
Polyurea Aerogels: Synthesis, Material Properties, and Applications

Abstract
:
1. Introduction
2. The Chemistry of Polyurea Aerogels
2.1. The Chemistry of the Isocyanate Group
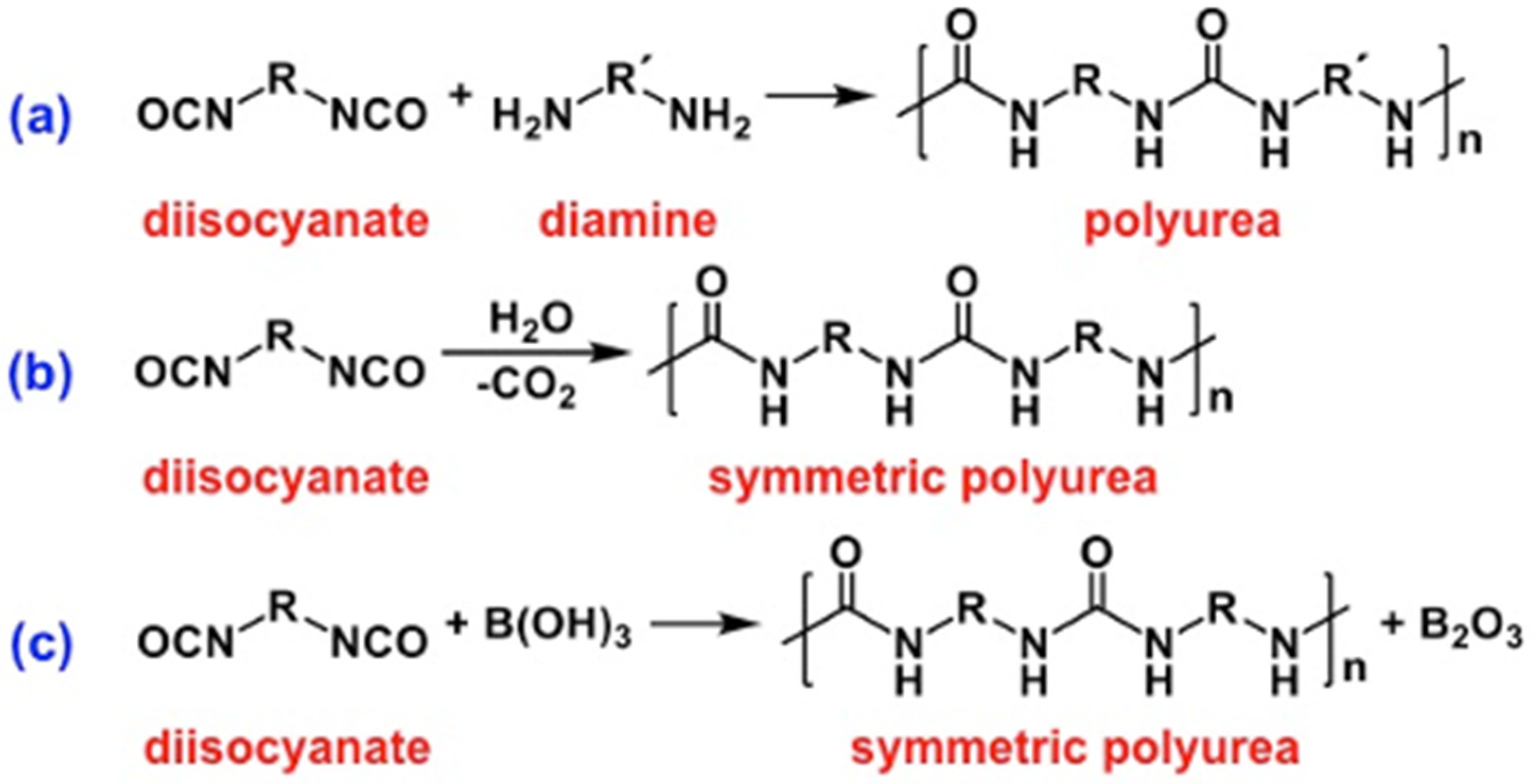
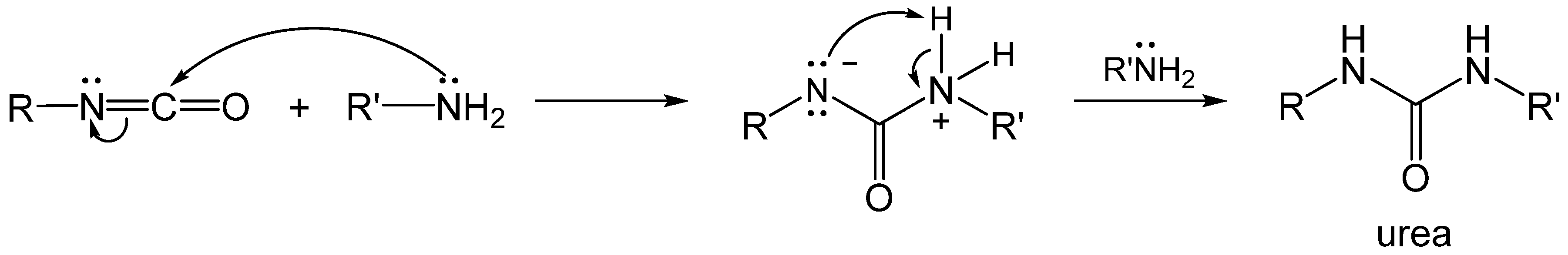
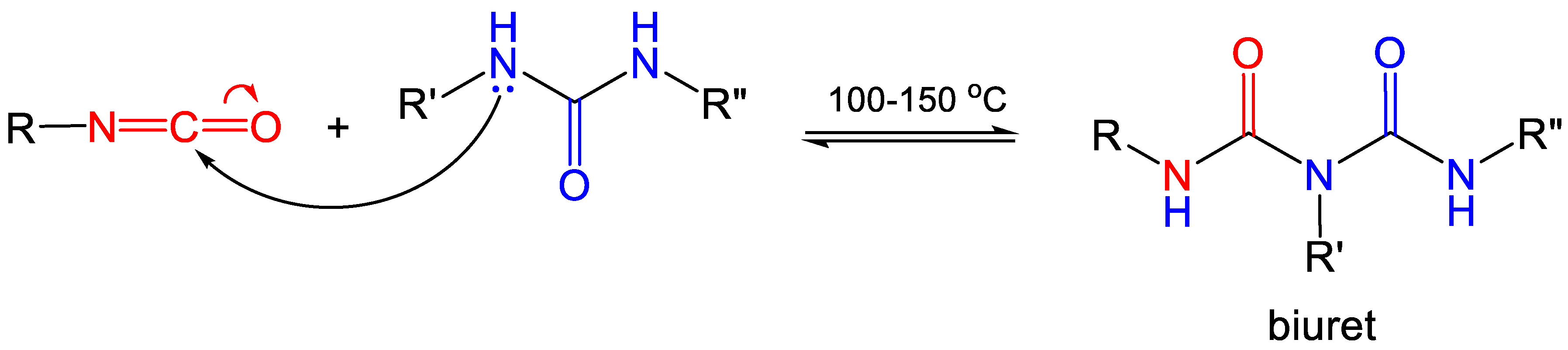
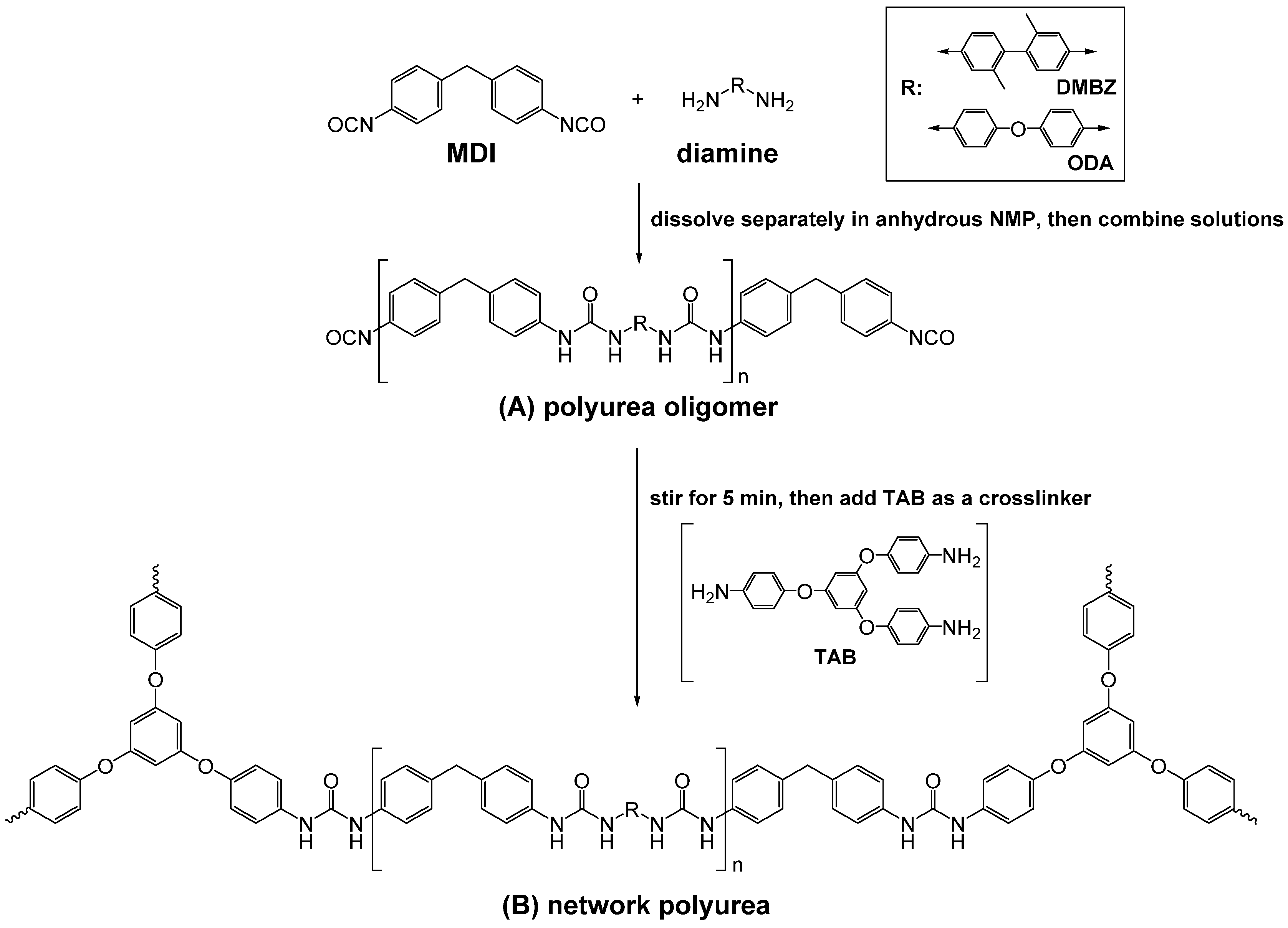
2.2. Formation of Polyurea Aerogels via Reaction of the –N=C=O with Amines
2.3. Formation of Polyurea Aerogels via Reaction of –N=C=O with Water
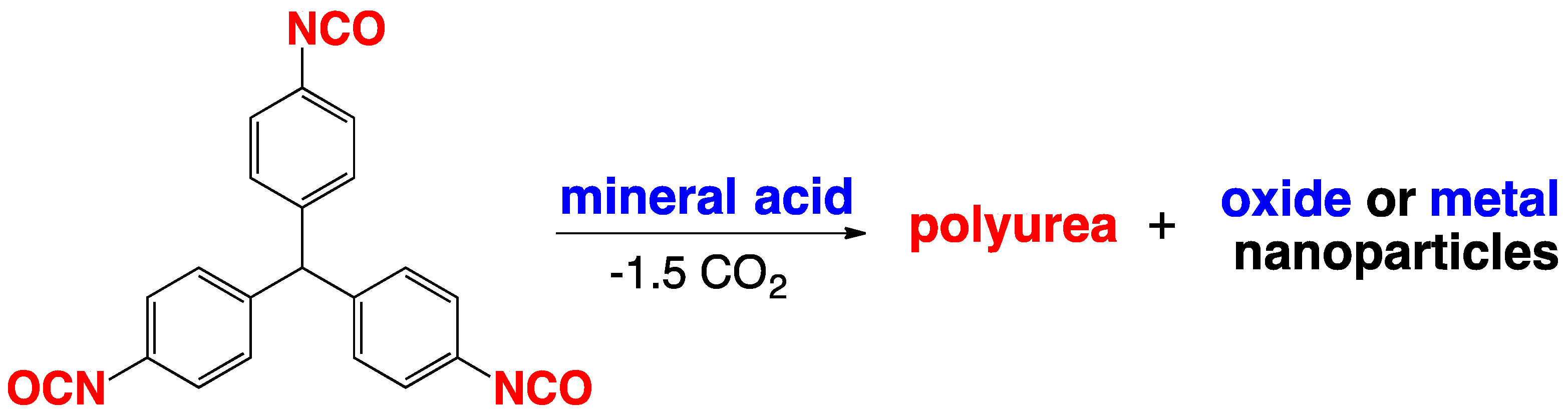
2.4. Formation of Polyurea Aerogels via Reaction of –N=C=O with Mineral Acids
3. Translating the Polymerization Chemistry into Aerogels
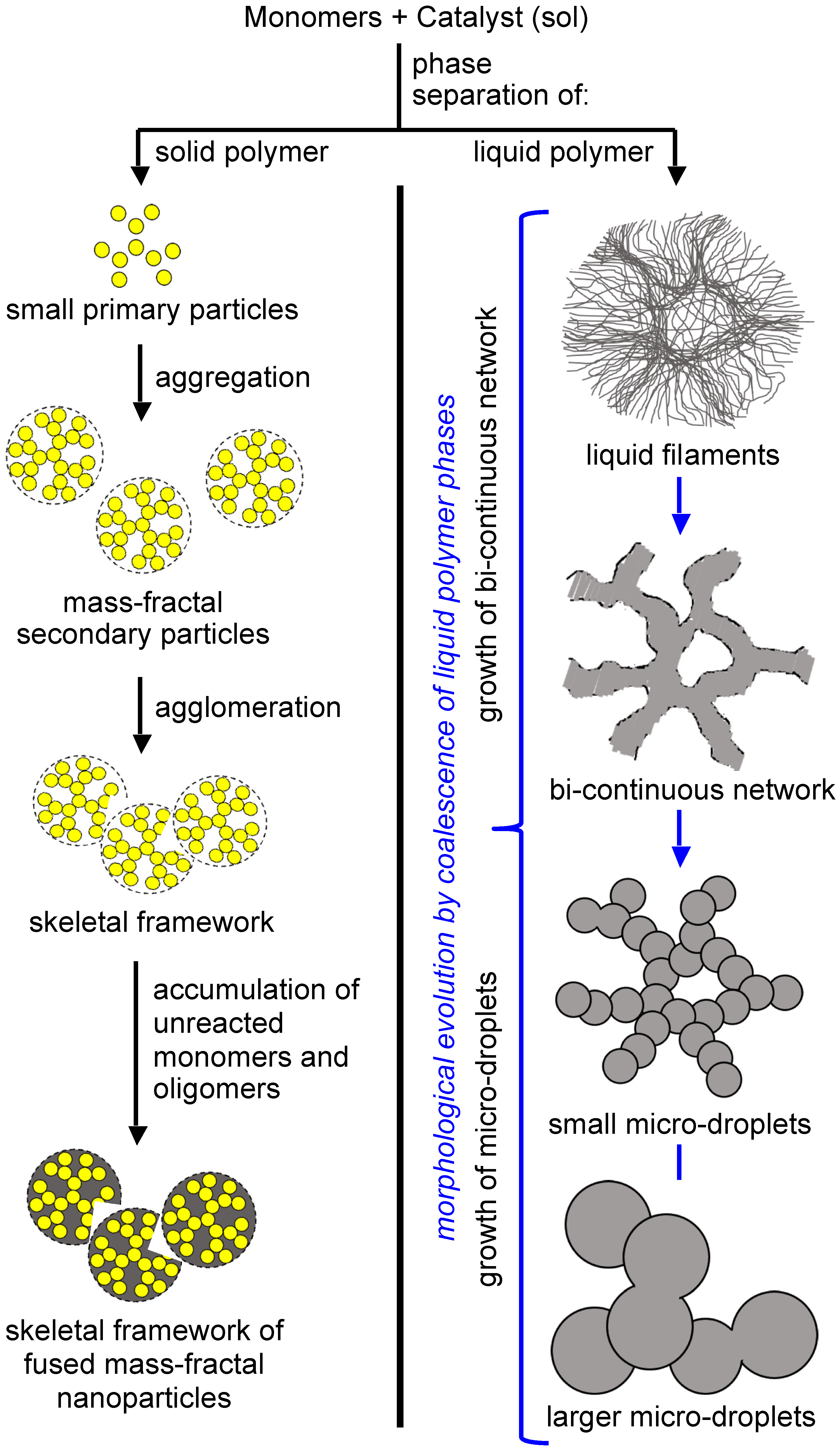
3.1. The Gelation Process and Nanomorphology
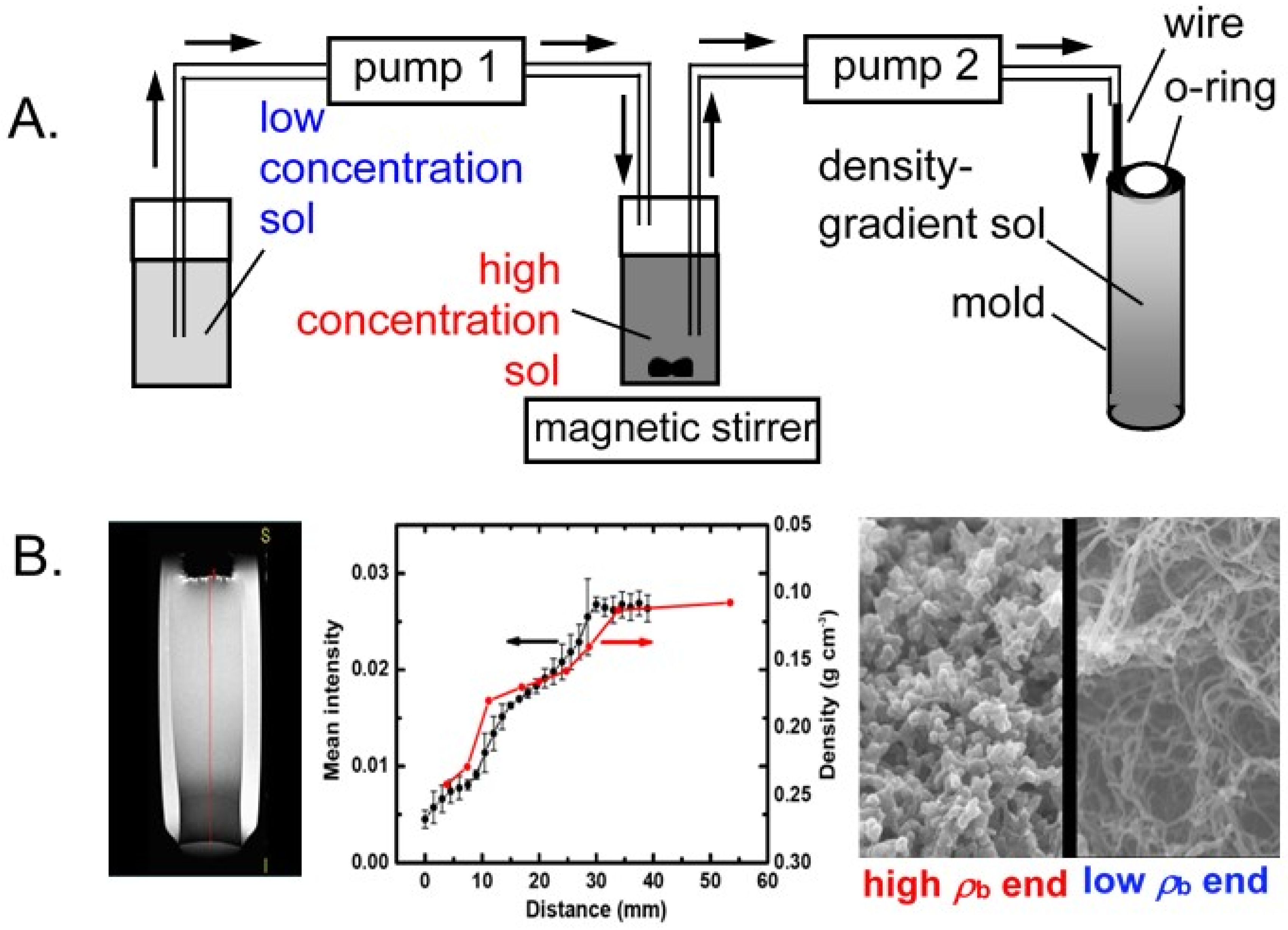
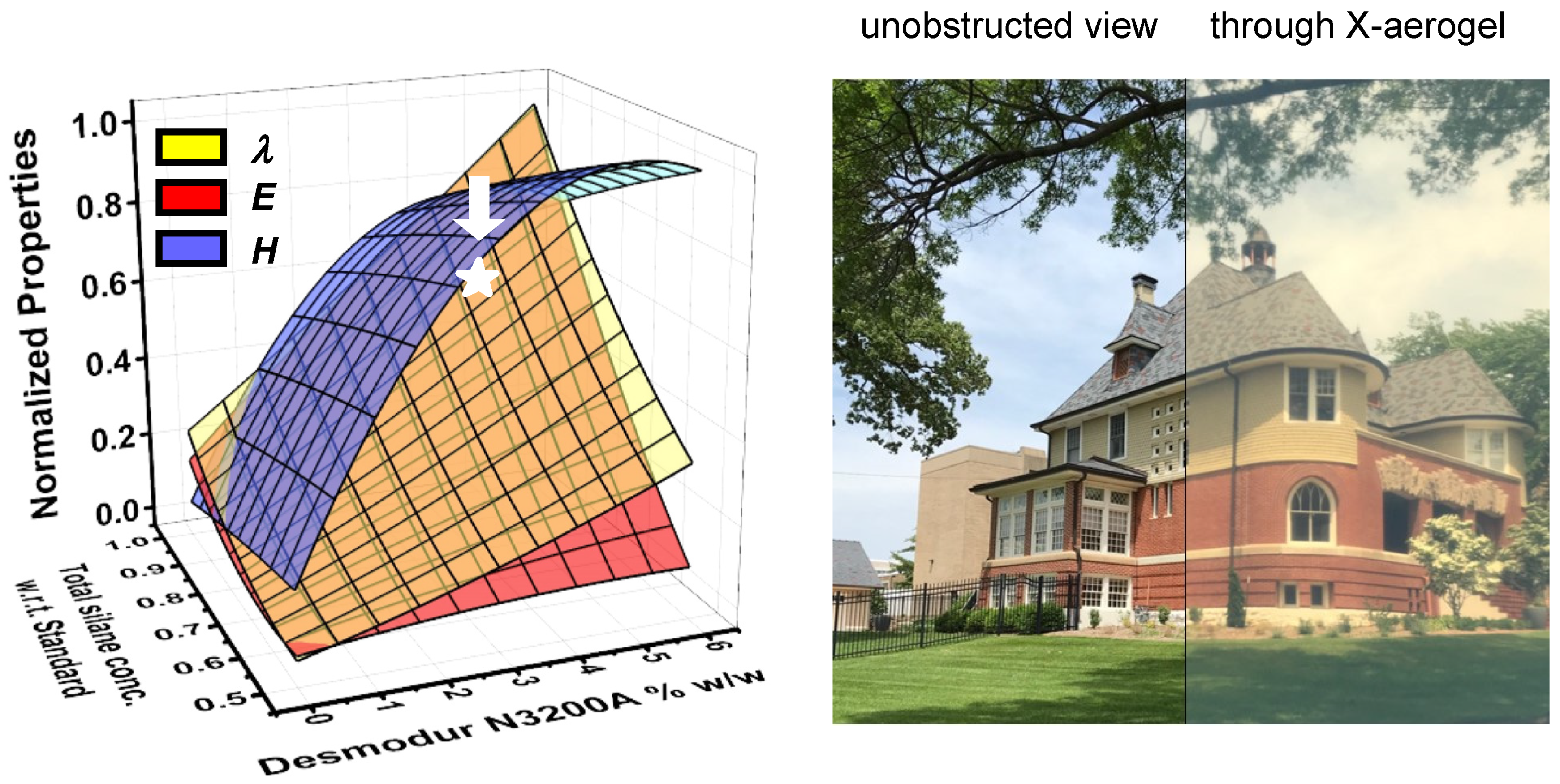
3.2. Molding, form Factors, and the Drying Process
4. The Literature on Polyurea Aerogels
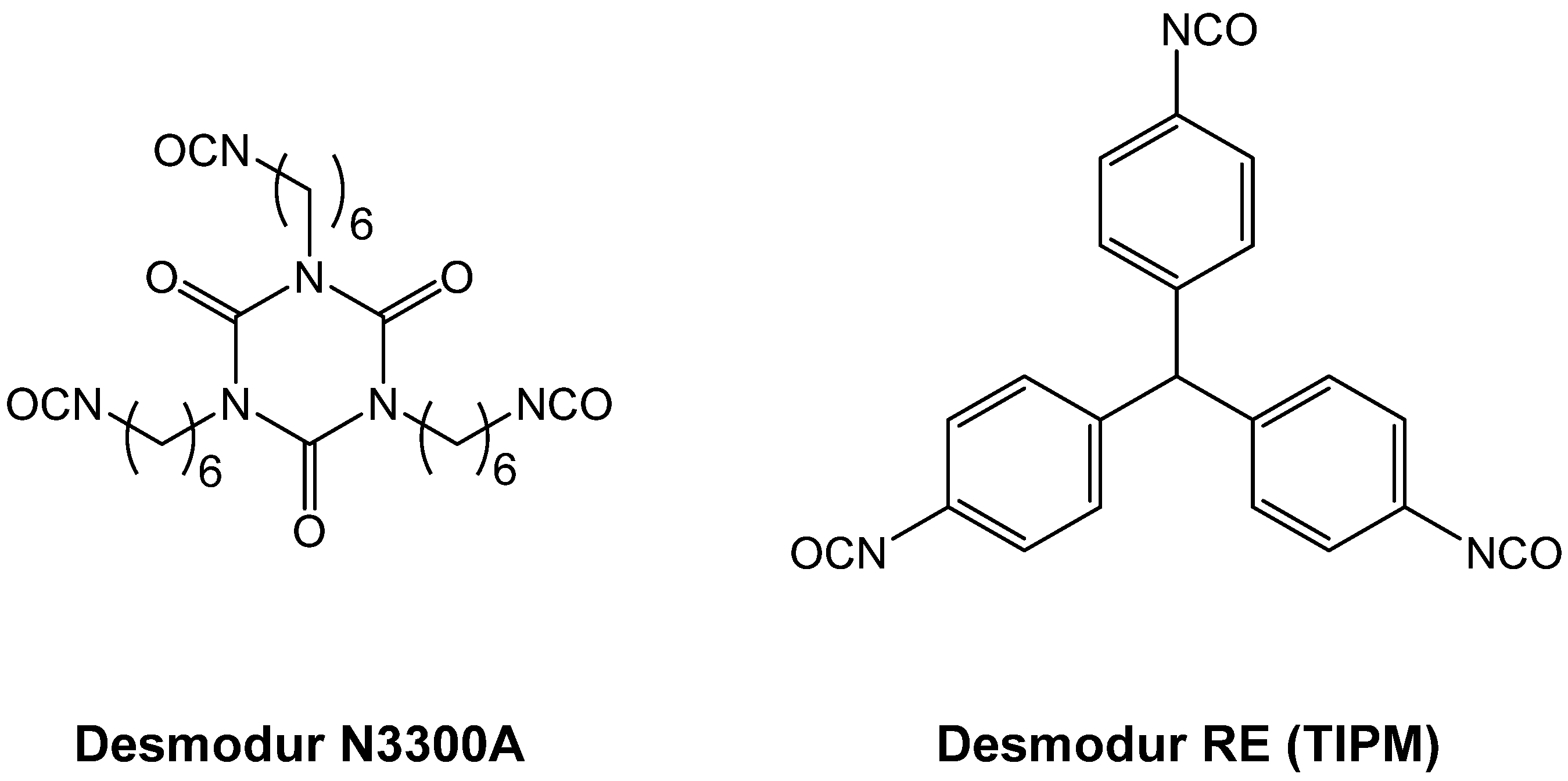
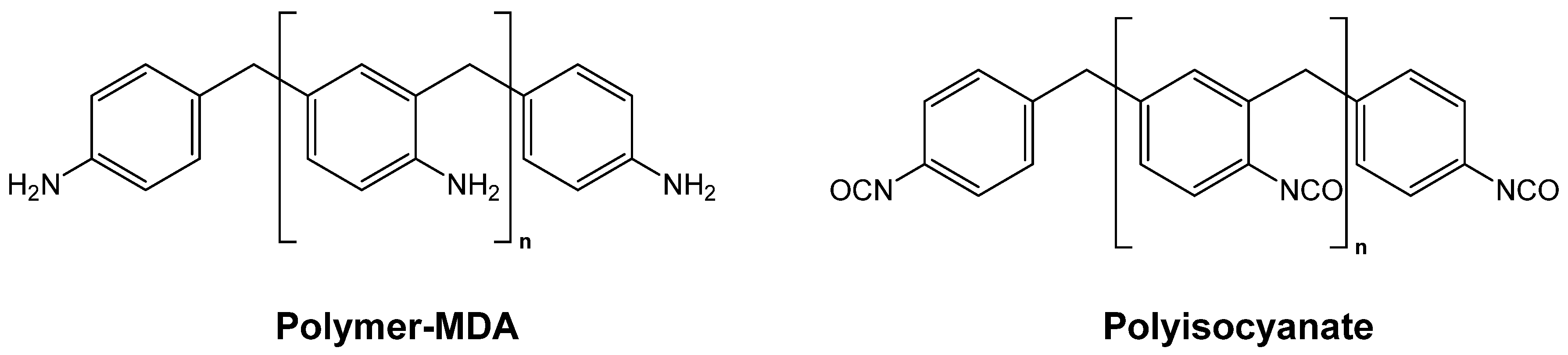
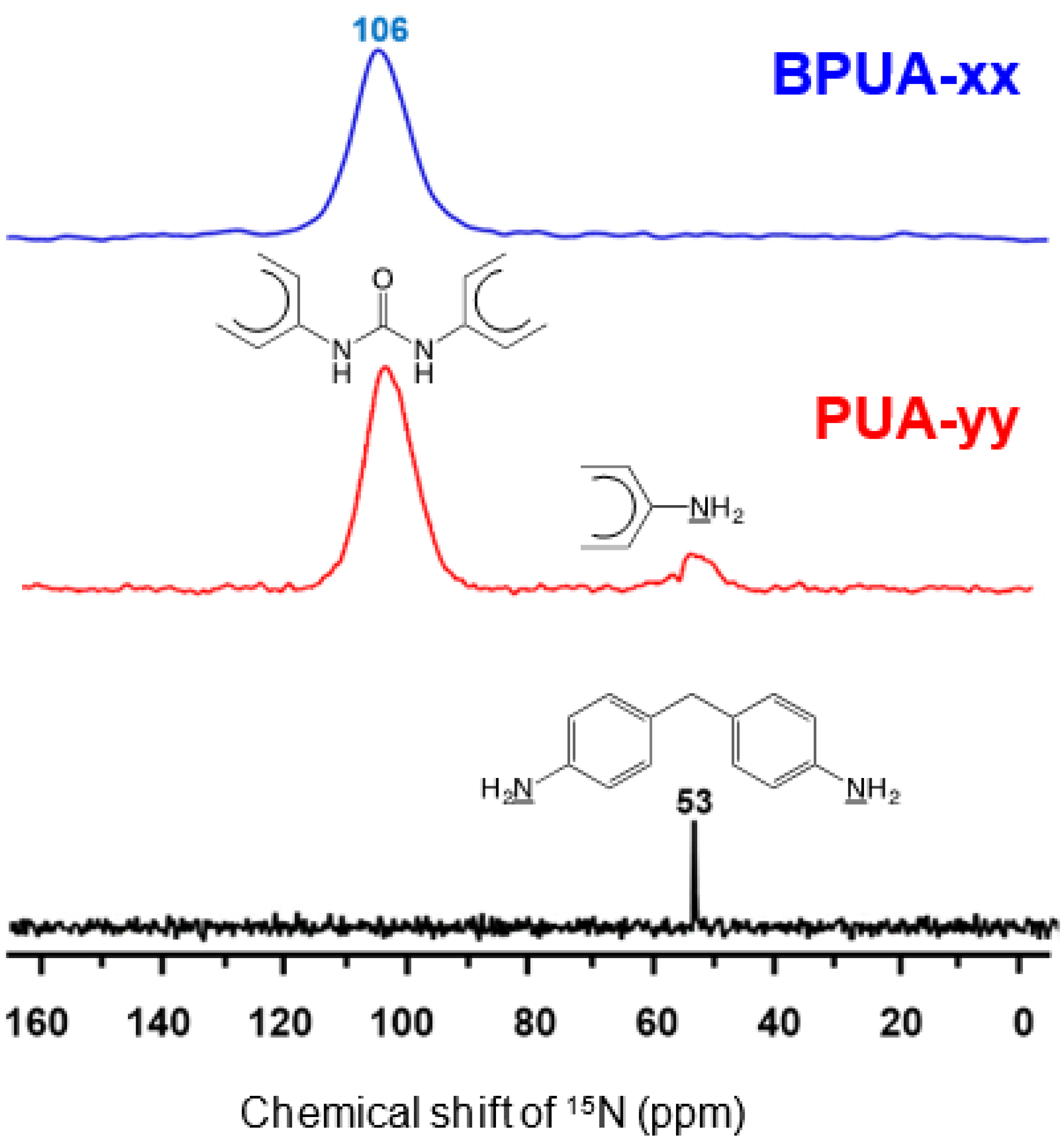
4.1. Polyurea Aerogels from Isocyanates and Amines
4.2. Polyurea Aerogels from Isocyanates and Water
4.3. Polyurea Aerogels from Isocyanates and Mineral Acids
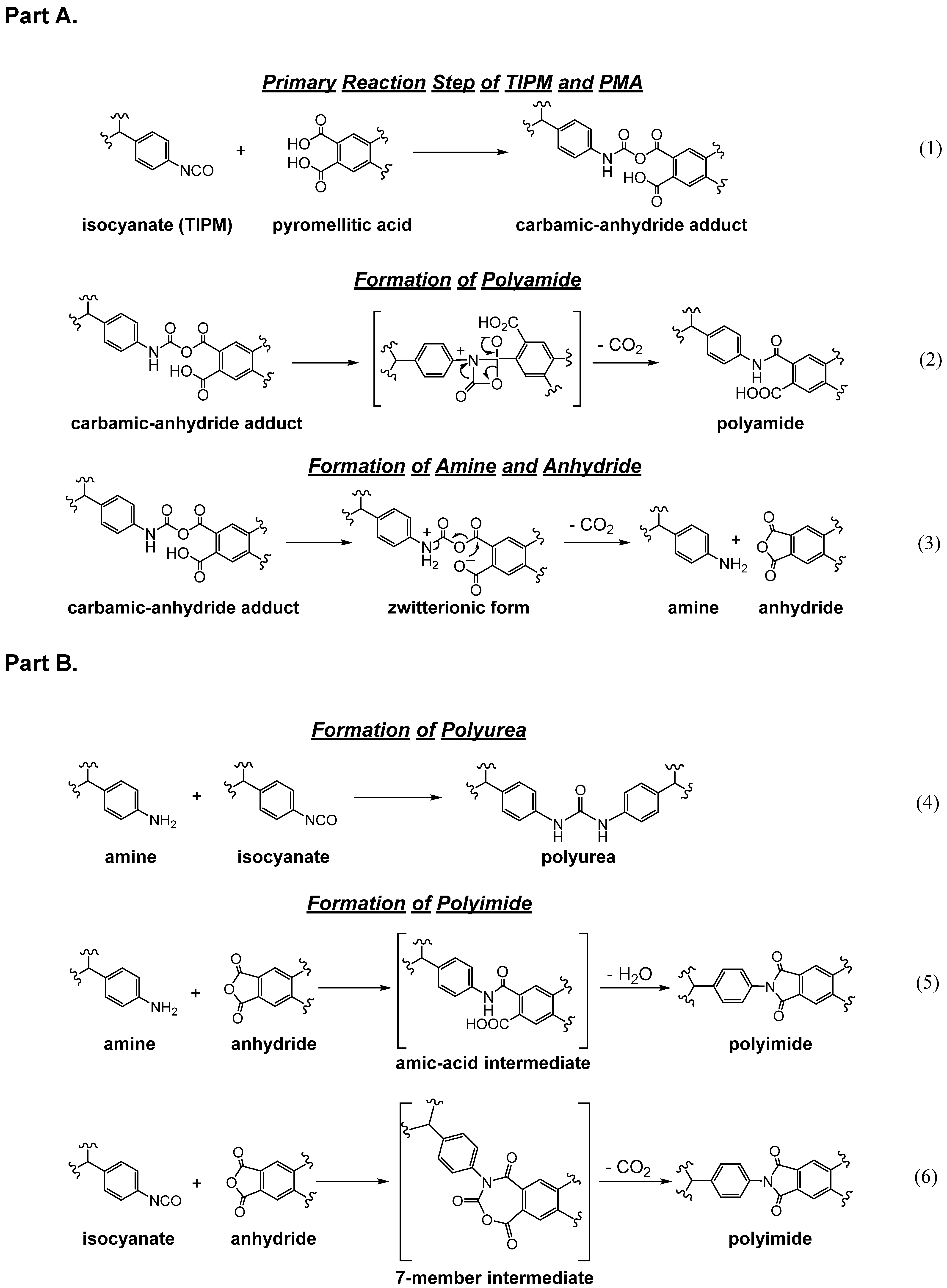
4.4. Polyurea Aerogels as Random Copolymers with Polyamides and Polyimides
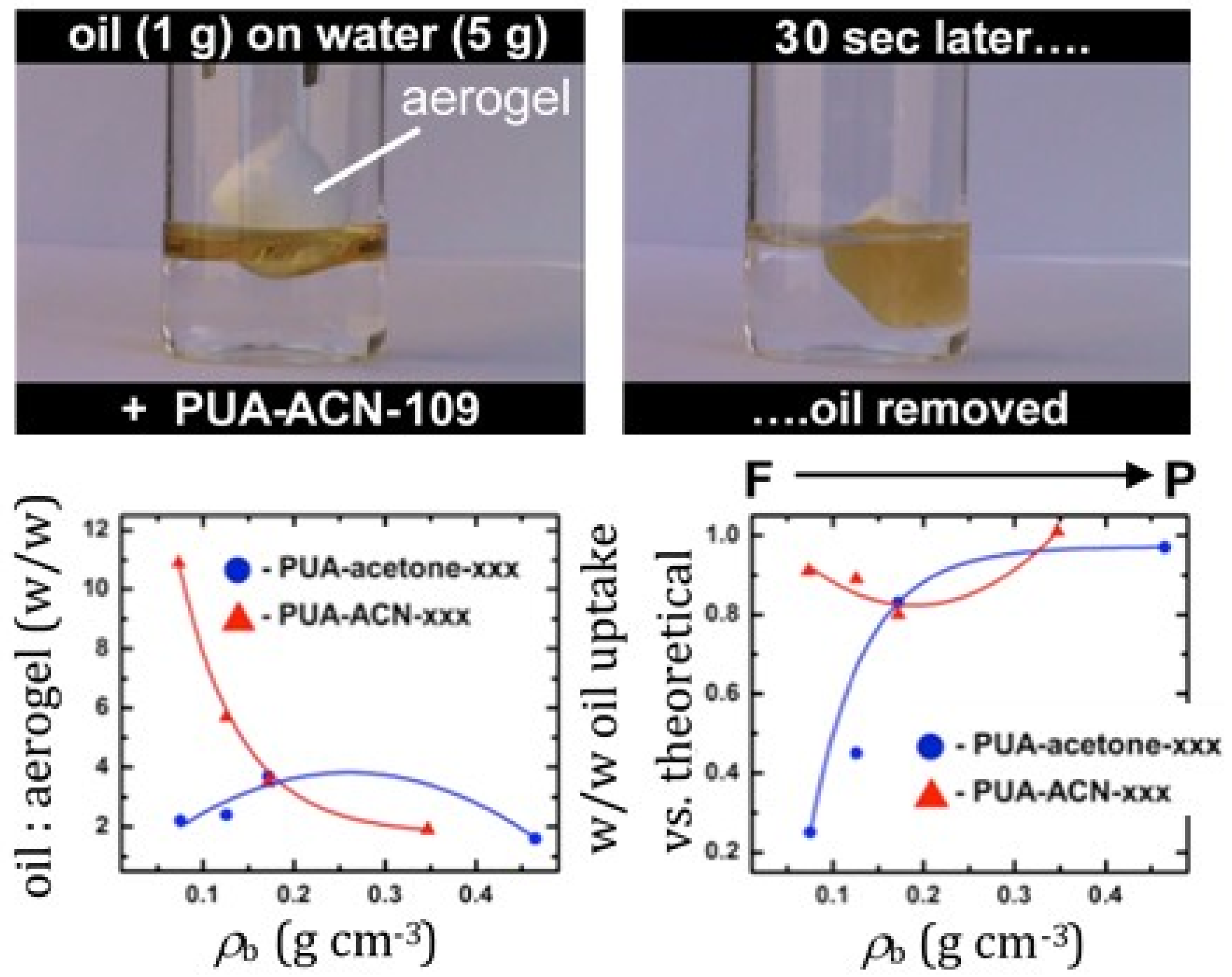
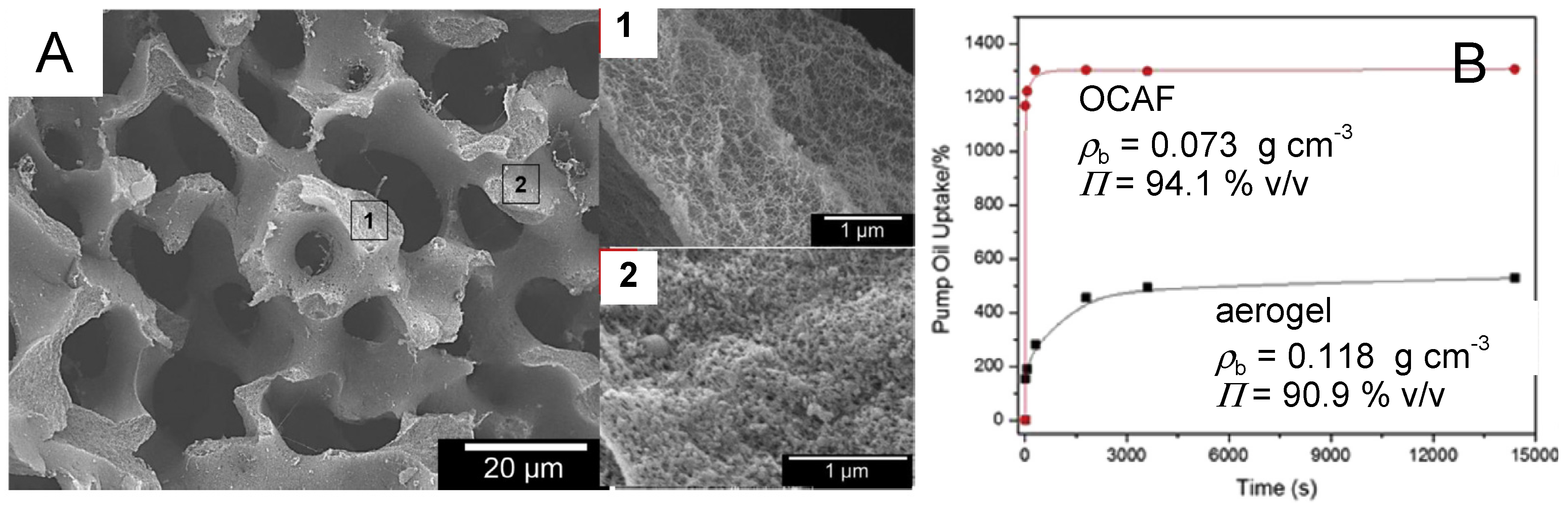
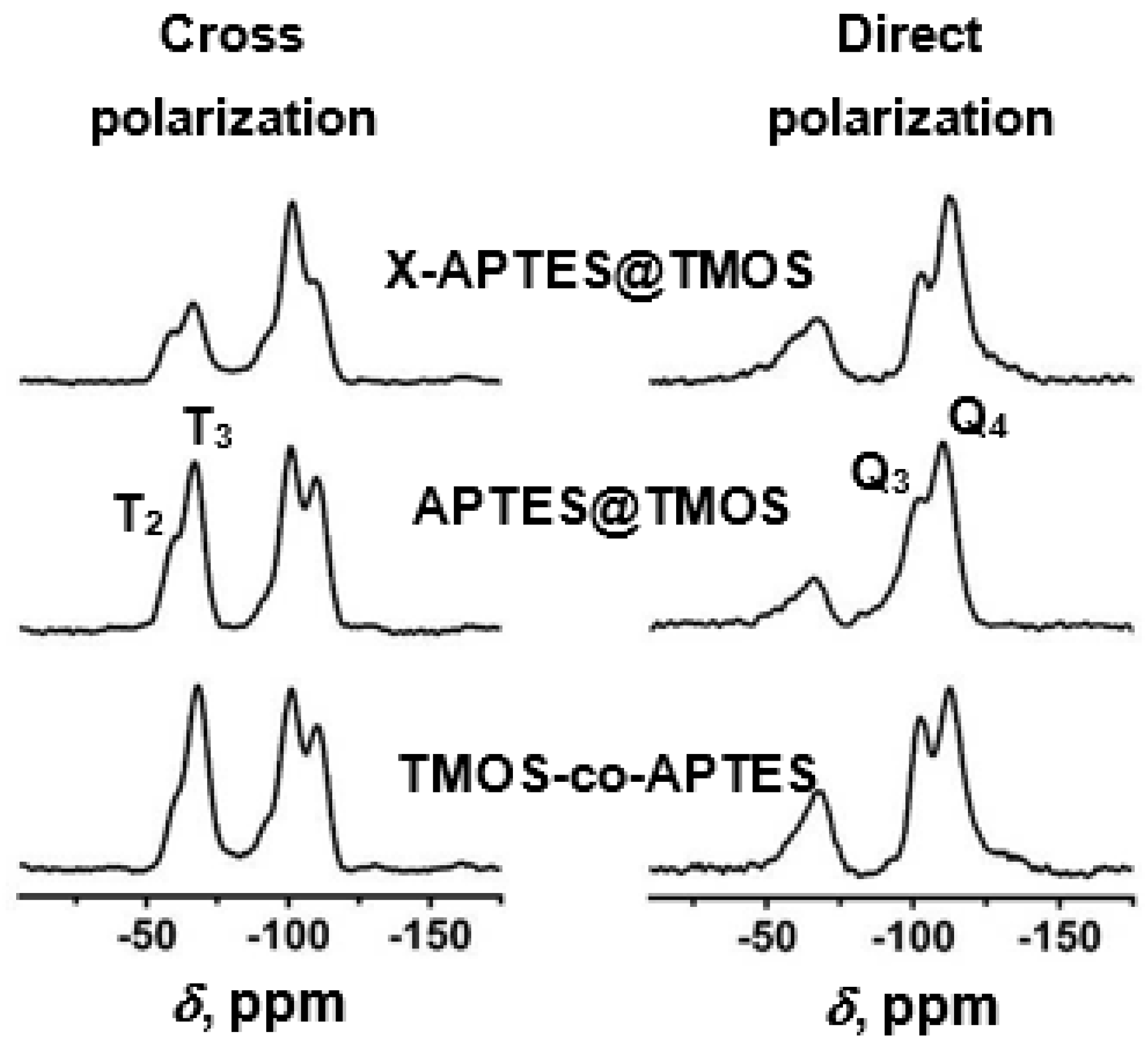
4.5. Polyurea-Crosslinked Oxide and Biopolymer Aerogels
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Leventis, N.; Sadekar, A.; Chandrasekaran, N.; Sotiriou-Leventis, C. Click synthesis of monolithic silicon carbide aerogels from polyacrylonitrile-crosslinked 3D silica networks. Chem. Mater. 2010, 22, 2790–2803. [Google Scholar] [CrossRef]
- Vareda, J.P.; Lamy-Mendes, A.; Durães, L. A reconsideration on the definition of the term aerogel based on current drying trends. Microporous Mesoporous Mater. 2018, 258, 211–216. [Google Scholar] [CrossRef]
- Kistler, S.S. Coherent expanded aerogels and jellies. Nature 1931, 127, 741. [Google Scholar] [CrossRef]
- Pierre, A.C.; Pajonk, G.M. Chemistry of aerogels and their applications. Chem. Rev. 2002, 102, 4243–4266. [Google Scholar] [CrossRef] [PubMed]
- Long, J.W.; Swider-Lyons, K.E.; Stroud, R.M.; Rolison, D.R. Design of pore and matter architectures in manganese oxide charge-storage materials. Electrochem. Solid-State Lett. 2000, 3, 453–456. [Google Scholar] [CrossRef]
- Mansour, A.N.; Smith, P.H.; Baker, W.M.; Balasubramanian, M.; McBreen, J. A comparative in situ X-ray absorption spectroscopy study of nanophase V2O5 aerogel and ambigel cathodes. J. Electrochem. Soc. 2003, 150, A403–A413. [Google Scholar] [CrossRef]
- Mandal, C.; Donthula, S.; Rewatkar, P.M.; Sotiriou-Leventis, C.; Leventis, N. Experimental deconvolution of depressurization from capillary shrinkage during drying of silica wet-gels with SCF CO2-Why aerogels shrink? J. Sol-Gel Sci. Technol. 2019, 92, 662–680. [Google Scholar] [CrossRef]
- Kistler, S.S. Coherent expanded aerogels. J. Phys. Chem. 1932, 36, 52–64. [Google Scholar] [CrossRef]
- Biesmans, G.; Randall, D.; Francais, E.; Perrut, M. Polyurethane-based organic aerogels’ thermal performance. J. Non-Cryst. Solids 1998, 225, 36–40. [Google Scholar] [CrossRef]
- Rhine, W.; Wang, J.; Begag, R. Polyimide Aerogels, Carbon Aerogels, and Metal Carbide Aerogels and Methods of Making Same. U.S. Patent No 7,074,880, 11 July 2006. [Google Scholar]
- Lee, J.K.; Gould, G.L. Polydicyclopentadiene based aerogel: A new insulation material. J. Sol-Gel Sci. Technol. 2007, 44, 29–40. [Google Scholar] [CrossRef]
- De Vos, R.; Biesmans, G.L.J.G. Organic Aerogels. U.S. Patent No 5,484,818, 16 January 1996. [Google Scholar]
- Lorjai, P.; Chaisuwan, T.; Wongkasemjit, S. Porous structure of polybenzoxazine-based organic aerogel prepared by sol-gel process and their carbon aerogels. J. Sol-Gel Sci. Technol. 2009, 52, 56–64. [Google Scholar] [CrossRef]
- An, H.; Wang, Y.; Wang, X.; Zheng, L.; Wang, X.; Yi, L.; Bai, L.; Zhang, X. Polypyrrole/carbon aerogel composite materials for supercapacitor. J. Power Sources 2010, 195, 6964–6969. [Google Scholar] [CrossRef]
- Leventis, N. Three Dimensional Core-Shell Superstructures: Mechanically Strong Aerogels. Acc. Chem. Res. 2007, 40, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Leventis, N.; Vassilaras, P.; Fabrizio, E.F.; Dass, A. Polymer nanoencapsulated rare earth aerogels: Chemically complex but stoichiometrically similar core-shell superstructures with skeletal properties of pure compounds. J. Mater. Chem. 2007, 17, 1502–1508. [Google Scholar] [CrossRef]
- Leventis, N.; Sotiriou-Leventis, C.; Mulik, S.; Dass, A.; Schnobrich, J.; Hobbs, A.; Fabrizio, E.F.; Luo, H.; Churu, G.; Zhang, Y.; et al. Polymer nanoencapsulated mesoporous vanadia with unusual ductility at cryogenic temperatures. J. Mater. Chem. 2008, 18, 2475–2482. [Google Scholar] [CrossRef]
- Leventis, N.; Sotiriou-Leventis, C.; Zhang, G.; Rawashdeh, A.-M.M. Nano Engineering Strong Silica Aerogels. NanoLetters 2002, 2, 957–960. [Google Scholar] [CrossRef]
- Leventis, N.; Mulik, N.S.; Sotiriou-Leventis, C. Macroporous electrically conducting carbon networks by pyrolysis of isocyanate cross-linked resorcinol-formaldehyde aerogels. Chem. Mater. 2008, 20, 6985–6997. [Google Scholar]
- Leventis, N.; Sotiriou-Leventis, C.; Chandrasekaran, N.; Mulik, S.; Larimore, Z.L.; Lu, H.; Churu, G.; Mang, J.T. Multifunctional polyurea aerogels from isocyanates and water. A structure-property case study. Chem. Mater. 2010, 22, 6692–6710. [Google Scholar] [CrossRef]
- Lee, S.T.; Ramesh, N.S. Polymeric Foams: Mechanism and Materials; CRC Press: Boca Raton, FL, USA, 2004; pp. 150–267. [Google Scholar]
- Saunders, J.H.; Frisch, K.C. Polyurethane Chemistry and Technology: Chemistry; Interscience Publishers: Hoboken, NJ, USA, 1964; pp. 63–118. [Google Scholar]
- Stevens, M.P. Polymer Chemistry. An Introduction; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Davis, T.L.; Ebersole, F. Relative velocities of reaction of amines with phenyl isocyanate. J. Am. Chem. Soc. 1934, 56, 885–886. [Google Scholar] [CrossRef]
- Neumann, W.; Fischer, P. Carbodiimide aus Isocyanaten. Angew. Chem. 1962, 74, 801–806. [Google Scholar] [CrossRef]
- Available online: https://coatings.specialchem.com/product/r-covestro-desmodur-n-3200 (accessed on 26 February 2022).
- Wicks, Z.W., Jr.; Jones, F.N.; Pappas, S.P.; Wicks, D.A. Organic Coatings, Science & Technology, 3rd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Odian, G. Principles of Polymerization; Wiley-Interscience: New York, NY, USA, 2004. [Google Scholar]
- Dodge, J. Polyurethanes and Polyureas. In Synthetic Methods in Step-Growth Polymers; Rogers, M.E., Long, T.E., Eds.; Wiley: New York, NY, USA, 2003; pp. 197–263. [Google Scholar]
- Leventis, N.; Sotiriou-Leventis, C.; Saeed, A.M.; Donthula, S.; Majedi Far, H.; Rewatkar, P.M.; Kaiser, H.; Robertson, J.D.; Lu, H.; Churu, G. Nanoporous polyurea from a triisocyanate and boric acid: A paradigm of a general reaction pathway for isocyanates and mineral acids. Chem. Mater. 2016, 28, 67–78. [Google Scholar] [CrossRef]
- Aries, R.S. Polymers from Boric Acid and Organic Diisocyanates. U.S. Patent No 2,945,841, 19 July 1960. [Google Scholar]
- Xiao, H.; Xiao, H.X.; Frisch, K.C.; Malwitz, N. Kinetic studies of the reactions between isocyanates and carboxylic acids. High Perform. Polym. 1994, 6, 235–239. [Google Scholar] [CrossRef]
- Sorenson, W.R. Reaction of an isocyanate and a carboxylic acid in dimethyl sulfoxide. J. Org. Chem. 1959, 24, 978–980. [Google Scholar] [CrossRef]
- Trappe, V.; Prasad, V.; Cipelletti, L.; Segre, P.N.; Weitz, D.A. Jamming phase diagrams for attractive particles. Nature 2001, 411, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Zaccarelli, E.; Ciulla, F.; Schofield, A.B.; Sciortino, F.; Weitz, D.A. Gelation of particles with short-range attraction. Nature 2008, 499, 453–503. [Google Scholar] [CrossRef] [PubMed]
- Rouwhorst, J.; Ness, C.; Stoyanov, S.; Zaccone, A.; Schall, P. Nonequilibrium continuous phase transition in colloidal gelation with short-range attraction. Nature Commun. 2020, 11, 3558. [Google Scholar] [CrossRef]
- Bang, A.; Buback, C.; Sotiriou-Leventis, C.; Leventis, N. Flexible aerogels from hyperbranched polyurethanes: Probing the role of molecular rigidity with poly(urethane acrylates) versus poly(urethane norbornenes). Chem. Mater. 2014, 26, 6979–6993. [Google Scholar] [CrossRef]
- Papastergiou, M.; Kanellou, A.; Chriti, D.; Raptopoulos, G.; Paraskevopoulou, P. Poly(urethane-acrylate) aerogels via radical polymerization of dendritic urethane-acrylate monomers. Materials 2018, 11, 2249. [Google Scholar] [CrossRef] [Green Version]
- Mohite, D.P.; Larimore, Z.J.; Lu, H.; Mang, J.T.; Sotiriou-Leventis, C.; Leventis, N. Monolithic hierarchical fractal assemblies of silica nanoparticles cross-linked with polynorbornene via ROMP: A structure-property correlation from molecular to bulk through nano. Chem. Mater. 2012, 24, 3434–3448. [Google Scholar] [CrossRef]
- Mohite, D.P.; Mahadik-Khanolkar, S.; Luo, H.; Lu, H.; Sotiriou-Leventis, C.; Leventis, N. Polydicyclopentadiene aerogels grafted with PMMA: II. Nanoscopic characterization and origin of macroscopic deformation. Soft Matter 2013, 9, 1531–1539. [Google Scholar] [CrossRef]
- Chidambareswarapattar, C.; McCarver, P.M.; Luo, H.; Lu, H.; Sotiriou-Leventis, C.; Leventis, N. Fractal multiscale nanoporous polyurethanes: Flexible to extremely rigid aerogels from multifunctional small molecules. Chem. Mater. 2013, 25, 3205–3224. [Google Scholar] [CrossRef]
- Malakooti, S.; ud Doulah, A.B.M.S.; Ren, Y.; Kulkarni, V.N.; Soni, R.U.; Edlabadkar, V.A.; Zhang, R.; Vivod, S.L.; Chariklia Sotiriou-Leventis, C.; Leventis, N.; et al. Meta-aerogels: Auxetic shape-memory polyurethane aerogels. ACS Appl. Polym. Mater. 2021, 3, 5727–5738. [Google Scholar] [CrossRef]
- Członka, S.; Bertino, M.F.; Kośny, J.; Shukla, N. Freeze-drying method as a new approach to the synthesis of polyurea aerogels from isocyanate and water. J. Sol-Gel Sci. Technol. 2018, 87, 685–695. [Google Scholar] [CrossRef]
- Steiner, S.A., III; Griffin, J.S.; Wunsch, B.H.; Schneider, J.N. Systems and Methods for Producing Aerogel Materials. U.S. Patent No 10,563,035 B2, 18 February 2020. [Google Scholar]
- Lee, J.K.; Gould, G.L.; Rhine, W. Polyurea based aerogels for a high performance thermal insulation material. J. Sol-Gel Sci. Technol. 2009, 49, 209–220. [Google Scholar] [CrossRef]
- Available online: https://azelisamericascase.com/wp-content/uploads/2020/03/JEFFAMINE-amines-Brochure.pdf (accessed on 26 February 2022).
- Available online: https://solutions.covestro.com/en/products/multranol/multranol-9185_05213088-12942099?SelectedCountry=US (accessed on 26 February 2022).
- Trifu, R.; Gould, G.; White, S. Flexible polyisocyanate based aerogels. MRS Adv. 2017, 2, 3479–3484. [Google Scholar] [CrossRef]
- Shinko, A.; Jana, S.C.; Meador, M.A. Crosslinked polyurea aerogels with controlled porosity. RSC Adv. 2015, 5, 105329–105338. [Google Scholar] [CrossRef]
- Shinko, A.; Jana, S.C.; Meador, M.A. Crosslinked polyurea-co-polyurethane aerogels with hierarchical structures and low stiffness. J. Non-Cryst. Solids 2018, 487, 19–27. [Google Scholar] [CrossRef]
- Wu, X.; Wu, Y.; Zou, W.; Wang, X.; Du, A.; Zhang, Z.; Shen, J. Synthesis of highly cross-linked uniform polyurea aerogels. J. Supercrit. Fluids 2019, 151, 8–14. [Google Scholar] [CrossRef]
- Lu, X.; Caps, R.; Fricke, J.; Alviso, C.T.; Pekala, R.W. Correlation between structure and thermal conductivity of organic aerogels. J. Non-Cryst. Solids 1995, 188, 226–234. [Google Scholar] [CrossRef]
- Lu, X.; Nilsson, O.; Fricke, J.; Pekala, R.W. Thermal and electrical conductivity of monolithic carbon aerogels. J. Appl. Phys. 1993, 73, 581–584. [Google Scholar] [CrossRef]
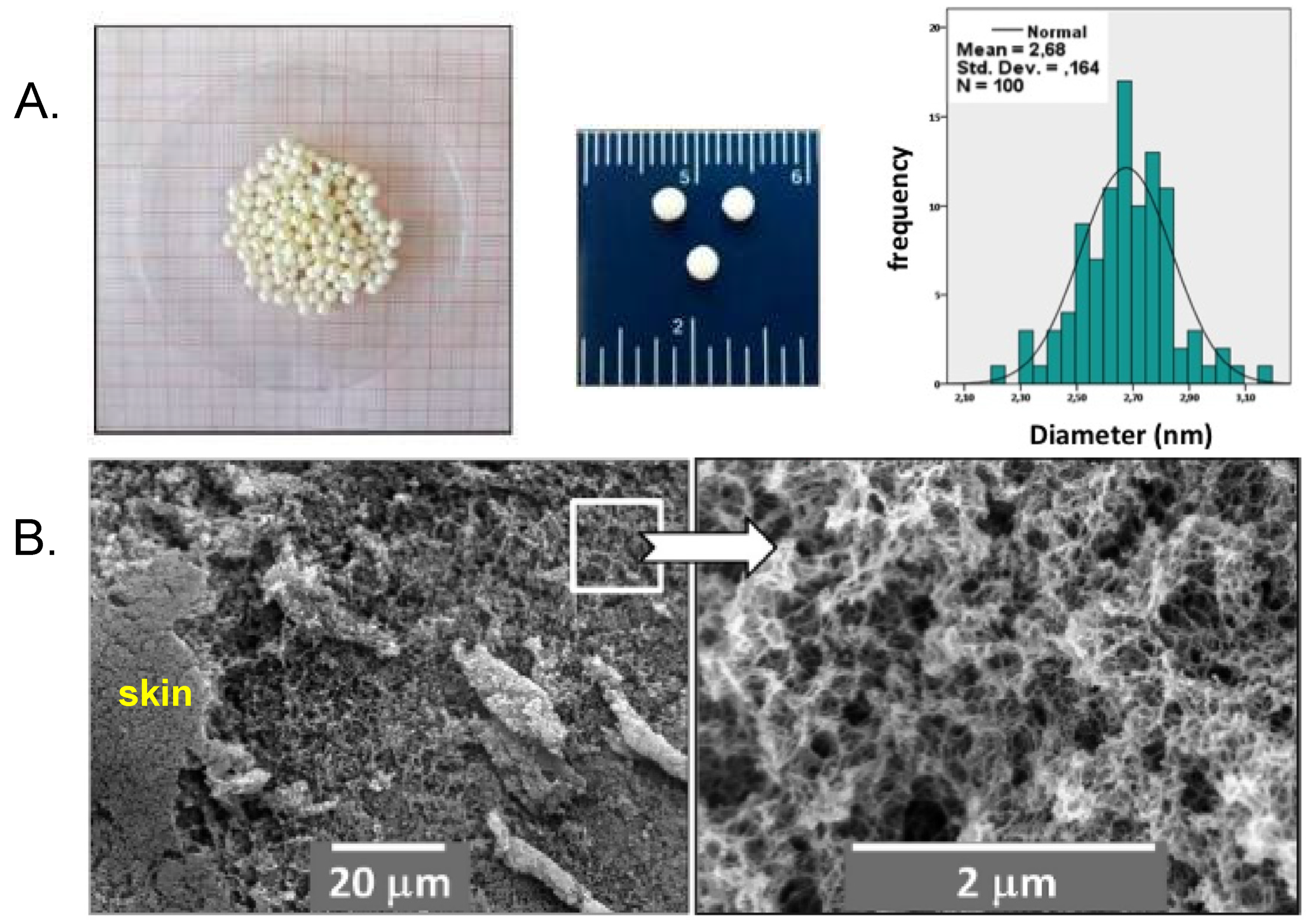
- Chriti, D.; Raptopoulos, G.; Papastergiou, M.; Paraskevopoulou, P. Millimeter-size spherical polyurea aerogel beads with narrow distribution. Gels 2018, 4, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petricevic, R.; Glora, M.; Fricke, J. Planar fibril reinforced carbon aerogels for application in PEM fuel cells. Carbon 2001, 39, 857–867. [Google Scholar] [CrossRef]
- Petricevic, R.; Glora, M.; Möginger, A.; Fricke, J. Skin formation on RF aerogel sheets. J. Non-Cryst. Solids 2001, 285, 272–276. [Google Scholar] [CrossRef]
- Available online: http://www.aerogeltechnologies.com/airloy/products (accessed on 26 February 2022).
- Leventis, N.; Sotiriou-Leventis, C.; Mulik, S. Three-Dimensional Porous Polyurea Networks and Methods of Manufacture. U.S. Patent No 10,301,445 B2, 28 May 2019. [Google Scholar]
- American Society for Metals. ASM Engineering Materials Handbook, Composites Volume 1; ASM International: Materials Park, OH, USA, 1998; p. 178. [Google Scholar]
- Jones, S.M. A method for producing gradient density aerogel. J. Sol-Gel Sci. Technol. 2007, 44, 255–258. [Google Scholar] [CrossRef]
- Leventis, N.; Sotiriou-Leventis, C.; Chandrasekaran, N.; Mulik, S.; Chidambareswarapattar, C.; Sadekar, A.; Mohite, D.; Mahadik, S.S.; Larimore, Z.J.; Lu, H.; et al. Isocyanate-derived organic aerogels: Polyureas, polyimides, polyamides. Mater. Res. Soc. Symp. Proc. Mater. Res. Soc. 2011, 1306, mrsf10-1306-bb03-01. [Google Scholar] [CrossRef]
- Bian, Q.; Chen, S.; Kim, B.-T.; Leventis, N.; Lu, H.; Chang, Z.; Lei, S. Micromachining of polyurea aerogel using femtosecond laser pulses. J. Non Cryst. Solids 2011, 357, 186–193. [Google Scholar] [CrossRef]
- Leventis, N.; Chidambareswarapattar, C.; Bang, A.; Sotiriou-Leventis, C. Cocoon-in-web-like superhydrophobic aerogels from hydrophilic polyurea and use in environmental remediation. ACS Appl. Mater. Interfaces 2014, 6, 6872–6882. [Google Scholar] [CrossRef]
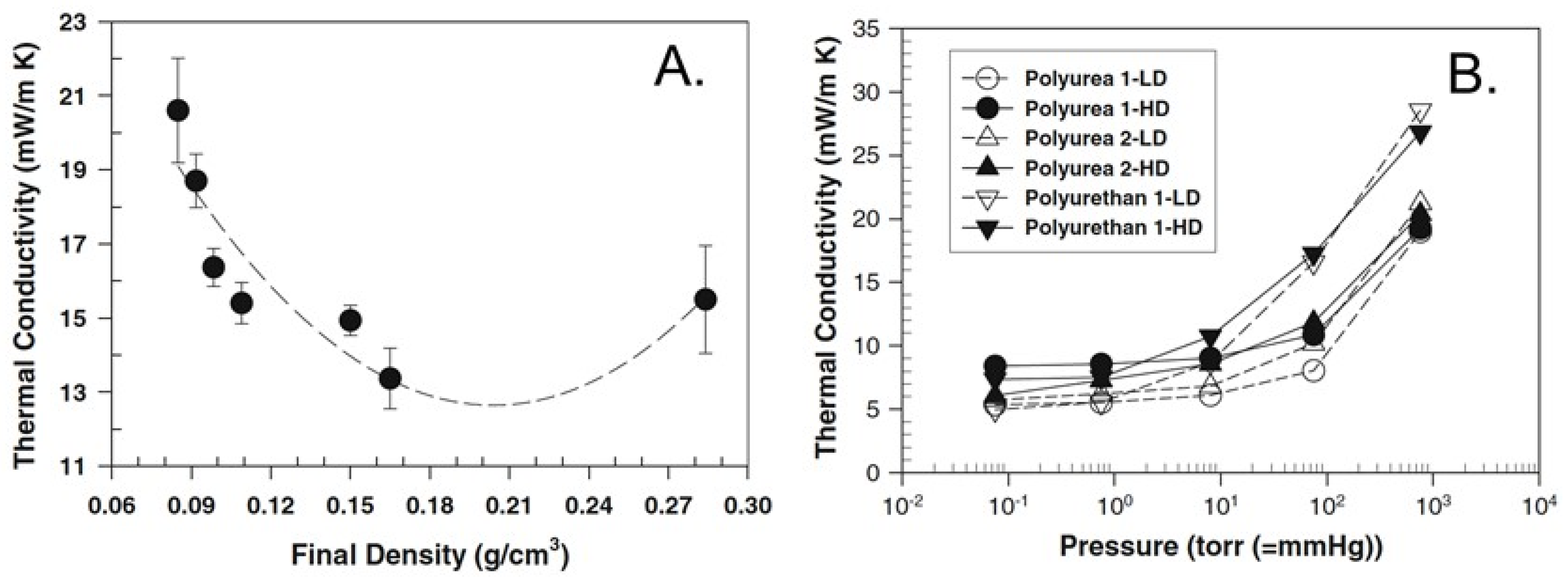
- Weigold, L.; Mohite, D.P.; Mahadik-Khanolkar, S.; Leventis, N.; Reichenauer, G. Correlation of microstructure and thermal conductivity in nanoporous solids: The case of polyurea aerogels synthesized from an aliphatic tri-isocyanate and water. J. Non-Cryst. Solids 2013, 368, 105–111. [Google Scholar] [CrossRef]
- Mohite, D.P.; Mahadik-Khanolkar, S.; Luo, H.; Lu, H.; Sotiriou-Leventis, C.; Leventis, N. Polydicyclopentadiene aerogels grafted with PMMA: I. Molecular and interparticle crosslinking. Soft Matter 2013, 9, 1516–1530. [Google Scholar] [CrossRef]
- Fricke, J.; Lu, X.; Wang, P.; Buettner, D.; Heinemann, U. Optimization of monolithic silica aerogel insulants. Int. J. Heat Mass Transf. 1992, 35, 2305–2309. [Google Scholar] [CrossRef]
- Gibson, L.J.; Ashby, M.F. Cellular Solids—Structure and Properties, 2nd ed.; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Weigold, L.; Reichenauer, G. Correlation between mechanical stiffness and thermal transport along the solid framework of a uniaxially compressed polyurea aerogel. J. Non-Cryst. Solids 2014, 406, 73–78. [Google Scholar] [CrossRef]
- Gould, G.L.; Lee, J.K.; Stepanian, C.J.; Lee, K.P. High Strength Nanoporous Bodies Reinforced with Fibrous Materials. U.S. Patent No 7,560,062, 14 July 2009. [Google Scholar]
- Weigold, L.; Reichenauer, G. Correlation between the elastic modulus and heat transport along the solid phase in highly porous materials: Theoretical approaches and experimental validation using polyurea aerogels. J. Supercrit. Fluids 2015, 106, 69–75. [Google Scholar] [CrossRef]
- Malakooti, S.; Churu, H.G.; Lee, A.; Xu, T.; Luo, H.; Xiang, N.; Sotiriou-Leventis, C.; Leventis, N.; Lu, H. Sound insulation properties in low-density, mechanically strong and ductile nanoporous polyurea aerogels. J. Non-Cryst. Solids 2017, 476, 36–45. [Google Scholar] [CrossRef]
- Lu, H.; Xiang, H.; Leventis, N.; Sotiriou-Leventis, C. Acoustic Attenuators Based on Porous Nanostructured Materials. U.S. Patent No 9,068,346 B1, 30 June 2015. [Google Scholar]
- Malakooti, S.; Gitogo, C.H.; Lee, A.; Rostami, S.; May, S.J.; Ghidei, S.; Wang, F.; Lu, Q.; Luo, H.; Xiang, N.; et al. Sound transmission loss enhancement in an inorganic-organic laminated wall panel using multifunctional low-density nanoporous polyurea aerogels: Experiment and modeling. Adv. Eng. Mater. 2018, 20, 1700937. [Google Scholar] [CrossRef]
- Price, M.A.; Aslam, T.D.; Quirk, J.J. Analysis of steady compaction waves in polyurea aerogel. AIP Conf. Proc. 2018, 1979, 110016. [Google Scholar]
- Whitworth, N.; Lambourn, B. A single-phase analytic equation of state for solid polyurea and polyurea aerogels. AIP Conf. Proc. 2018, 1979, 30007. [Google Scholar]
- Pacheco, A.H.; Gustavsen, R.L.; Aslam, T.D.; Bartram, B.D. Hugoniot based equation of state for solid polyurea and polyurea aerogels. AIP Conf. Proc. 2017, 1793, 120029. [Google Scholar]
- Aslam, T.D.; Gustavsen, R.L.; Bartram, B.D. An equation of state for polyurea aerogel based on multi-shock response. J. Phys. Conf. Series 2014, 500, 32001. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Taghvaee, T.; Wei, C.; Ghasemi, A.; Chen, G.; Leventis, N.; Gao, W. Multi-scale progressive failure mechanism and mechanical properties of nanofibrous polyurea aerogels. Soft Matter 2018, 14, 7801–7808. [Google Scholar] [CrossRef]
- Fakharifar, M.; Lin, Z.; Wu, C.; Mahadik-Khanolkar, S.; Leventis, N.; Chen, G. Microstructural characteristics of polyurea and polyurethane xerogels for concrete confinement with FRP system. Adv. Mater. Res. 2013, 742, 237–242. [Google Scholar] [CrossRef]
- Li, Y.; Liao, W.; Taghvaee, T.; Wu, C.; Ma, H.; Leventis, N. Bioinspired strong nanocellular composite prepared with magnesium phosphate cement and polyurea aerogel. Mater. Lett. 2019, 237, 274–277. [Google Scholar] [CrossRef]
- Yin, W.; Lu, H.; Leventis, N.; Rubenstein, D.A. Characterization of the Biocompatibility and Mechanical Properties of Polyurea Organic Aerogels with the Vascular System: Potential as a Blood Implantable Material. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 109–118. [Google Scholar] [CrossRef]
- Gu, S.; Jana, S.C. Open cell aerogel foams with hierarchical pore structures. Polymer 2017, 125, 1–9. [Google Scholar] [CrossRef]
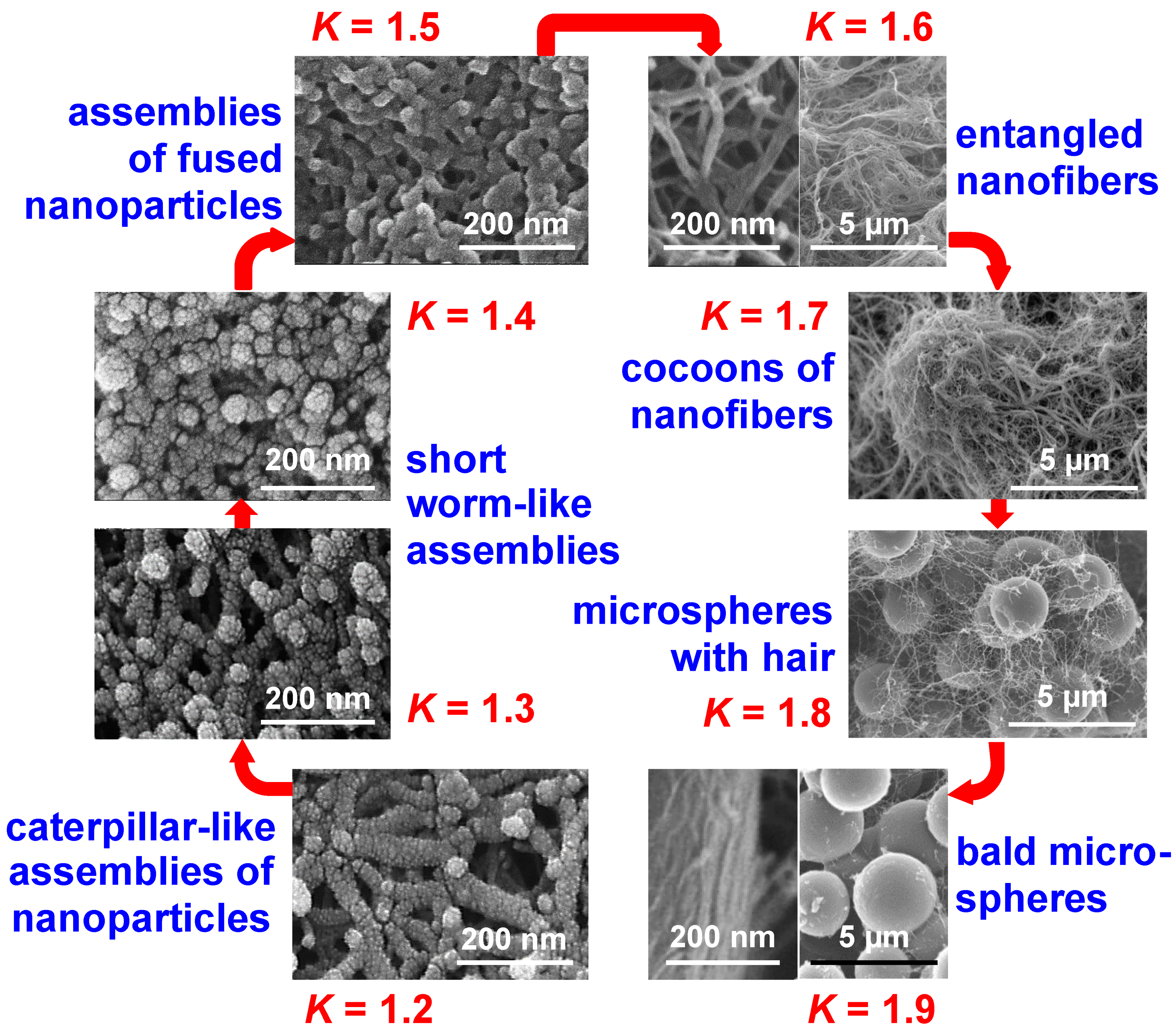
- Taghvaee, T.; Donthula, S.; Rewatkar, P.M.; Majedi Far, H.; Sotiriou-Leventis, C.; Leventis, N. K-index: Descriptor, predictor, and correlator of complex nanomorphology to other material properties. ACS Nano 2019, 13, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Rege, A.; Preibisch, I.; Schestakow, M.; Ganesan, K.; Gurikov, P.; Milow, B.; Smirnova, I.; Itskov, M. Correlating Synthesis Parameters to Morphological Entities: Predictive Modeling of Biopolymer Aerogels. Materials 2018, 11, 1670. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Parsons, G.N.; Khan, S.A.; Rojas, O.J. Synthesis of organic aerogels with tailorable morphology and strength by controlled solvents swelling following Hansen solubility. Sci. Rep. 2018, 8, 2106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Snellings, G.M.B.F.; Koebel, M.M.; Malfait, W.J. Superinsulating polyisocyanate based aerogels: A targeted search for the optimum solvent system. ACS Appl. Mater. Interfaces 2017, 9, 18222–18230. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Kawamura, K. Polymeric Carbons, Carbon Fibre, Glass and Char; Cambridge University Press: New York, NY, USA, 1976. [Google Scholar]
- McKenzie, D.R.; Muller, D.; Pailthorpe, B.A. Compressive-stress-induced formation of thin-film tetrahedral amorphous carbon. Phys. Rev. Lett. 1991, 67, 773–776. [Google Scholar] [CrossRef]
- Loelsberg, W.; Fricke, M.; Weinrich, D.; Leventis, N.; Sotiriou-Leventis, C.; Saeed, A.M. Process for Producing Isocyanate-Based Xerogels and Aerogels with Mineral acids. U.S. Patent No. 10,759,893 B2, 1 September 2020. [Google Scholar]
- Leventis, N.; Chidambareswarapattar, C.; Mohite, D.P.; Larimore, Z.J.; Lu, H.; C Sotiriou-Leventis, C. Multifunctional porous aramids (aerogels) by efficient reaction of carboxylic acids and isocyanates. J. Mater. Chem. 2011, 21, 11981–11986. [Google Scholar] [CrossRef]
- Leventis, N.; Sotiriou-Leventis, C.; Chidambareswarapattar, C. Multifunctional Porous Aramids (Aerogels) and Fabrication Thereof. U.S. Patent No 8,877,824, 11 April 2014. [Google Scholar]
- Leventis, N.; Sotiriou-Leventis, C.; Saeed, M.A. Multifunctional Porous Aramids (Aerogels) and Fabrication Thereof. U.S. Patent No 9,260,581, 16 February 2016. [Google Scholar]
- Leventis, N.; Sotiriou-Leventis, C.; Saeed, M.A. Multifunctional Porous Aramids (Aerogels), Fabrication Thereof, and Catalytic Compositions Derived Therefrom. U.S. Patent No 9,593,225, 14 March 2017. [Google Scholar]
- Saeed, A.M.; Wisner, C.A.; Donthula, S.; Majedi Far, H.; Sotiriou-Leventis, C.; Leventis, N. Reuseable monolithic nanoporous graphite-supported nanocatalysts (Fe, Au, Pt, Pd, Ni, and Rh) from pyrolysis and galvanic transmetalation of ferrocene-based polyamide aerogels. Chem. Mater. 2016, 28, 4867–4877. [Google Scholar] [CrossRef]
- Saeed, A.M.; Rewatkar, P.M.; Majedi Far, H.; Taghvaee, T.; Donthula, S.; Mandal, C.; Sotiriou-Leventis, C.; Leventis, N. Selective CO2 sequestration with monolithic bimodal micro/macroporous carbon aerogels derived from stepwise pyrolytic decomposition of polyamide-polyimide-polyurea random co-polymers. ACS Appl. Mater. Interfaces 2017, 9, 13520–13536. [Google Scholar] [CrossRef] [PubMed]
- Leventis, N.; Sotiriou-Leventis, C.; Saeed, M.A. Novel Porous Polymer Compositions for the Synthesis of Monolithic Bimodal Microporous/Macroporous Carbon Compositions Useful for Selective CO2 Sequestration. U.S. Patent Application Publication No 2018/0272312 A1, 5 October 2021. [Google Scholar]
- Chidambareswarapattar, C.; Larimore, Z.; Sotiriou-Leventis, C.; Mang, J.T.; Leventis, N. One-step room-temperature synthesis of fibrous polyimide aerogels from anhydrides and isocyanates and conversion to isomorphic carbons. J. Mater. Chem. 2010, 20, 9666–9678. [Google Scholar] [CrossRef]
- Chidambareswarapattar, C.; Sotiriou-Leventis, C.; Leventis, N. One-step polyimide aerogels from anhydrides and isocyanates. Polym. Prepr. 2010, 51, 638–639. [Google Scholar]
- Chidambareswarapattar, C.; Xu, L.; Sotiriou-Leventis, C.; Leventis, N. Robust monolithic multiscale nanoporous polyimides and conversion to isomorphic carbons. RSC Adv. 2013, 3, 26459–26469. [Google Scholar] [CrossRef]
- Leventis, N.; Sotiriou-Leventis, C.; Chidambareswarapattar, C. Porous Nanostructured Polyimide Networks and Methods of Manufacture. U.S. Patent No 9,745,198, 29 August 2017. [Google Scholar]
- Majedi Far, H.; Rewatkar, P.M.; Donthula, S.; Taghvaee, T.; Saeed, A.M.; Sotiriou-Leventis, C.; Leventis, N. Exceptionally high CO2 adsorption at 273 K by microporous carbons from phenolic aerogels: The role of heteroatoms in comparison with carbons from polybenzoxazine and other organic aerogels. Macromol. Chem. Phys. 2019, 220, 1800333. [Google Scholar] [CrossRef]
- Zhang, G.; Dass, A.; Rawashdeh, A.-M.M.; Thomas, J.; Counsil, J.A.; Sotiriou-Leventis, C.; Fabrizio, E.F.; Ilhan, F.; Vassilaras, P.; Scheiman, D.A.; et al. Isocyanate cross-linked silica aerogel monoliths: Preparation and characterization. J. Non-Cryst. Solids 2004, 350, 152–164. [Google Scholar] [CrossRef]
- Leventis, N.; Lu, H. Polymer Crosslinked Aerogels. In Aerogels Handbook—Advances in Sol-Gel Derived Materials and Technologies; Aegerter, M., Leventis, N., Koebel, M., Eds.; Springer: New York, NY, USA, 2011; pp. 251–285. [Google Scholar]
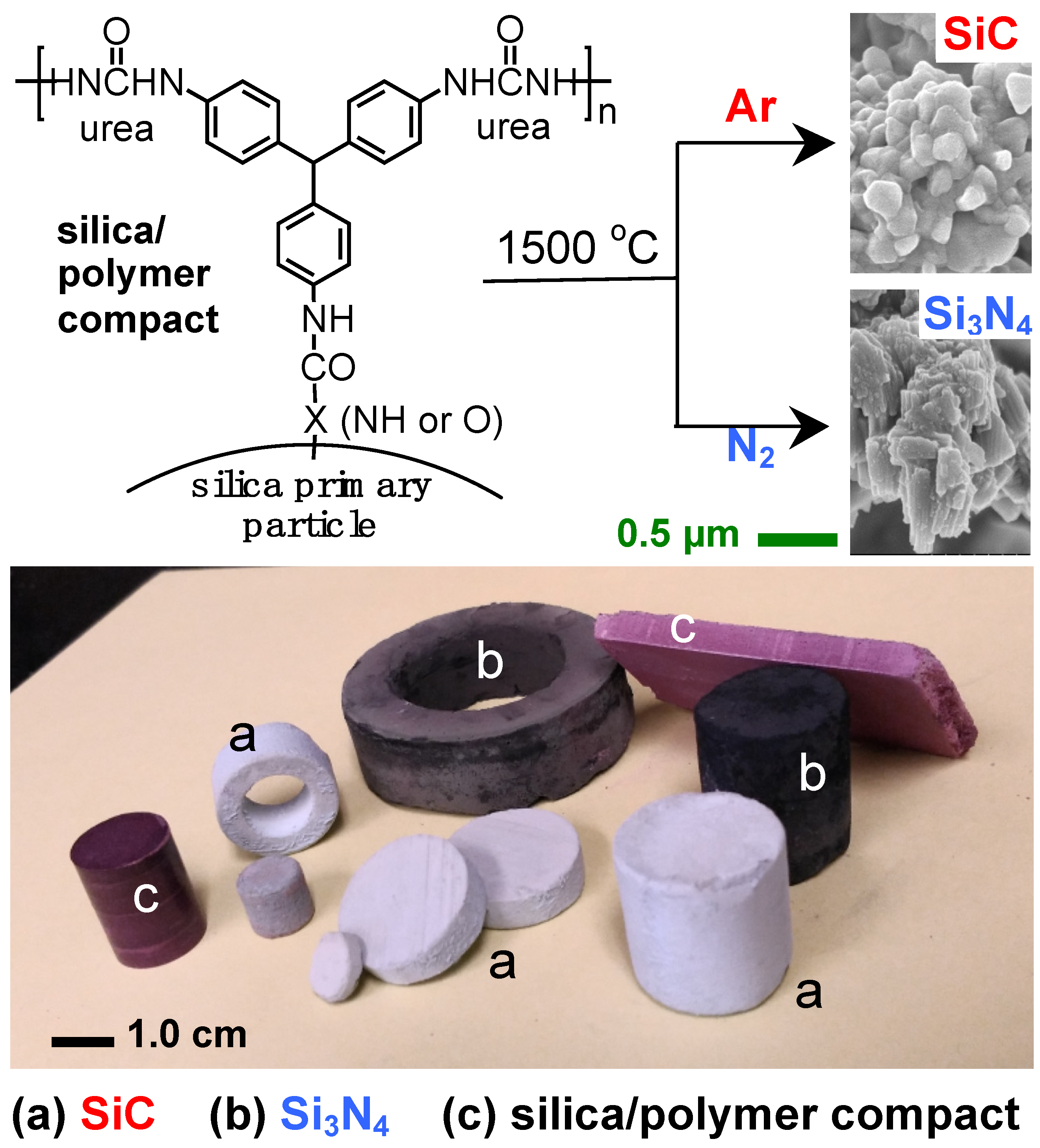
- Rewatkar, P.M.; Taghvaee, T.; Saeed, A.M.; Donthula, S.; Mandal, C.; Chandrasekaran, N.; Leventis, T.; Shruthi, T.K.; Sotiriou-Leventis, C.; Leventis, N. Sturdy, monolithic SiC and Si3N4 aerogels from compressed polymer-cross-linked silica xerogel powders. Chem. Mater. 2018, 30, 1635–1647. [Google Scholar] [CrossRef]
- Katti, A.; Shimpi, N.; Roy, S.; Lu, H.; Fabrizio, E.F.; Dass, A.; Capadona, L.A.; Leventis, N. Chemical, physical and mechanical characterization of isocyanate-crosslinked amine-modified silica aerogels. Chem. Mater. 2006, 18, 285–296. [Google Scholar] [CrossRef]
- Mandal, C.; Donthula, S.; Majedi Far, H.; Saeed, A.M.; Sotiriou-Leventis, C.; Leventis, N. Transparent, mechanically strong, thermally insulating cross-linked silica aerogels for energy-efficient windows. J. Sol-Gel Sci. Technol. 2019, 92, 84–100. [Google Scholar] [CrossRef]
- Rewatkar, P.M.; Soni, R.U.; Sotiriou-Leventis, C.; Leventis, N. A cobalt sunrise: Thermites based on LiClO4-filled Co(0) aerogels prepared from polymer-cross-linked cobaltia xerogel powders. ACS Appl. Mater. Interfaces 2019, 11, 22668–22676. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, P.; Smirnova, I.; Athamneh, T.; Papastergiou, M.; Chriti, D.; Mali, G.; Čendak, T.; Chatzichristidi, M.; Raptopoulos, G.; Gurikov, P. Mechanically strong polyurea/polyurethane-cross-linked alginate aerogels. ACS Appl. Polym. Mater. 2020, 2, 1974–1988. [Google Scholar] [CrossRef]
- Paraskevopoulou, P.; Smirnova, I.; Athamneh, T.; Papastergiou, M.; Chriti, D.; Mali, G.; Čendak, T.; Raptopoulos, G.; Gurikov, P. Polyurea-crosslinked biopolymer aerogel beads. RSC Adv. 2020, 10, 40843–40852. [Google Scholar] [CrossRef]
- Paraskevopoulou, P.; Raptopoulos, G.; Len, A.; Dudás, Z.; Fábián, I.; Kalmár, J. Fundamental skeletal nanostructure of nanoporous polymer-cross-linked alginate aerogels and its relevance to environmental remediation. ACS Appl. Nano Mater. 2021, 4, 10575–10583. [Google Scholar] [CrossRef]
- Paraskevopoulou, P.; Raptopoulos, G.; Leontaridou, F.; Papastergiou, M.; Sakellari, A.; Karavoltsos, S. Evaluation of polyurea-crosslinked alginate aerogels for seawater decontamination. Gels 2021, 7, 27. [Google Scholar] [CrossRef]
- Georgiou, E.; Raptopoulos, G.; Papastergiou, M.; Paraskevopoulou, P.; Pashalidis, I. Extremely efficient uranium removal from aqueous environments with polyurea-crosslinked alginate aerogel beads. ACS Appl. Polym. Mater. 2022, 4, 920–928. [Google Scholar] [CrossRef]
- Raptopoulos, G.; Papastergiou, M.; Chriti, D.; Effraimopoulou, E.; Čendak, T.; Samartzis, N.; Mali, G.; Ioannides, T.; Gurikov, P.; Smirnova, I.; et al. Metal-doped carbons from polyurea-crosslinked alginate aerogel beads. Mater. Adv. 2021, 2, 2684–2699. [Google Scholar] [CrossRef]
- Wang, P.; Ma, X.; Li, Q.; Yang, B.; Shang, J.; Deng, Y. Green synthesis of polyureas from CO2 and diamines with a functional ionic liquid as the catalyst. RSC Adv. 2016, 6, 54013–54019. [Google Scholar] [CrossRef]
- Qaroush, A.; Al-Hamayda, A.S.; Khashman, Y.K.; Vagin, S. Highly efficient isocyanate-free microwave-assited synthesis of [6]-oligourea. Catal. Sci. Technol. 2013, 3, 2221–2226. [Google Scholar] [CrossRef]
- Tiwari, L.; Kumar, V.; Kumar, B.; Majajan, D. A practiclly simple, catalyst free and scalablesynthesis of N-substitutedureas in water. RSC Adv. 2018, 8, 21585–21595. [Google Scholar] [CrossRef] [Green Version]
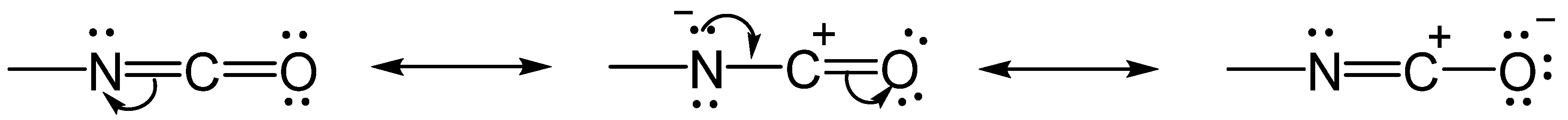
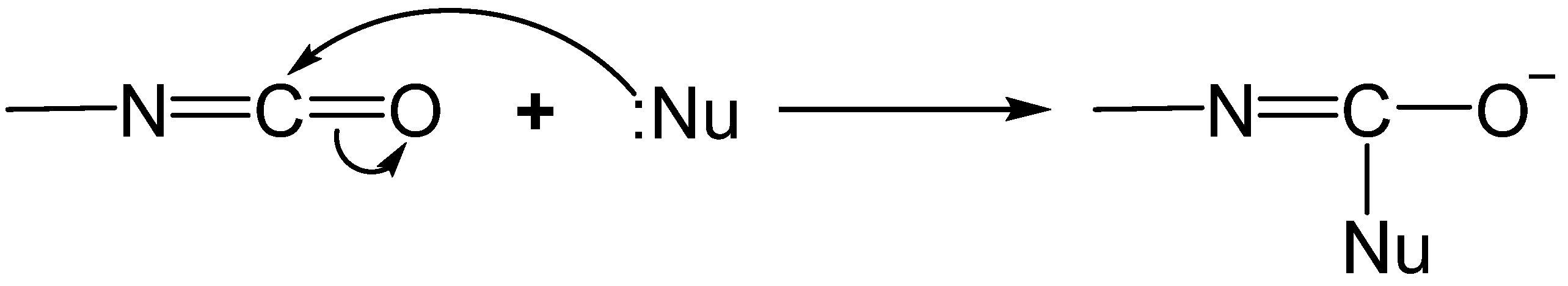























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active-Hydrogen Nucleophile | Relative Reaction Rates (Uncatalyzed at 25 °C) | Product Classification |
|---|---|---|
| Primary aliphatic amines (R–NH2) | 100,000 | Urea |
| Secondary aliphatic amines (R–NH–R’) | 20,000–50,000 | Urea |
| Primary aromatic amines (Ar–NH2) | 200–300 | Urea |
| Primary hydroxyl (e.g., R–OH) | 100 | Urethane |
| Water (H–OH) | 100 | Urea |
| Carboxylic acids (R(C=O)–OH) | 40 | Amide |
| Secondary hydroxyls (e.g., RCH(OH)R’) | 30 | Urethane |
| Ureas (–NH(C=O)NH–) | 15 | Biuret |
| Tertiary hydroxyls (R3C–OH) | 0.5 | Urethane |
| Urethane (–NH(C=O)O–) | 0.3 | Allophanate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leventis, N. Polyurea Aerogels: Synthesis, Material Properties, and Applications. Polymers 2022, 14, 969. https://doi.org/10.3390/polym14050969
Leventis N. Polyurea Aerogels: Synthesis, Material Properties, and Applications. Polymers. 2022; 14(5):969. https://doi.org/10.3390/polym14050969
Chicago/Turabian StyleLeventis, Nicholas. 2022. "Polyurea Aerogels: Synthesis, Material Properties, and Applications" Polymers 14, no. 5: 969. https://doi.org/10.3390/polym14050969
APA StyleLeventis, N. (2022). Polyurea Aerogels: Synthesis, Material Properties, and Applications. Polymers, 14(5), 969. https://doi.org/10.3390/polym14050969