
Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications



Abstract
:1. Introduction
- The characterization of PBCA as a defined pharmaceutical excipient to ensure quality prior to the NP formation is not possible;
- The solvent-dependent molecular weight (MW) control insufficiency of PCBA during polymerization.
2. Materials and Methods
2.1. Chemicals
2.2. Characterization Techniques
2.2.1. Advanced Polymer Chromatography™ (APC™)
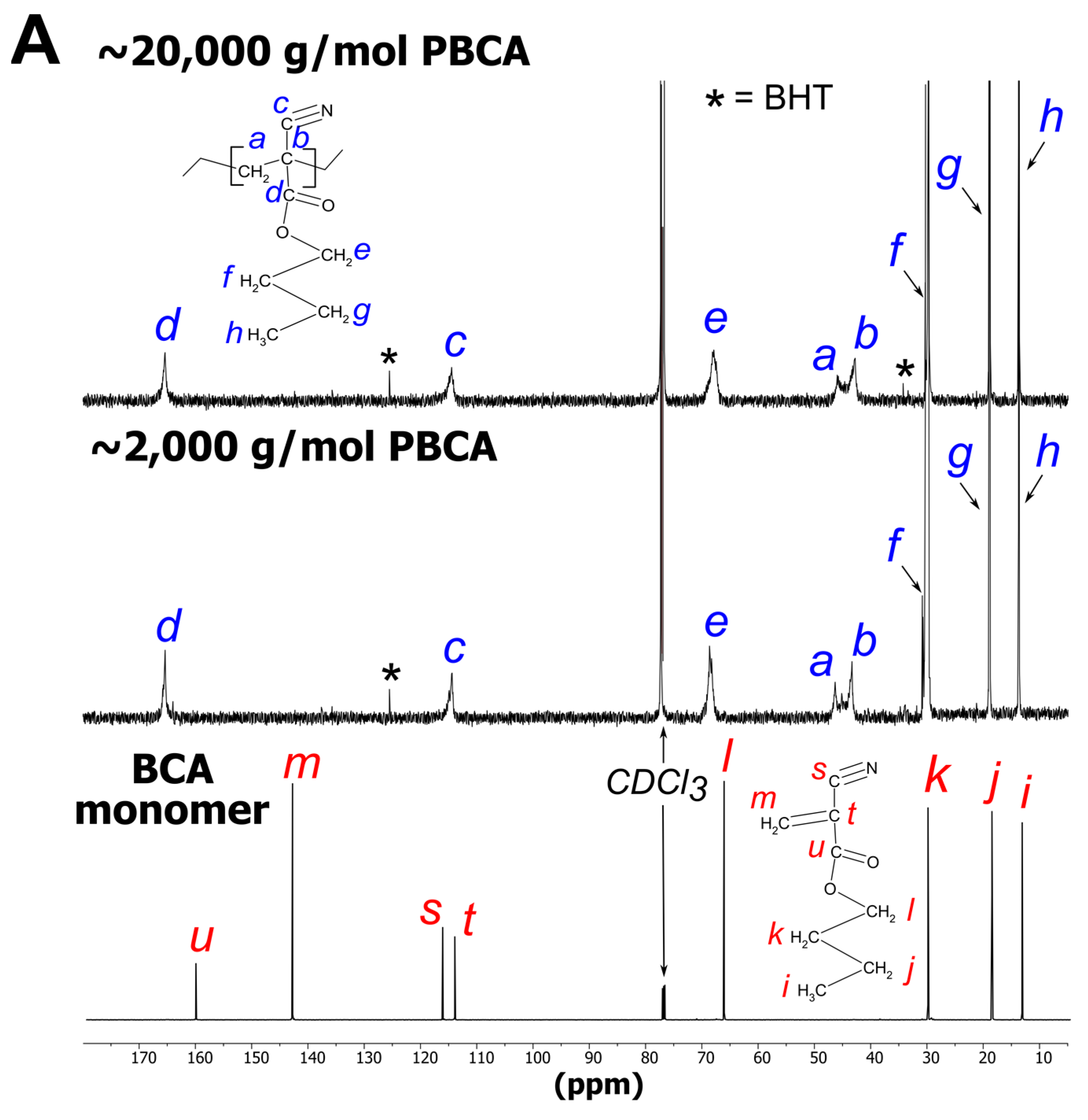
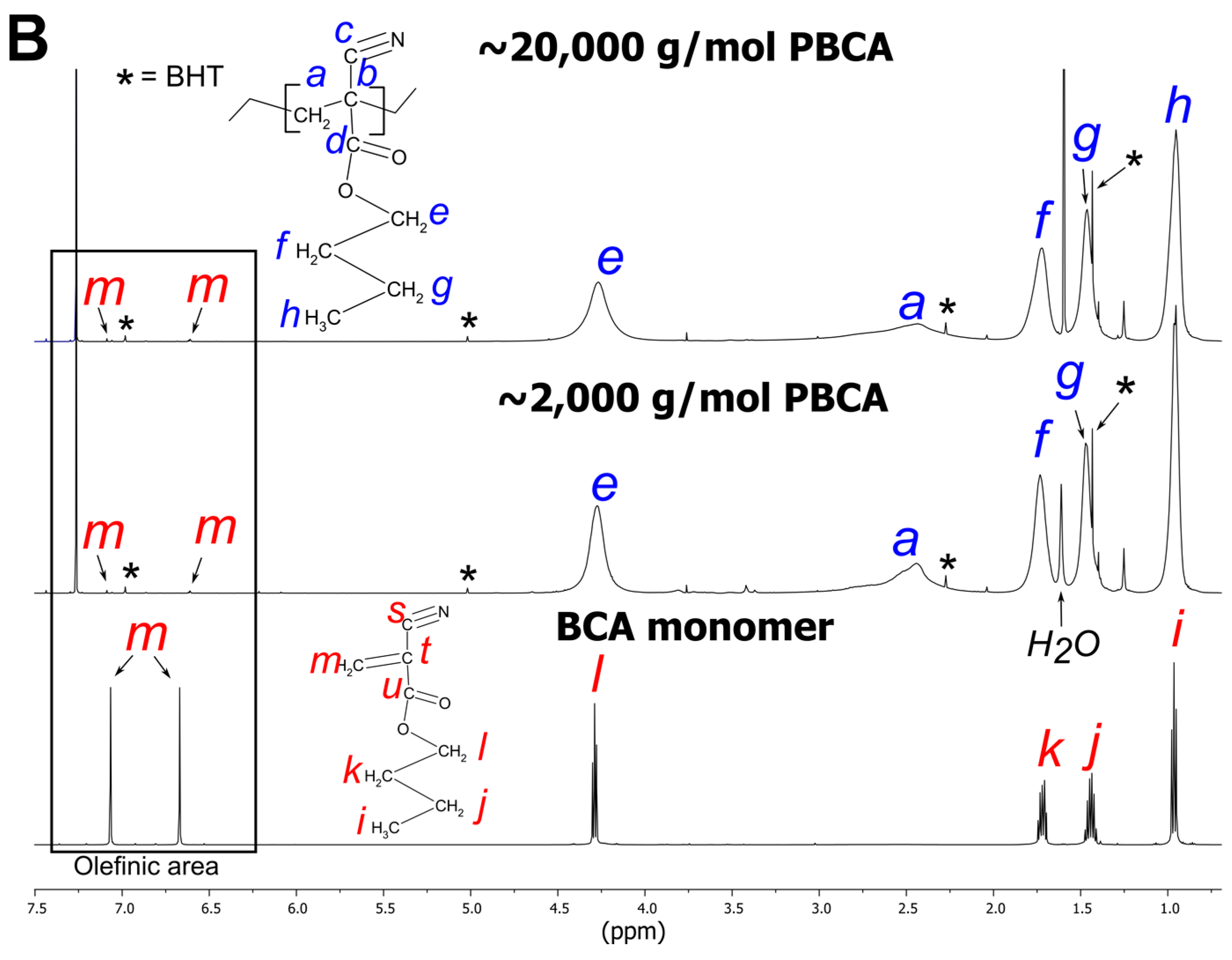
2.2.2. NMR
2.3. Synthesis
2.3.1. Anionic Polymerization in THF
2.3.2. PBCA End-Capping with EBMA in THF
2.3.3. Polymer Stability in Solution
3. Results
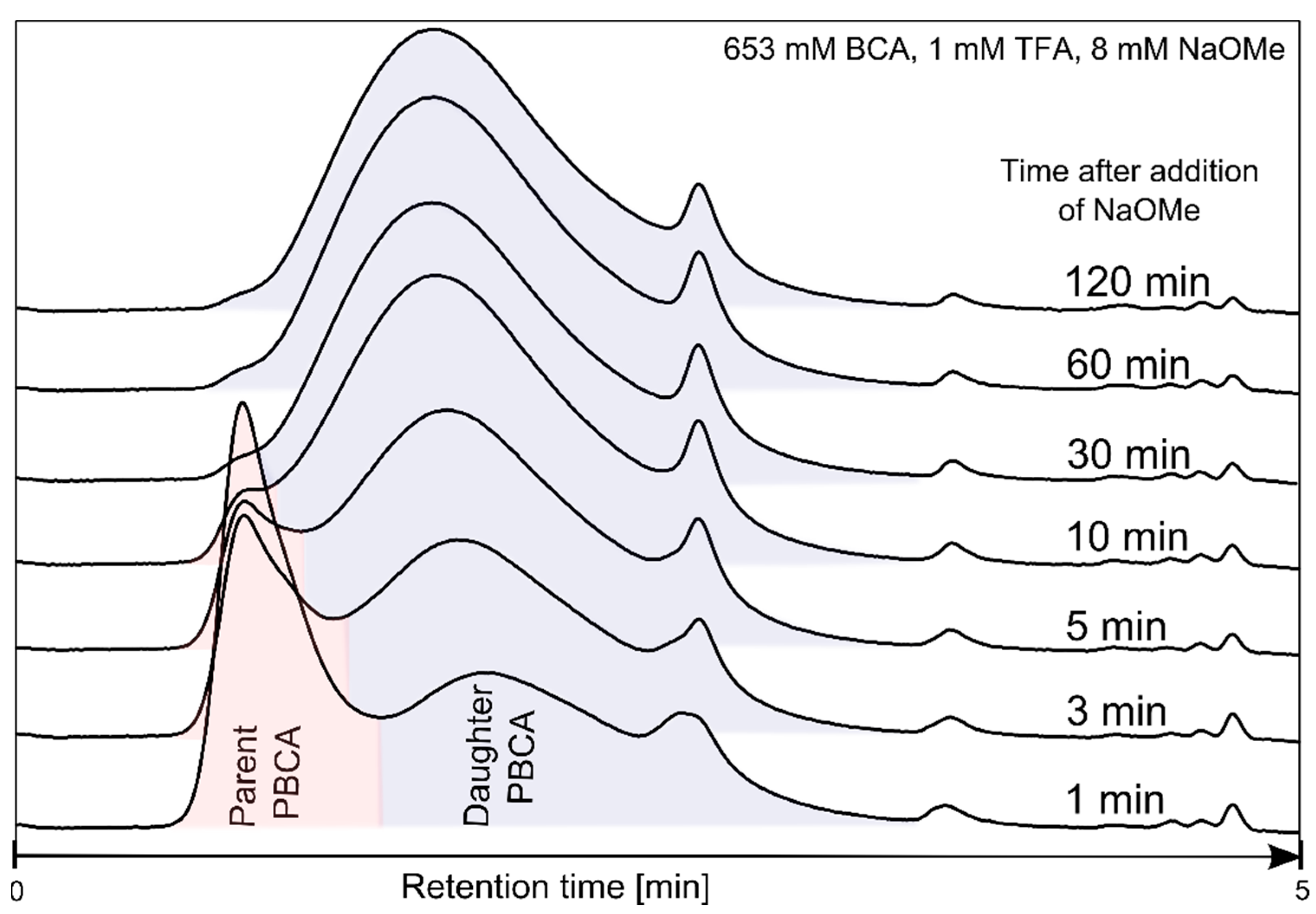
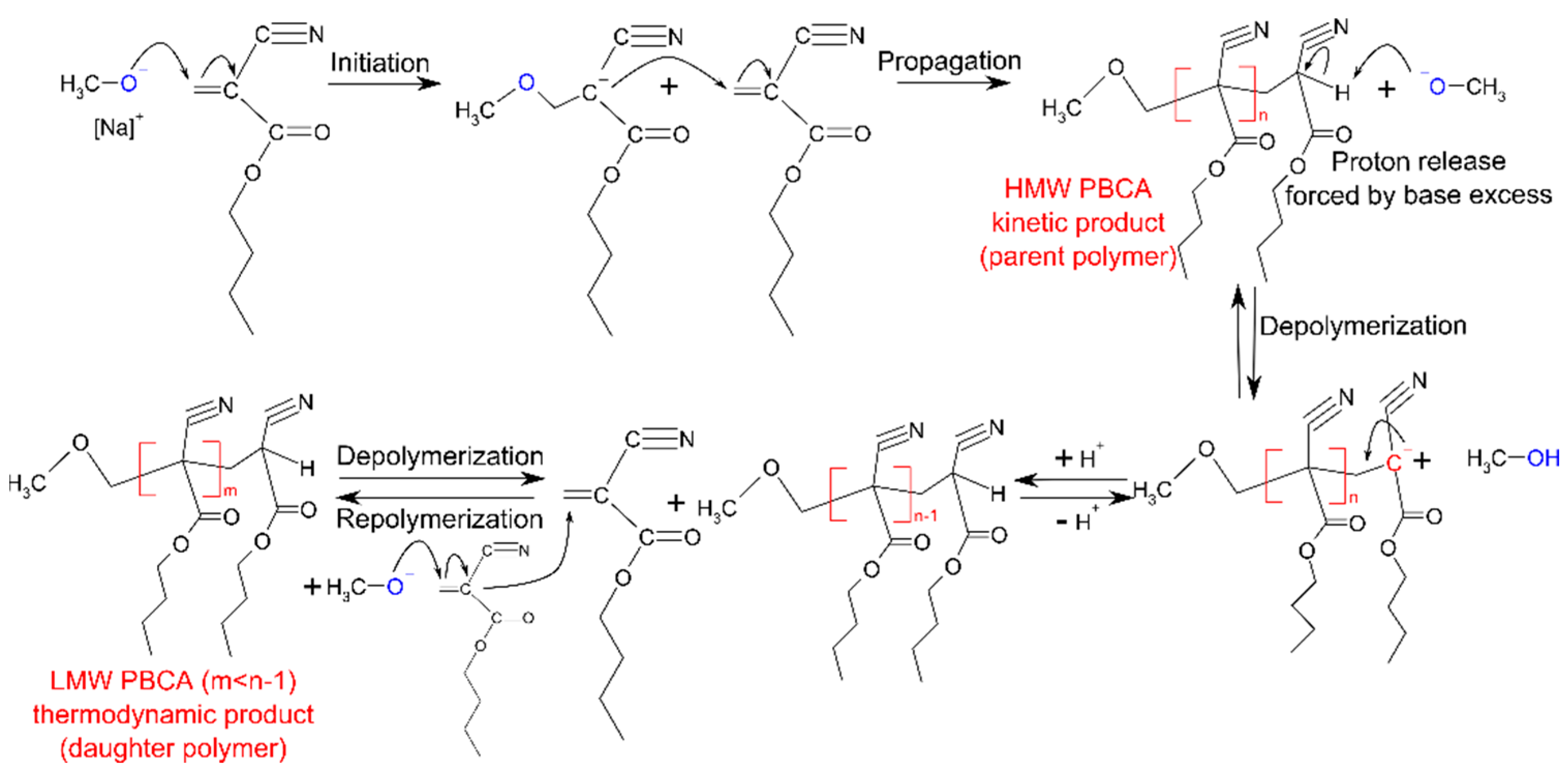
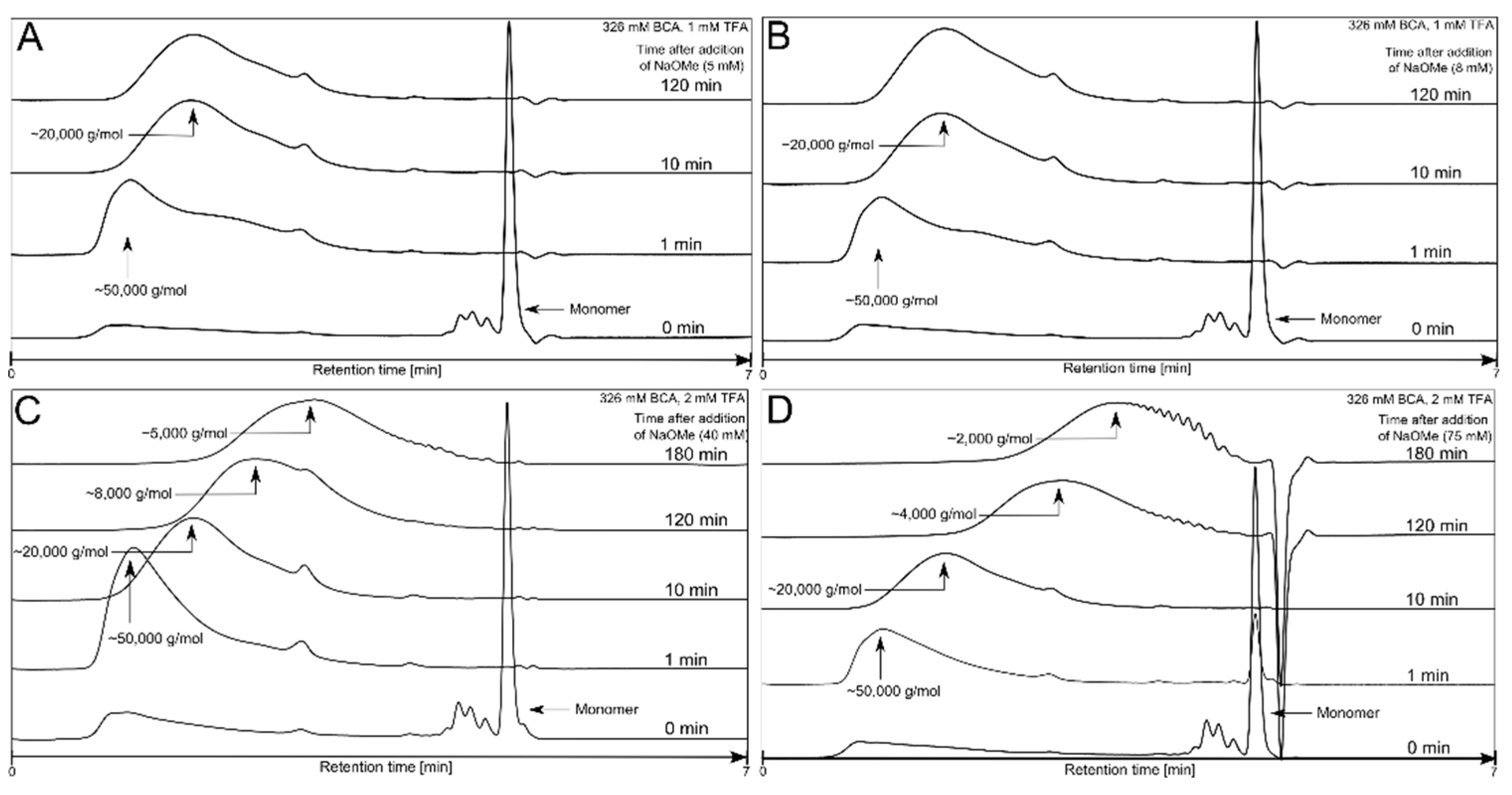
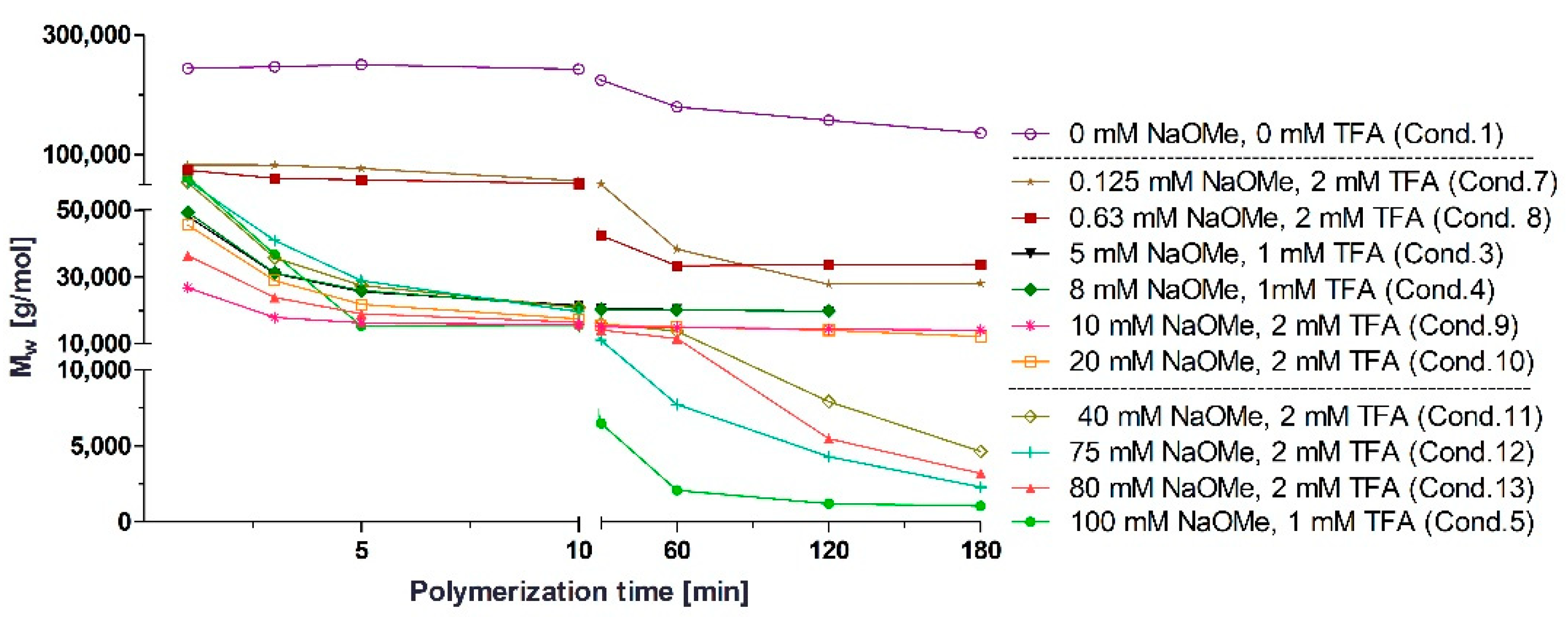
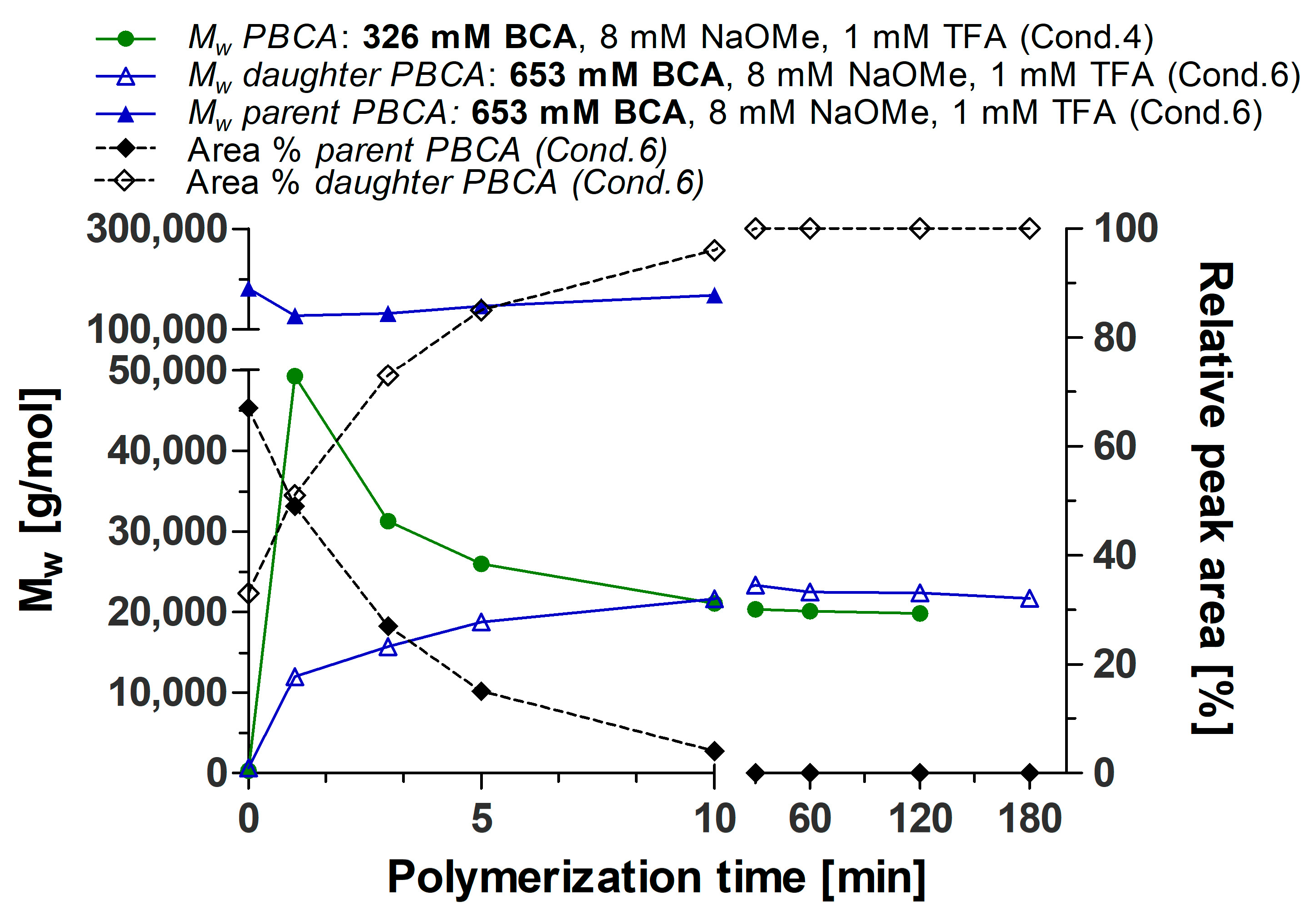
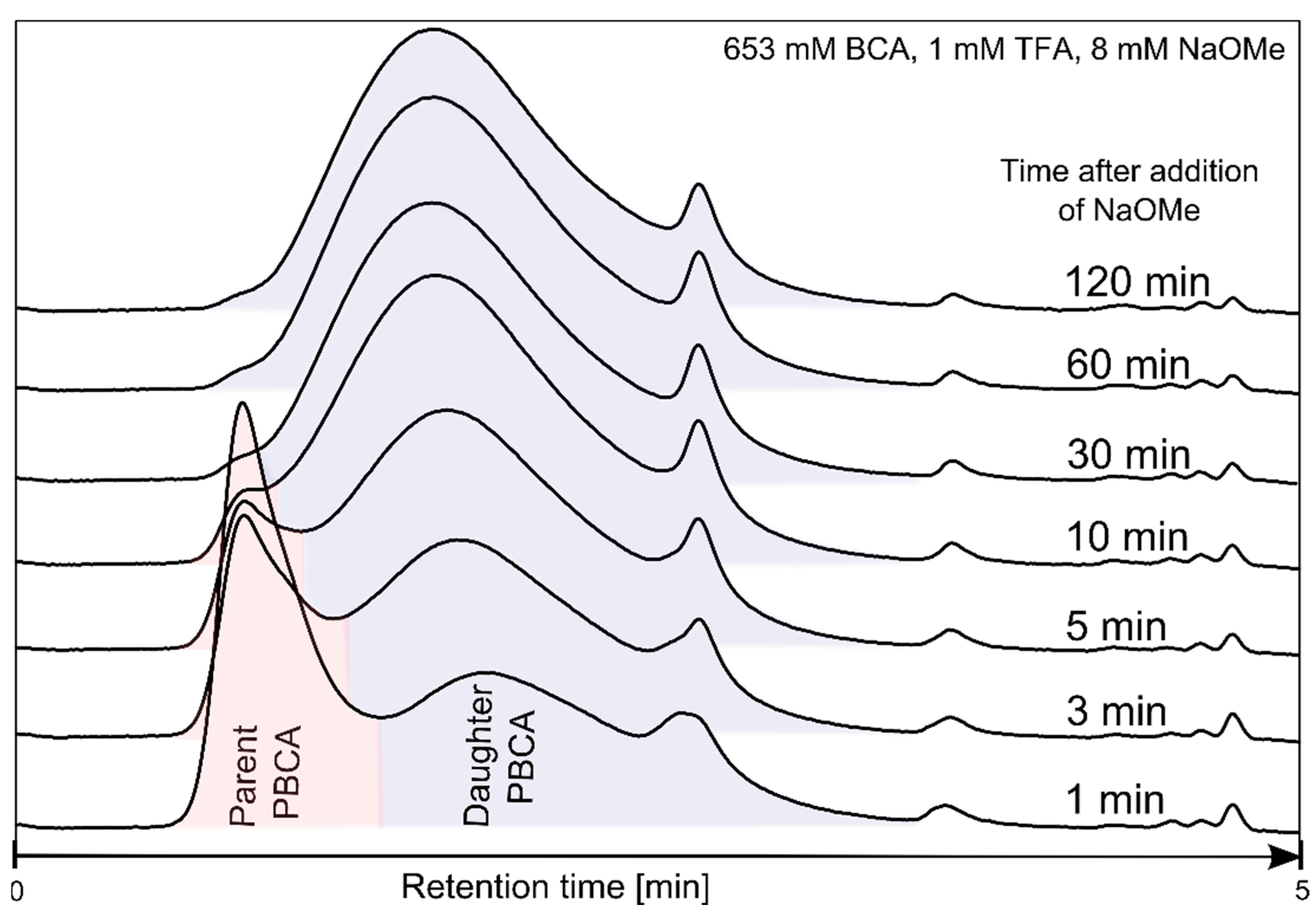
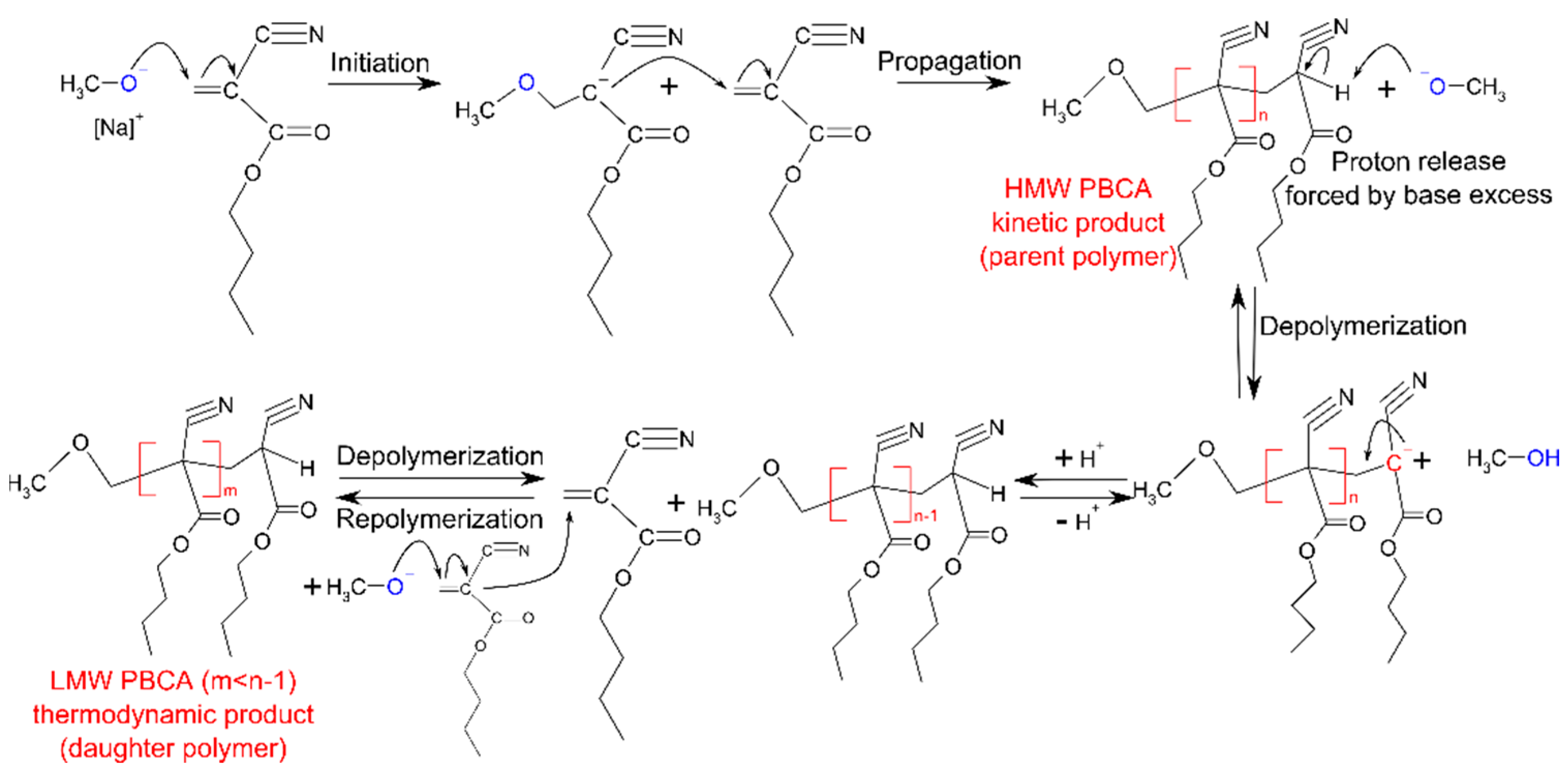
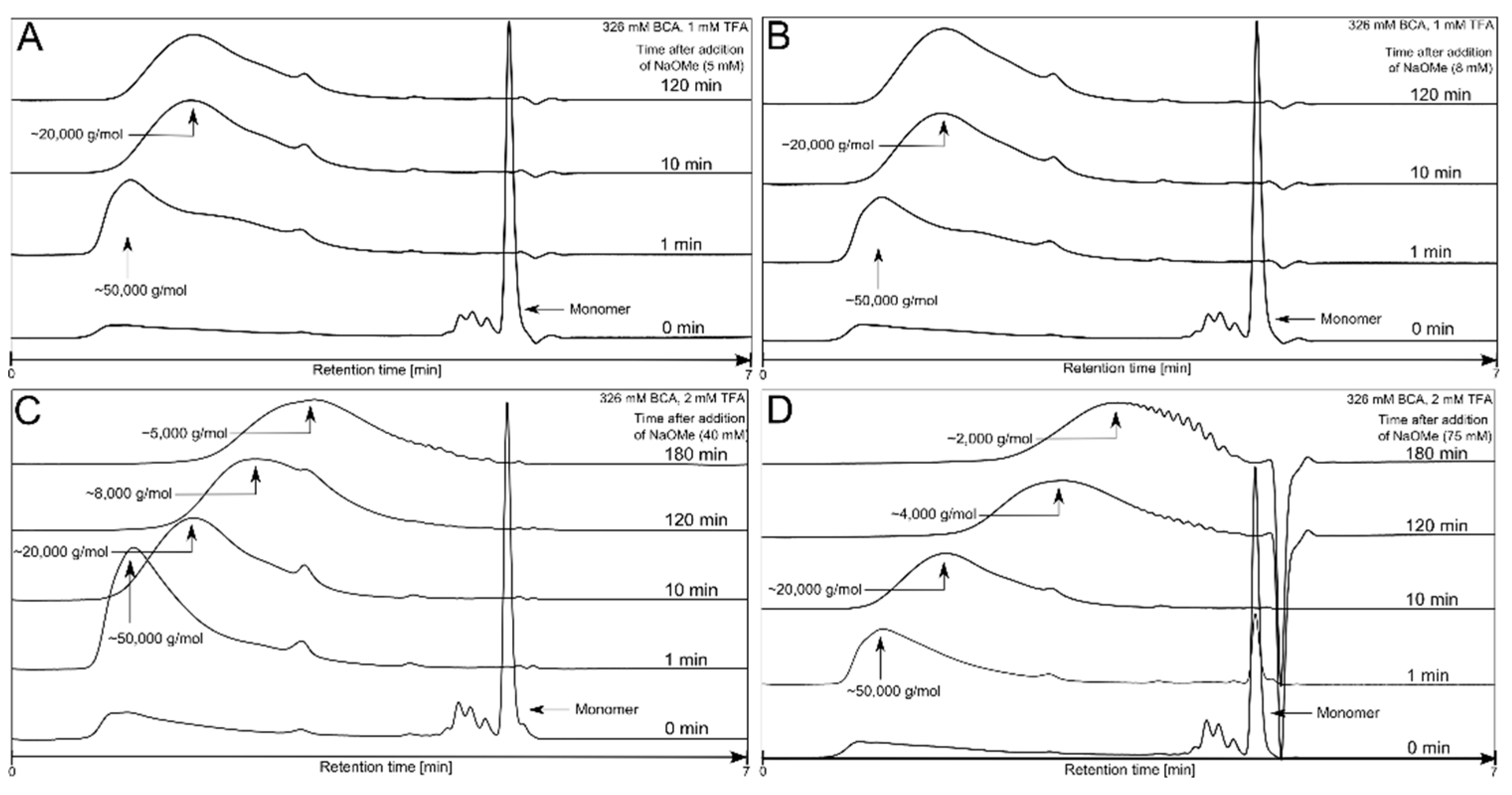
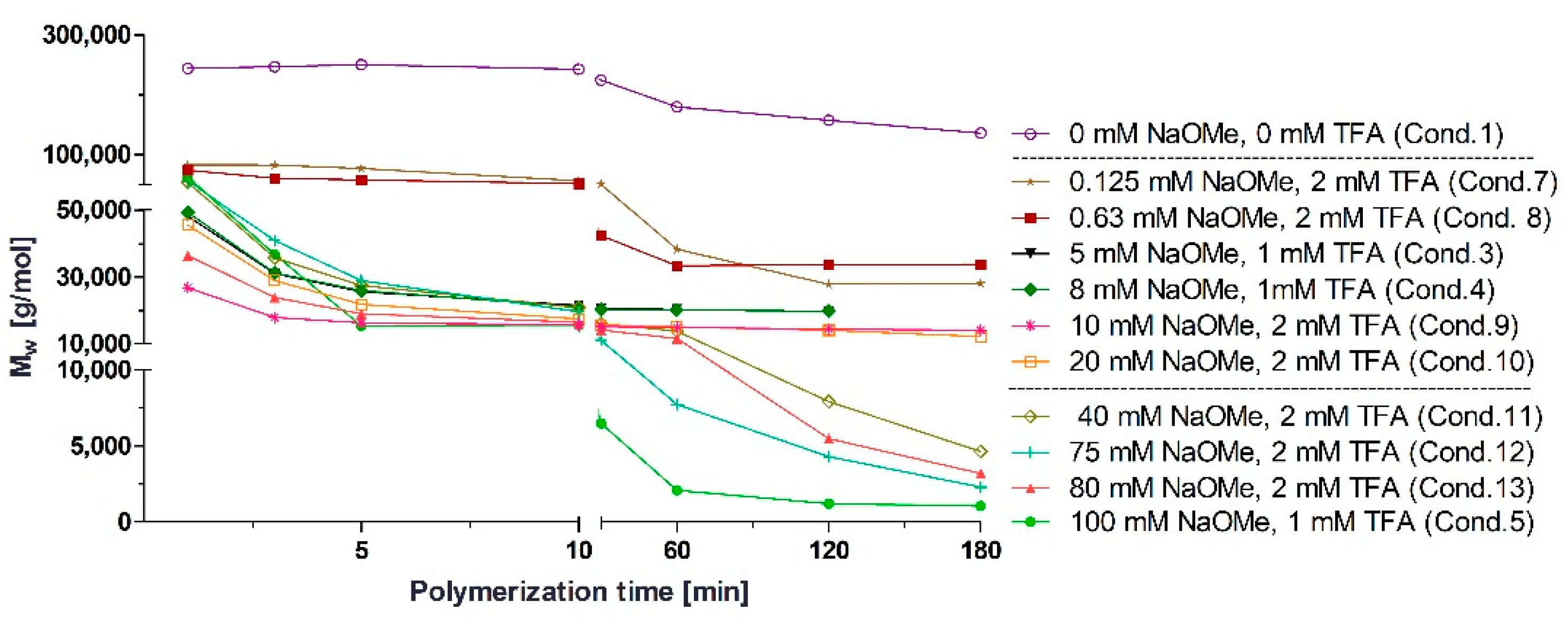
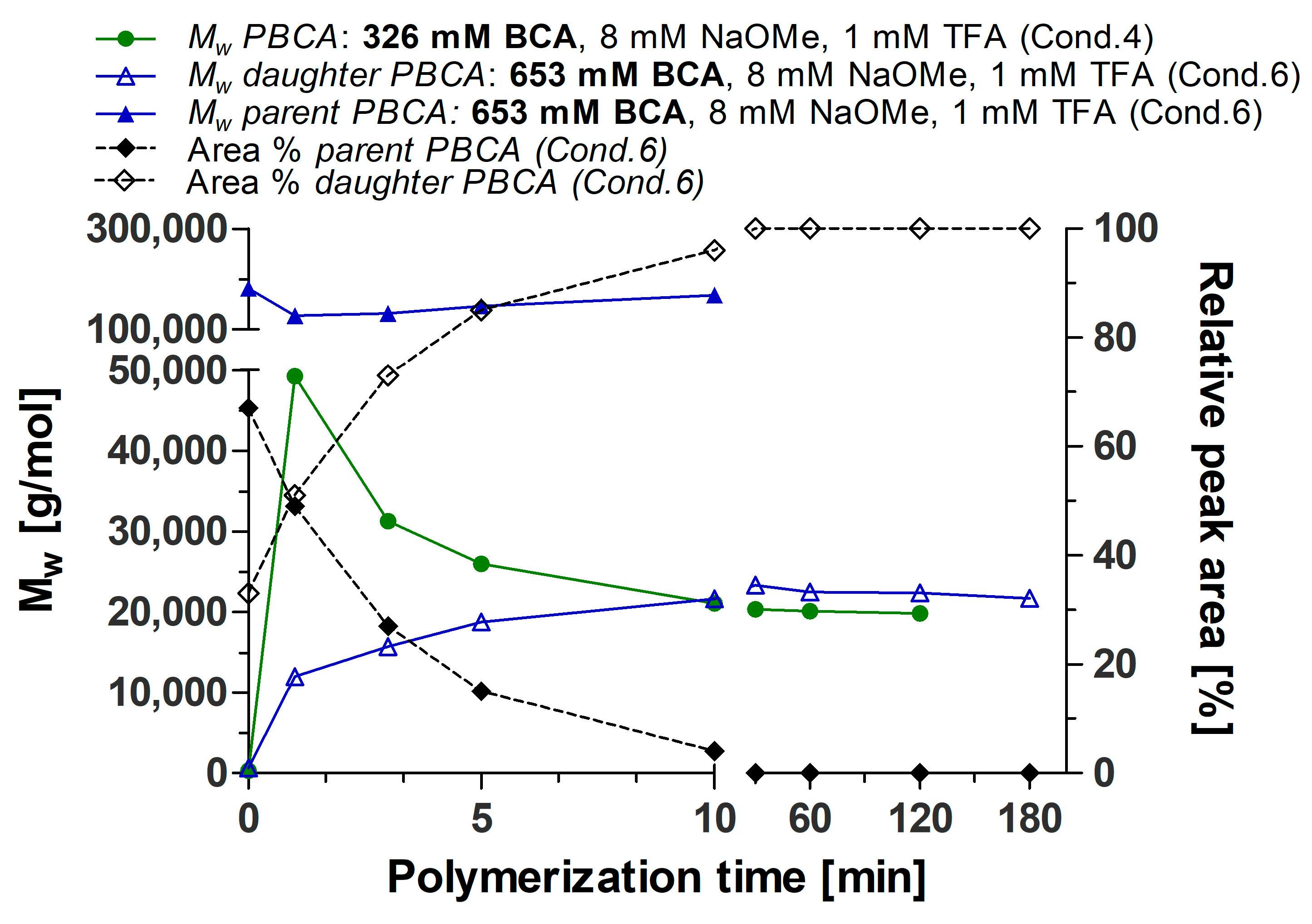
3.1. Polymerization Process Monitoring by APC™
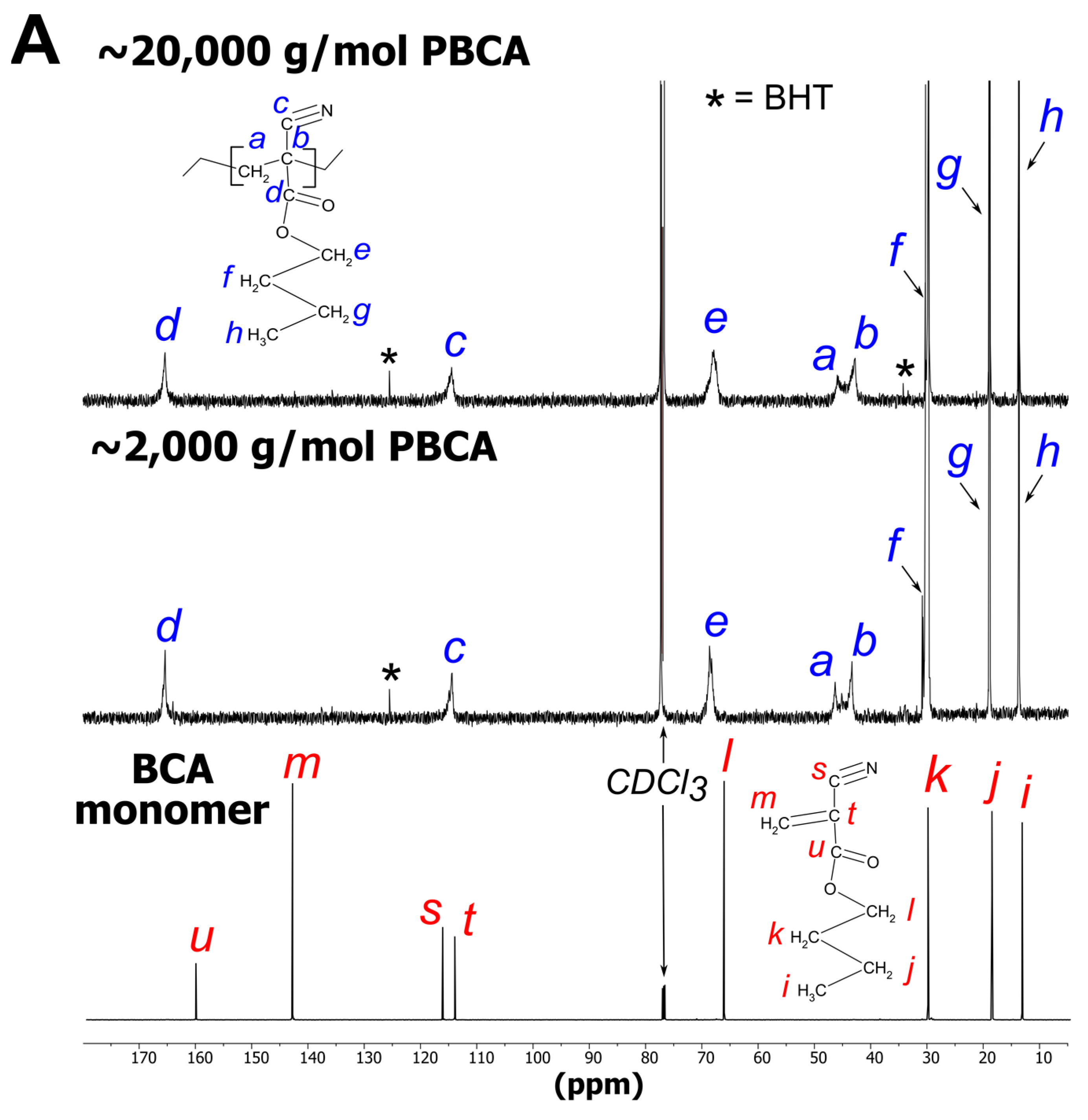
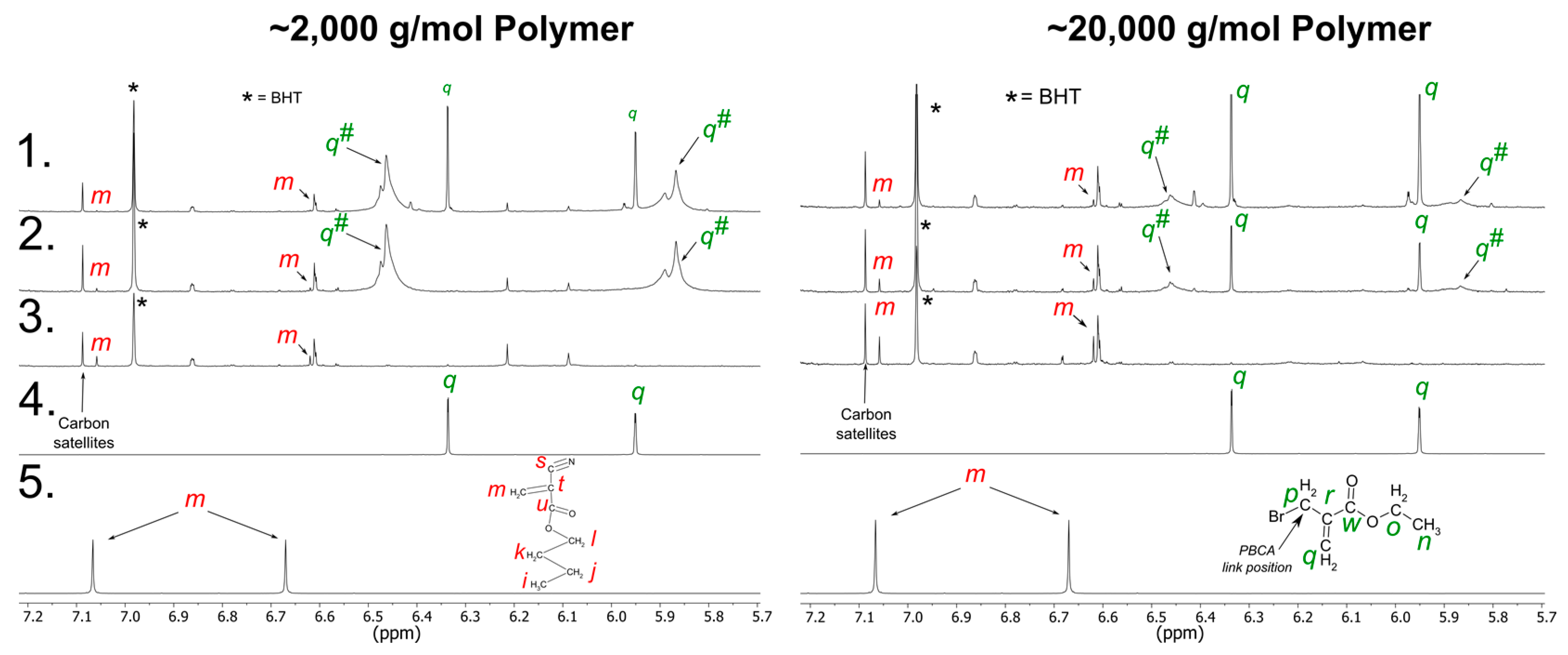
3.2. NMR
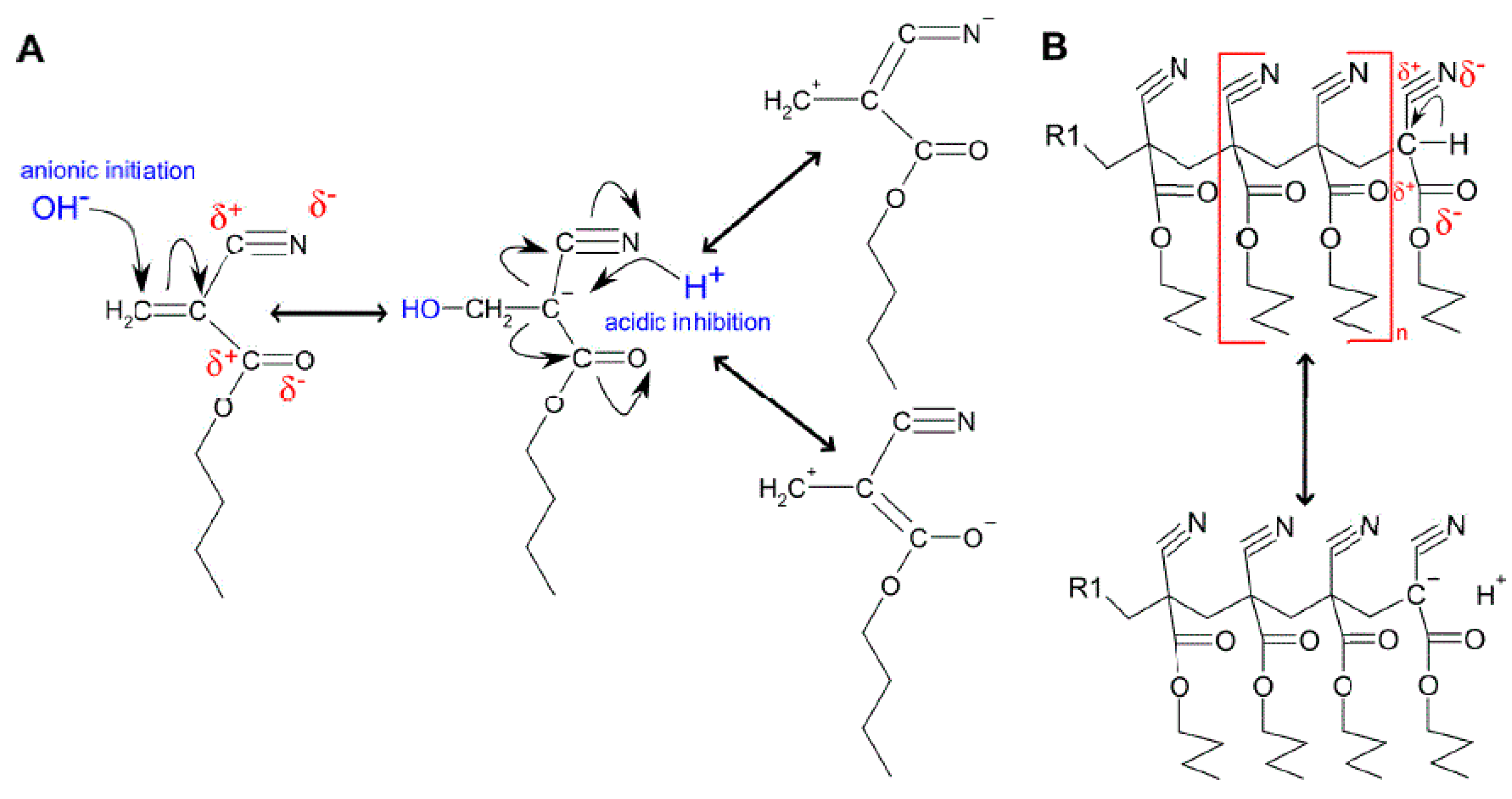
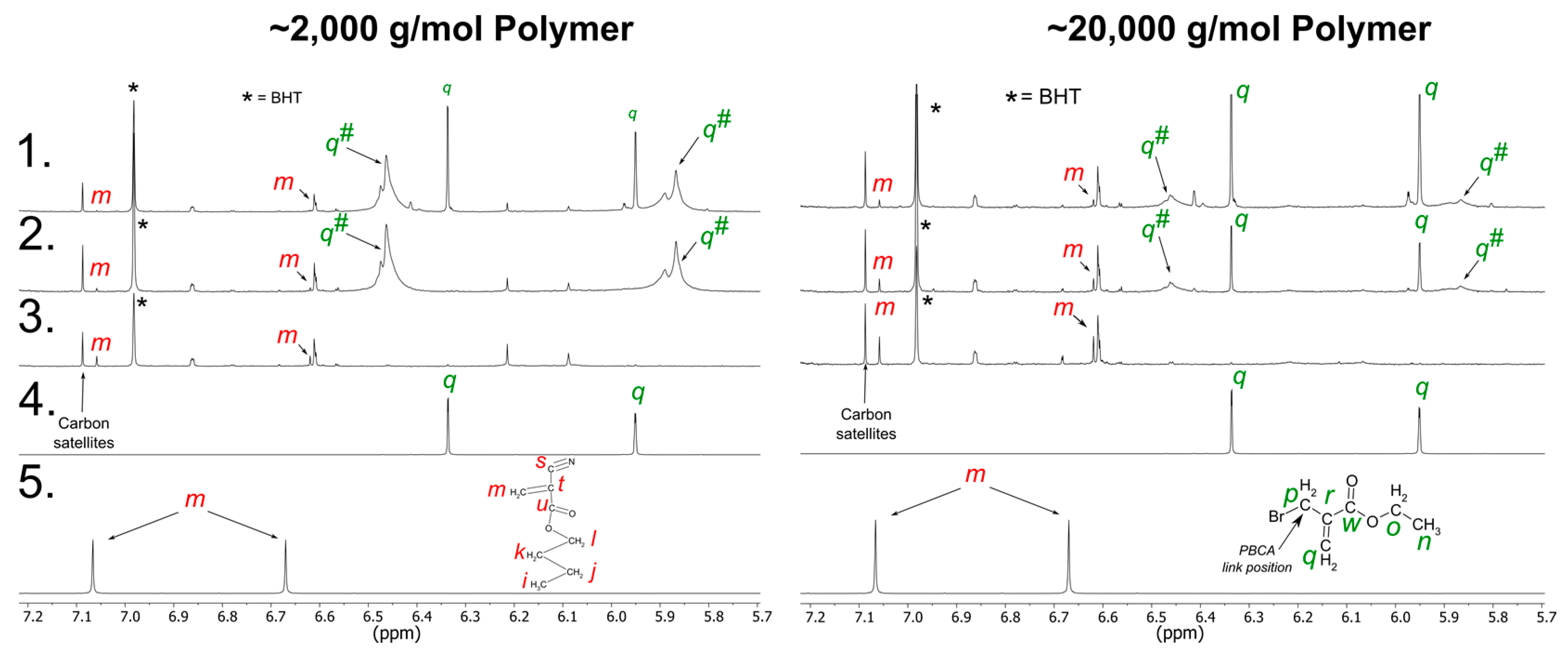
3.2.1. PBCA Structure Elucidation
3.2.2. tPBCA Structure Elucidation
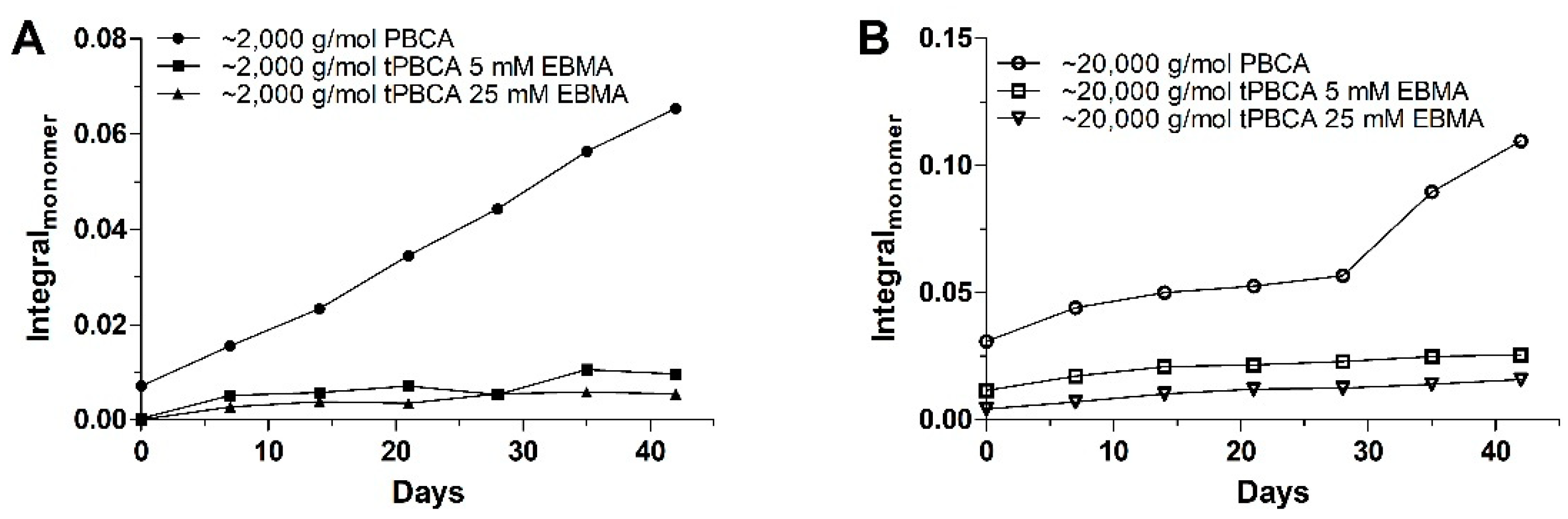
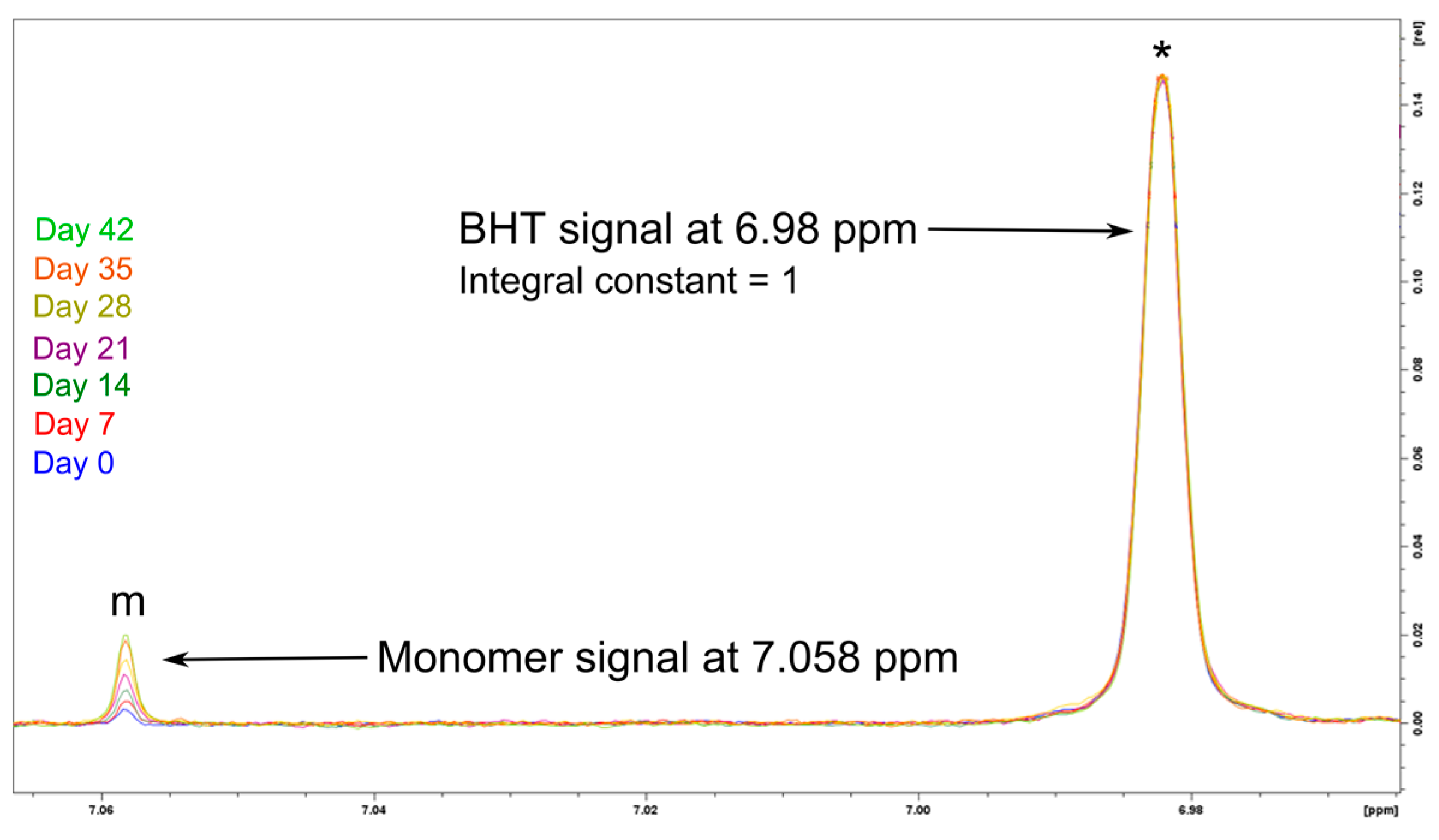
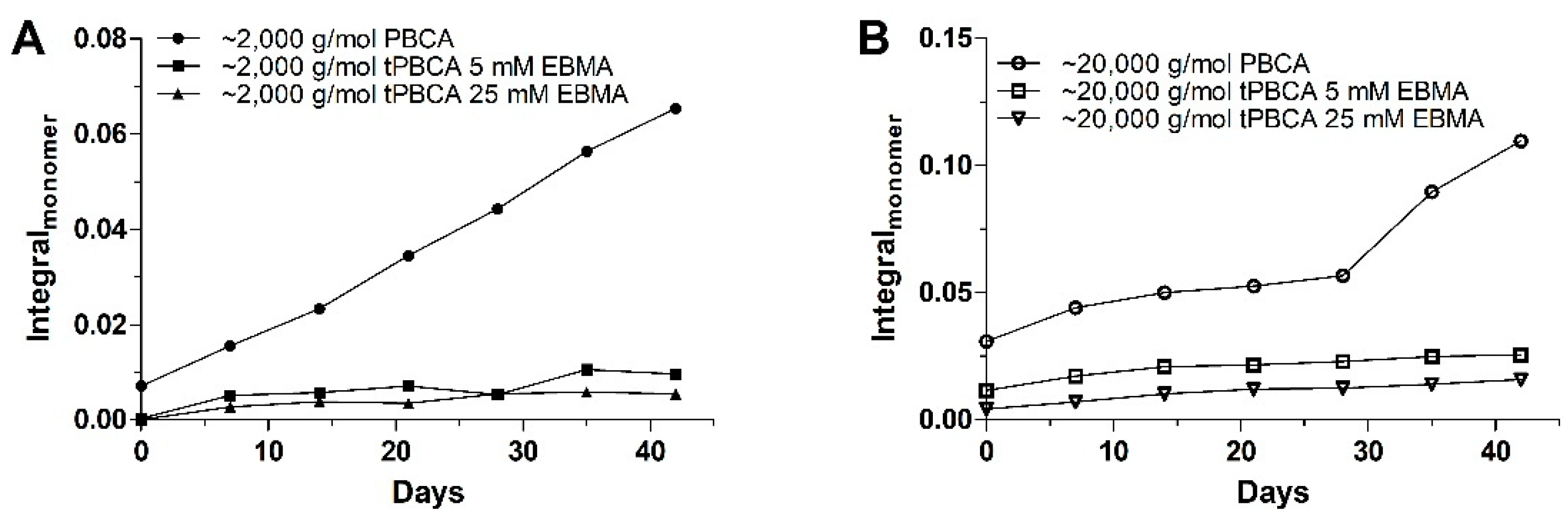
3.2.3. Polymer Stability in Solution
- The monomer release is indeed a progressive phenomenon for both PBCA batches, as the monomer amount clearly increased during storage;
- The EBMA end-capping of the PBCA chains (tPBCA) inhibited the progression of the monomer release.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Khatak, S.; Sara, U.V.S. Solid lipid nanoparticles—A review. Int. J. Appl. Pharma 2013, 5, 8–18. [Google Scholar]
- Ling, D.; Hyeon, T. Chemical design of biocompatible iron oxide nanoparticles for medical applications. Small 2013, 9, 1450–1466. [Google Scholar] [CrossRef] [PubMed]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal drug delivery systems: An update review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, L.; Bianco, A.; Prato, M.; Kostarelos, K. Carbon nanotubes as nanomedicines: From toxicology to pharmacology. Adv. Drug Deliv. Rev. 2006, 58, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Van, E.B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles-based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Kante, B.; Roland, M.; Guiot, P.; Bauduin, P.; Speiser, P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: Preparation, morphological and sorptive properties. J. Pharm. Pharmacol. 1979, 5, 331–332. [Google Scholar] [CrossRef]
- Vauthier, C.; Dubernet, C.; Fattal, E.; Pinto-Alphandary, H.; Couvreur, P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv. Drug Deliv. Rev. 2003, 55, 519–548. [Google Scholar] [CrossRef]
- Nicolas, J.; Couvreur, P. Synthesis of poly(alkyl cyanoacrylate)-based colloidal nanomedicines. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 111–127. [Google Scholar] [CrossRef]
- Gao, S.; Xu, Y.; Asghar, S.; Chen, M.; Zou, L.; Eltayeb, S.; Huo, M.; Ping, Q.; Xiao, Y. Polybutylcyanoacrylate nanocarriers as promising targeted drug delivery systems. J. Drug Target 2015, 23, 481–496. [Google Scholar] [CrossRef] [PubMed]
- P010002; Indermil™ Tissue Adhesive. U.S. Food and Drug Administration: Silver Spring, MD, USA, 2002.
- Pifferi, G.; Santoro, P.; Pedrani, M. Quality and functionality of excipients. Farmaco 1999, 54, 1–14. [Google Scholar] [CrossRef]
- Pifferi, G.; Restani, P. The safety of pharmaceutical excipients. Farmaco 2003, 58, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Eromosele, I.C.; Pepper., D.C. Anionic polymerization of butyl cyanoacrylate by tetrabutylammonium salts, Propagation rate constants. Macromol. Chem. Phys. 1989, 190, 3095–3103. [Google Scholar] [CrossRef]
- Limouzin, C.; Caviggia, A.; Ganachaud, F.; Hemery, P. Anionic Polymerization of n-Butyl Cyanoacrylate in Emulsion and Miniemulsion. Macromolecules 2003, 36, 667–674. [Google Scholar] [CrossRef]
- Almalik, A.; Donno, R.; Cadman, C.J.; Cellesi, F.; Day, P.J.; Tirelli, N. Hyaluronic acid-coated chitosan nanoparticles: Molecular weight-dependent effects on morphology and hyaluronic acid presentation. J. Control. Release 2013, 172, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Mittal, G.; Sahana, D.K.; Bhardwaj, V.; Kumar, M.N.V.R. Estradiol loaded PLGA nanoparticles for oral administration: Effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J. Control. Release 2007, 119, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Gorner, T.; Gref, R.; Michenot, D.; Sommer, F.; Tran, M.N.; Dellacherie, E. Lidocaine-loaded biodegradable nanospheres. I. Optimization Of the drug incorporation into the polymer matrix. J. Control. Release 1999, 57, 259–268. [Google Scholar] [CrossRef]
- Ryan, B.; McCann, G. Novel sub-ceiling temperature rapid depolymerization-repolymerization reactions of cyanoacrylate polymers. Macromol. Chem. Phys. 1996, 17, 217–227. [Google Scholar] [CrossRef]
- Pepper, D.C.; Ryan, B. Initiation Processes in Polymerization of Alkyl Cyanoacrylates by Tertiary Amines: Inhibition by Strong Acids. Macromol. Chem. Phys. 1983, 184, 383–394. [Google Scholar] [CrossRef]
- Costa, G.; Cronin, D.C.; Pepper, D.C.; Loonan, C. Termination and transfer by acids in the pyridineinitiated. polymerization of butyl cyanoacrylate. Euro Polym. J. 1983, 19, 939–945. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 412–436. [Google Scholar] [CrossRef]
- Donnelly, E.F.; Johnston, D.S.; Pepper, D.C. Ionic and zwitterionic polymerization of n-alkyl 2-cyanoacrylates. Polym. Lett. Ed. 1977, 15, 399–405. [Google Scholar] [CrossRef]
- Canale, A.J.; Goode, W.E.; Kinsinger, J.B.; Panchak, J.R.; Kelso, R.L.; Graham, R.K. Methyl α-cyanoacrylate I Free-radical homopolymerization. J. Appl. Polym. Sci. 1960, 4, 231–236. [Google Scholar] [CrossRef]
- Han, M.G.; Kim, S.; Liu, S.X. Synthesis and degradation behavior of poly(ethyl cyanoacrylate). Polym. Degrad. Stab. 2008, 93, 1243–1251. [Google Scholar] [CrossRef]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009, 4, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Robello, D.R.; Eldridge, T.D.; Swanson, M.T. Degradation and Stabilization of Polycyanoacrylates. J. Polym. Sci. 1999, 37, 4570–4581. [Google Scholar] [CrossRef]
- Leonard, F.; Kulkarni, R.K.; Brandes, J.N.; Cameron, J. Synthesis and Degradation of Poly(alkyl-cyanoacrylates. J. Appl. Polym. Sci. 1966, 10, 259–272. [Google Scholar] [CrossRef]
- Pirker, S.; Kruse, J.; Noe, C.; Langer, K.; Zimmer, A.; Kreuter, J. Characterization of polybutyleyanoacrylate nanoparticles. Part II: Determination of polymer content by NMR analysis. Int. J. Pharm. 1995, 128, 189–195. [Google Scholar] [CrossRef]
- Wohlgemuth, M.; Machtle, W.; Mayer, C. Improved preparation and physical studies of polybutylcyanoacrylate nanocapsules. J. Microencapsul. 2000, 17, 437–448. [Google Scholar] [CrossRef]
- Mulik, R.; Mahadik, K.; Paradkar, A. Development of curcuminoids loaded poly(butyl)cyanoacrylate nanoparticles: Physicochemical characterization and stability study. Eur. J. Pharm. Sci. 2009, 37, 395–404. [Google Scholar] [CrossRef]
- Mayer, C. NMR Studies of Nanoparticles. Annu. Rep. NMR Spectrosc. 2005, 55, 258. [Google Scholar] [CrossRef]
- Szanka, I.; Szanka, A.; Kennedy, J. Robbery Wound Closure Adhesives. II. Initiators for and Initiation of 2-Octyl Cyanoacrylate Polymerization. J. Polym. Sci. 2015, 53, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, Y.; Ishihara., S.; Kitayama, T. Termination of Living Anionic Polymerization of Butyl Acrylate with alpha-(Chloromethyl)acrylate for End-Functionalization and Application to Evaluation of Monomer Reactivity. Macromol. Chem. Phys. 2015, 216, 1534–1539. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | TFA (mM) | BCA (mM) | NaOMe (mM) |
|---|---|---|---|
| 1 | 0.00 | 326 | 0 |
| 2 | 0.01 | 326 | 5 |
| 3 | 1.00 | 326 | 5 |
| 4 | 1.00 | 326 | 8 |
| 5 | 1.00 | 326 | 100 |
| 6 | 1.00 | 653 | 8 |
| 7 | 2.00 | 326 | 0.125 |
| 8 | 2.00 | 326 | 0.630 |
| 9 | 2.00 | 326 | 10 |
| 10 | 2.00 | 326 | 20 |
| 11 | 2.00 | 326 | 40 |
| 12 | 2.00 | 326 | 75 |
| 13 | 2.00 | 326 | 80 |
| Time (min) | Parent PBCA | Daughter PBCA | ||
|---|---|---|---|---|
| MW (g/mol) | Relative Peak Area (%) | MW (g/mol) | Relative Peak Area (%) | |
| 0.5 | 181,488 | 67 | 645 | 33 |
| 1 | 127,605 | 49 | 12,023 | 51 |
| 3 | 131,730 | 27 | 15,751 | 73 |
| 5 | 145,970 | 15 | 18,775 | 85 |
| 10 | 168,314 | 4 | 21,623 | 96 |
| 30 | - | 0 | 23,328 | 100 |
| 60 | - | 0 | 22,496 | 100 |
| 120 | - | 0 | 22,388 | 100 |
| 180 | - | 0 | 21,713 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, B.-L.; Lohmann, C.A.; Kyeremateng, S.O.; Fricker, G. Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications. Polymers 2022, 14, 998. https://doi.org/10.3390/polym14050998
Keller B-L, Lohmann CA, Kyeremateng SO, Fricker G. Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications. Polymers. 2022; 14(5):998. https://doi.org/10.3390/polym14050998
Chicago/Turabian StyleKeller, Benjamin-Luca, Claudia A. Lohmann, Samuel O. Kyeremateng, and Gert Fricker. 2022. "Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications" Polymers 14, no. 5: 998. https://doi.org/10.3390/polym14050998
APA StyleKeller, B.-L., Lohmann, C. A., Kyeremateng, S. O., & Fricker, G. (2022). Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications. Polymers, 14(5), 998. https://doi.org/10.3390/polym14050998