing Molecular-Level Structuring and Thermo-Mechanical Properties. Polymers 2022, 14, 1451. https://doi.org/10.3390/polym14071451
Chen K, Demir B. A Computational Procedure for Atomistic Modelling of Polyphosphazenes towards Better Capturing Molecular-Level Structuring and Thermo-Mechanical Properties. Polymers. 2022; 14(7):1451. https://doi.org/10.3390/polym14071451
Chicago/Turabian StyleChen, Kay, and Baris Demir. 2022. "A Computational Procedure for Atomistic Modelling of Polyphosphazenes towards Better Capturing Molecular-Level Structuring and Thermo-Mechanical Properties" Polymers 14, no. 7: 1451. https://doi.org/10.3390/polym14071451
APA StyleChen, K., & Demir, B. (2022). A Computational Procedure for Atomistic Modelling of Polyphosphazenes towards Better Capturing Molecular-Level Structuring and Thermo-Mechanical Properties. Polymers, 14(7), 1451. https://doi.org/10.3390/polym14071451