
The 3-Phase Structure of Polyesters (PBT, PET) after Isothermal and Non-Isothermal Crystallization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fast Scanning Calorimetry (FSC)
2.3. Differential Scanning Calorimetry (DSC)
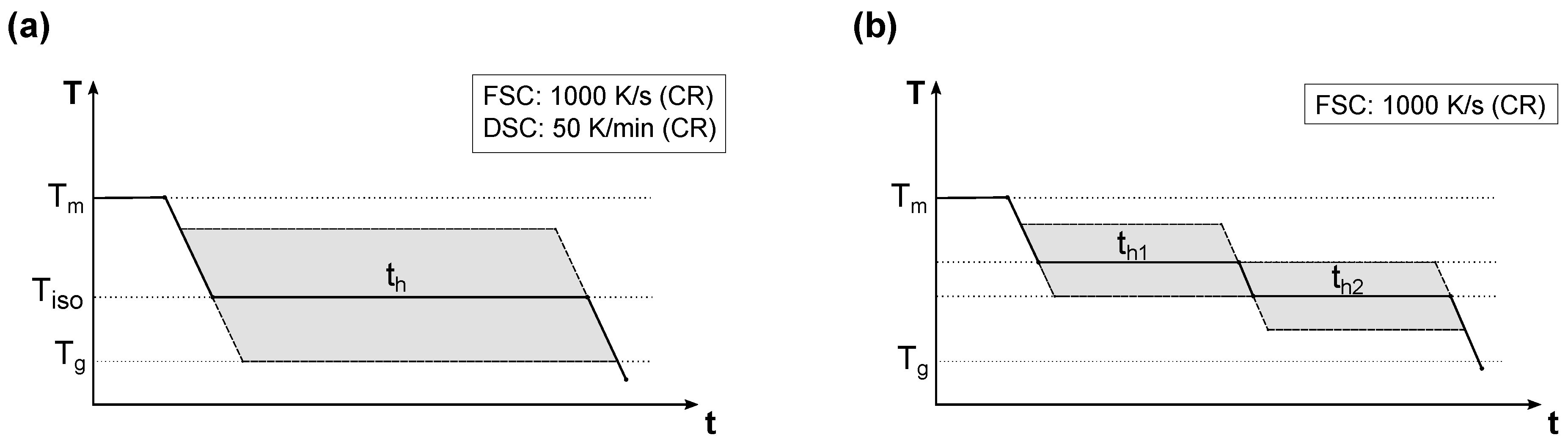
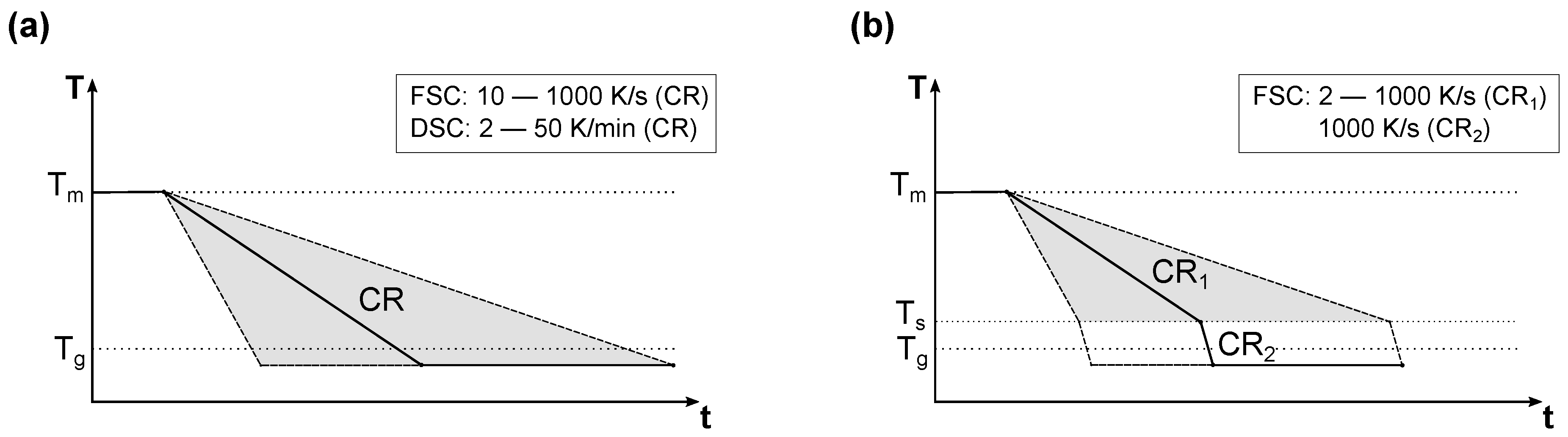
2.4. Experimental Procedure
3. Results and Discussion
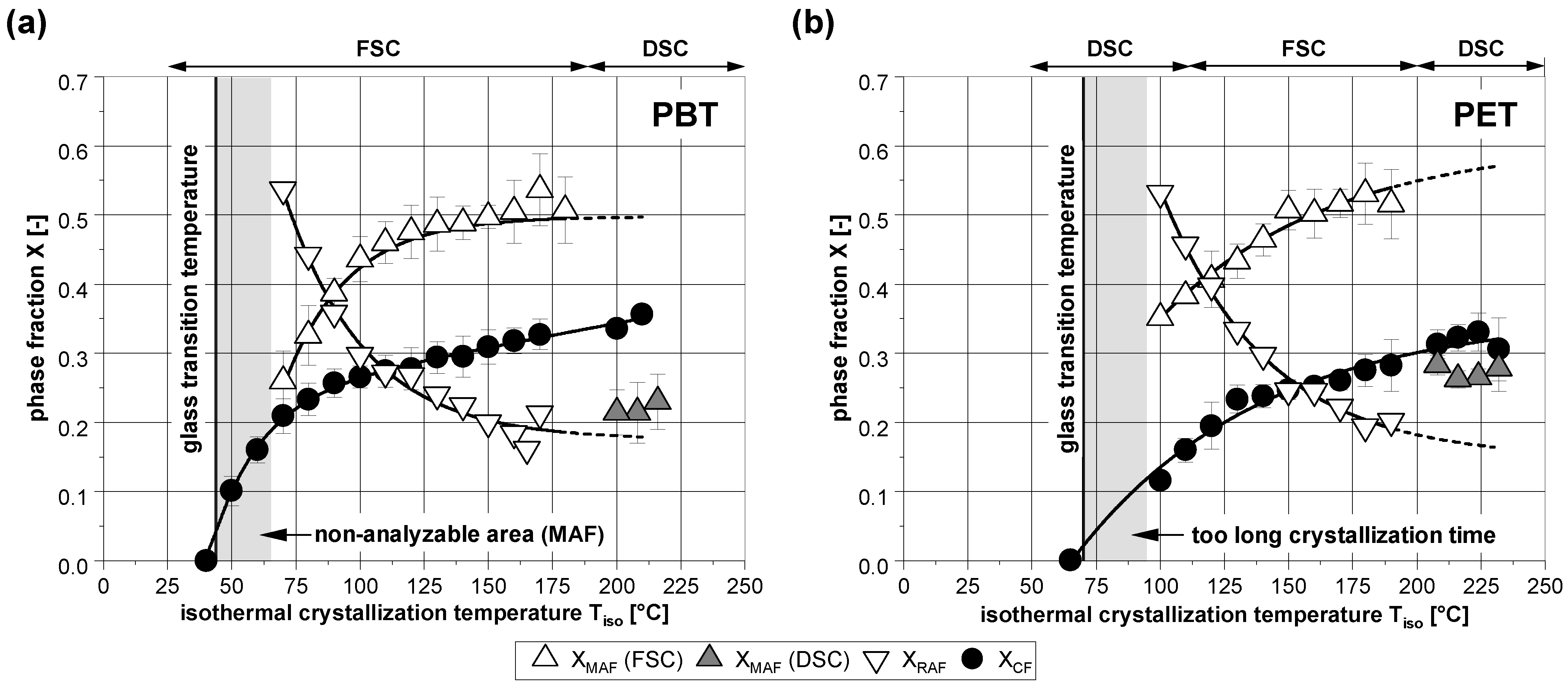
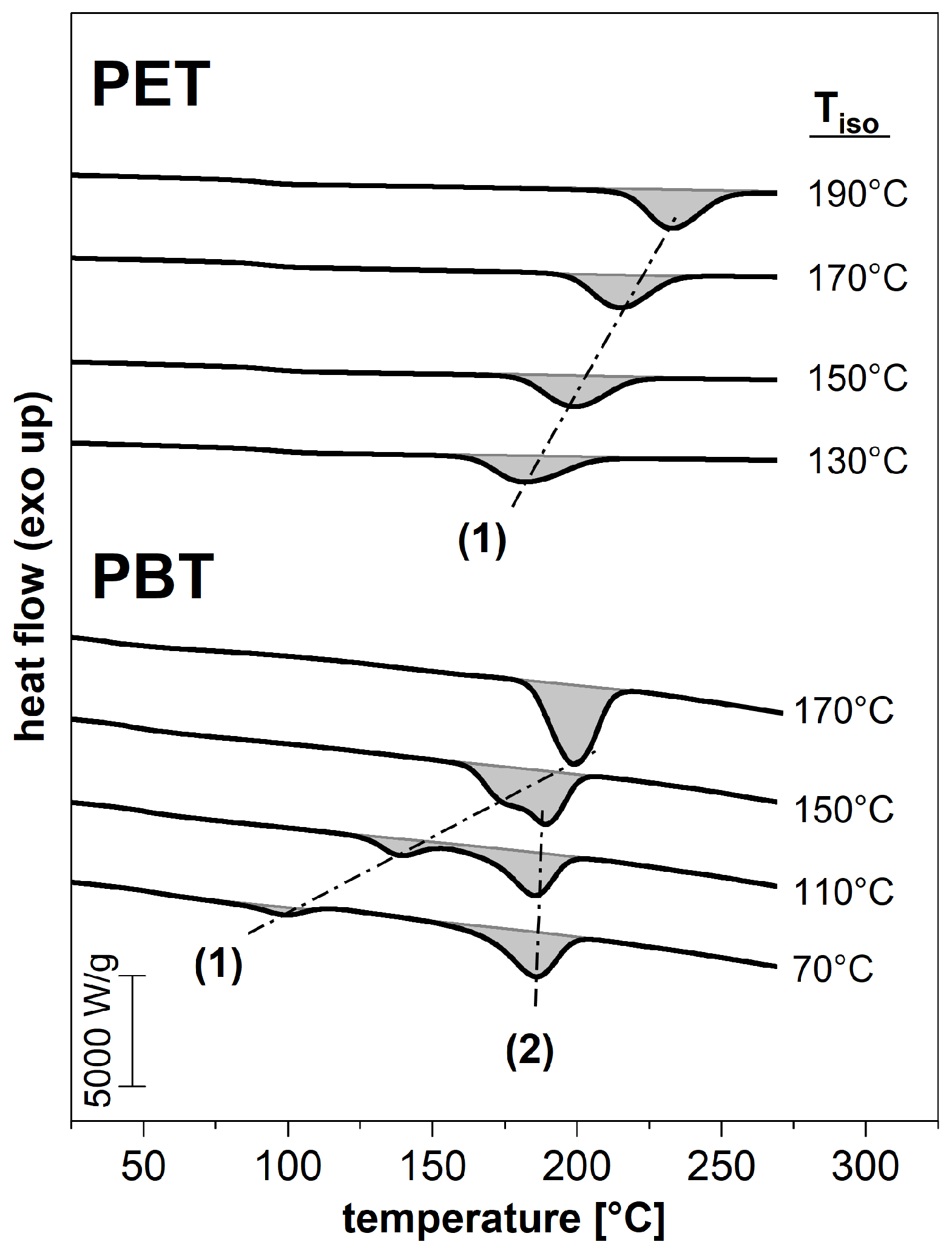
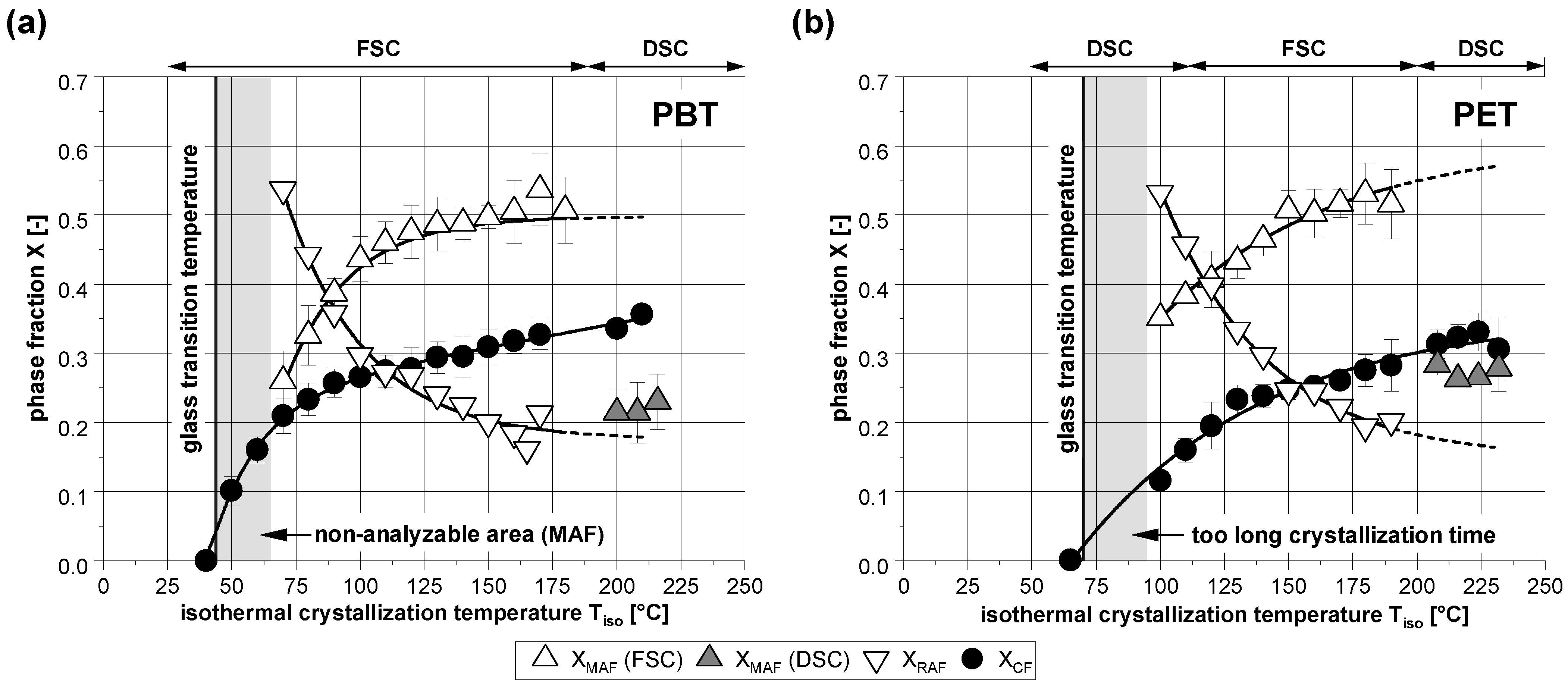
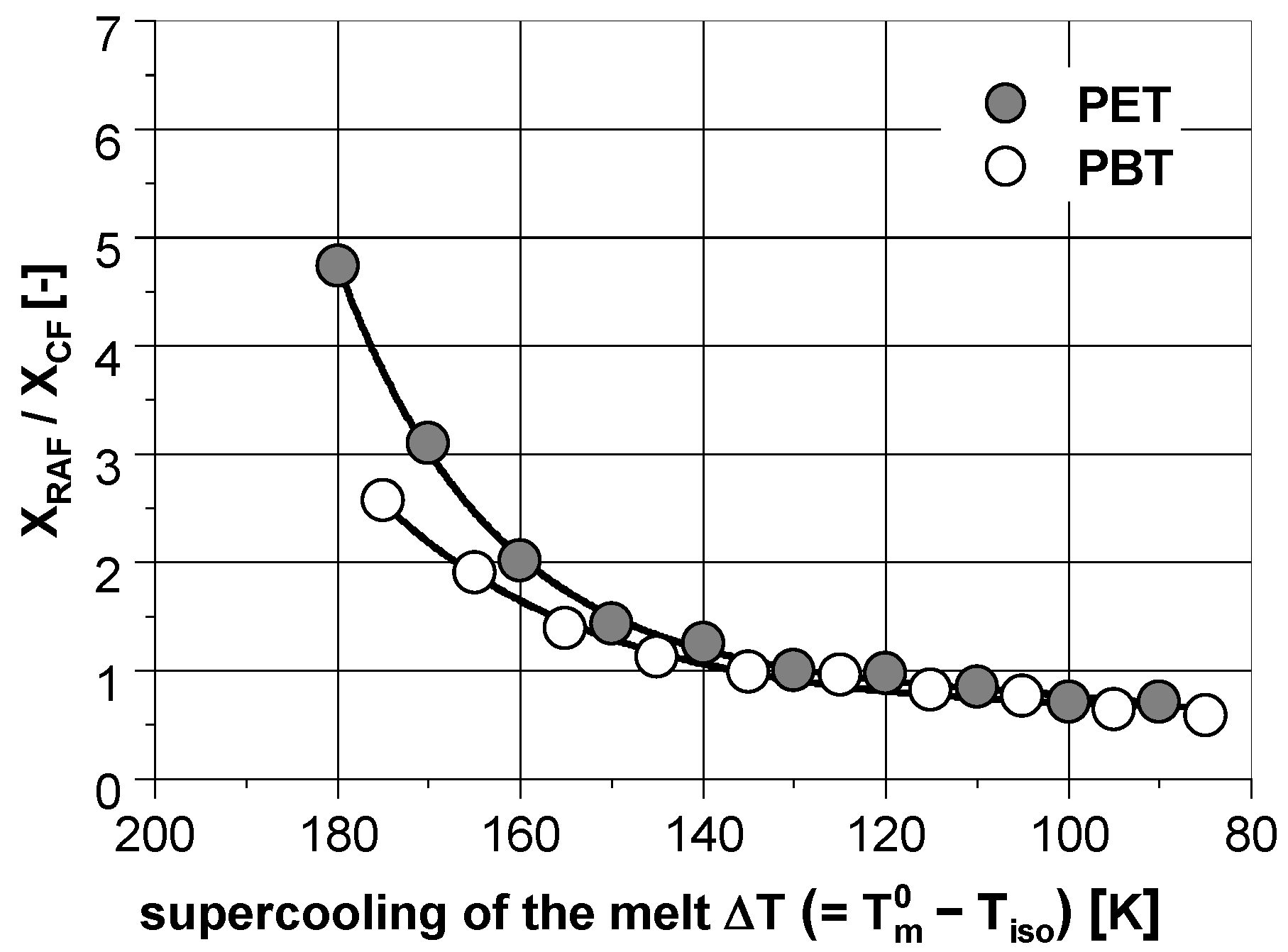
3.1. Phase Composition after Isothermal Crystallization
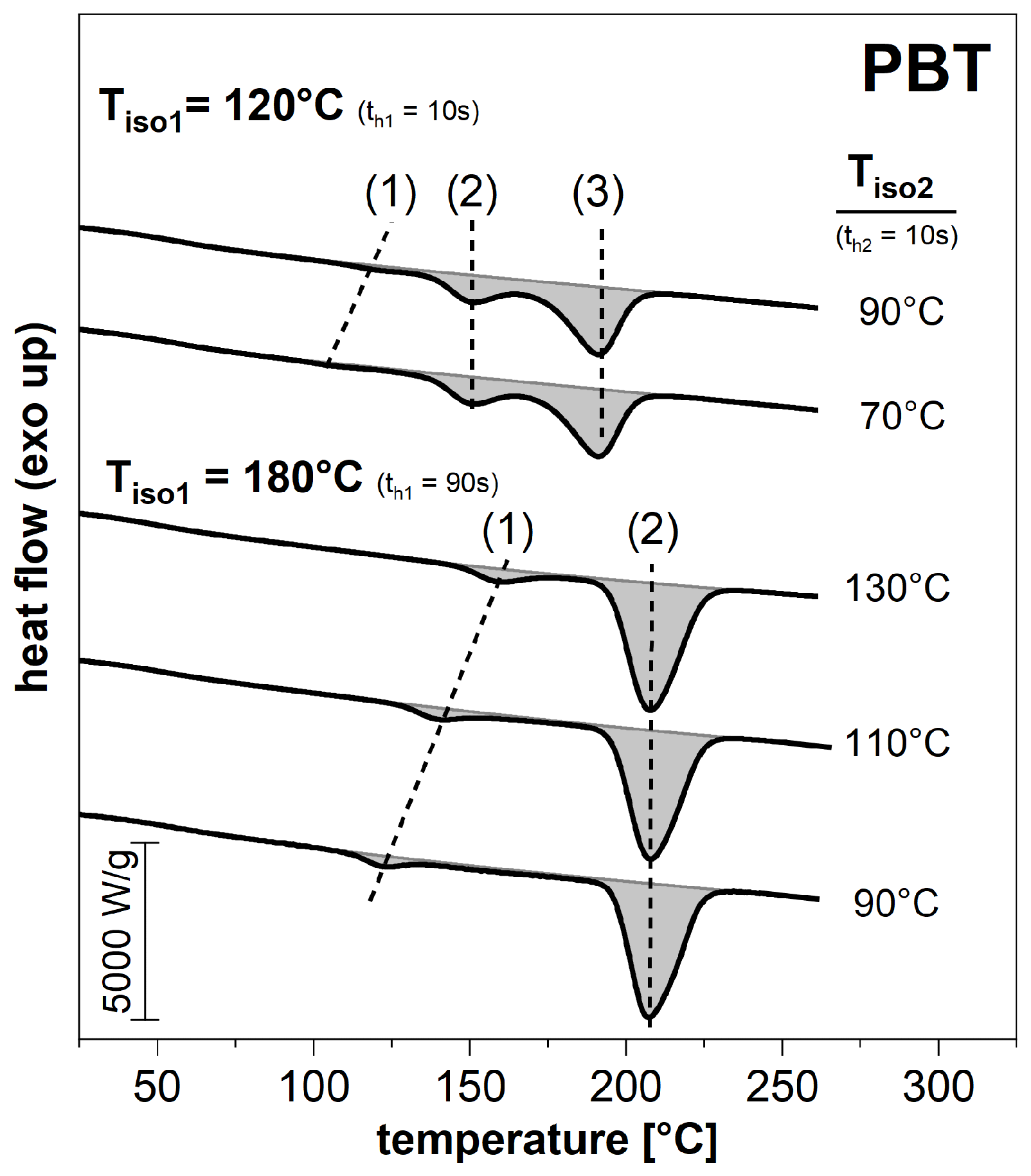
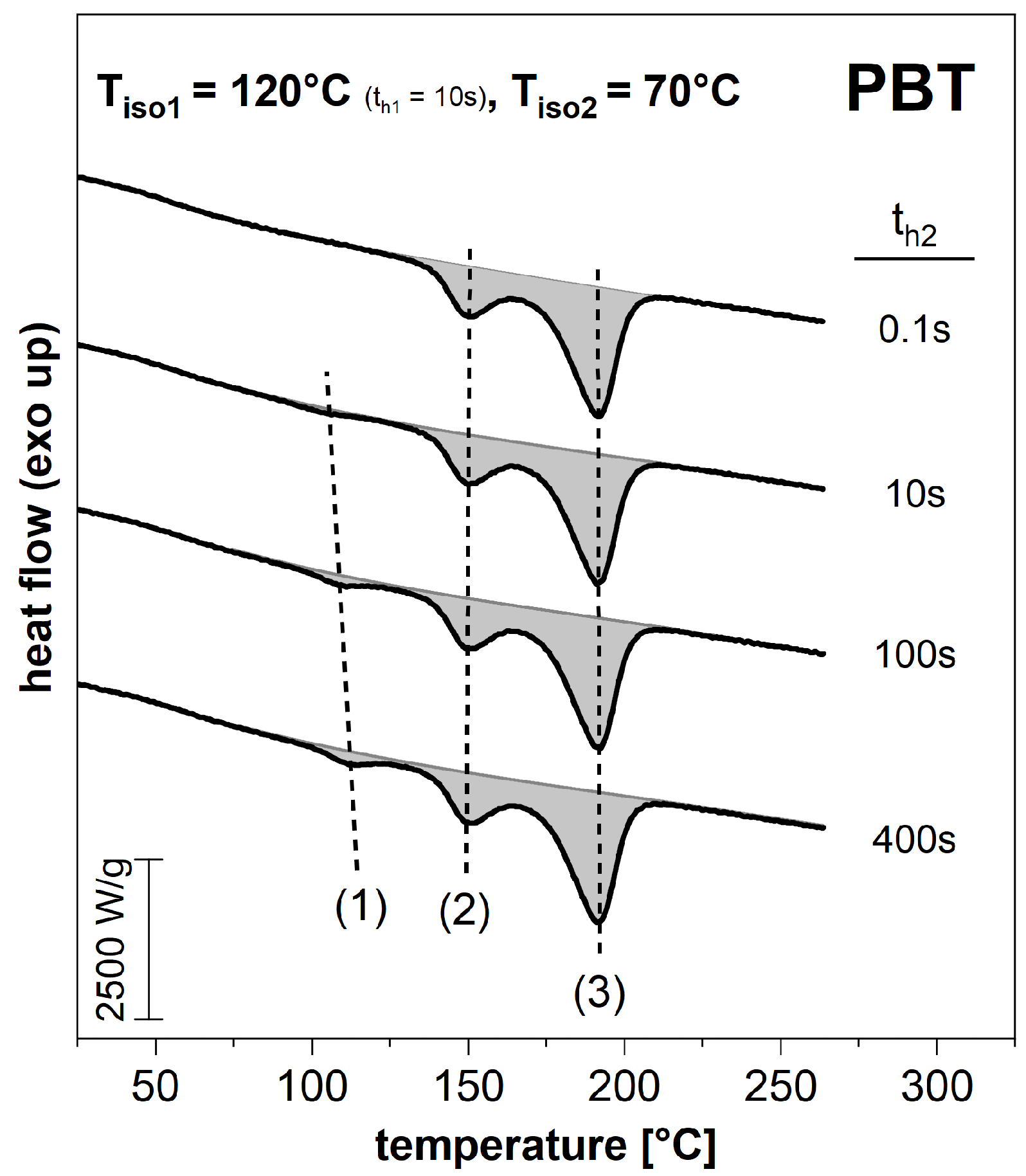
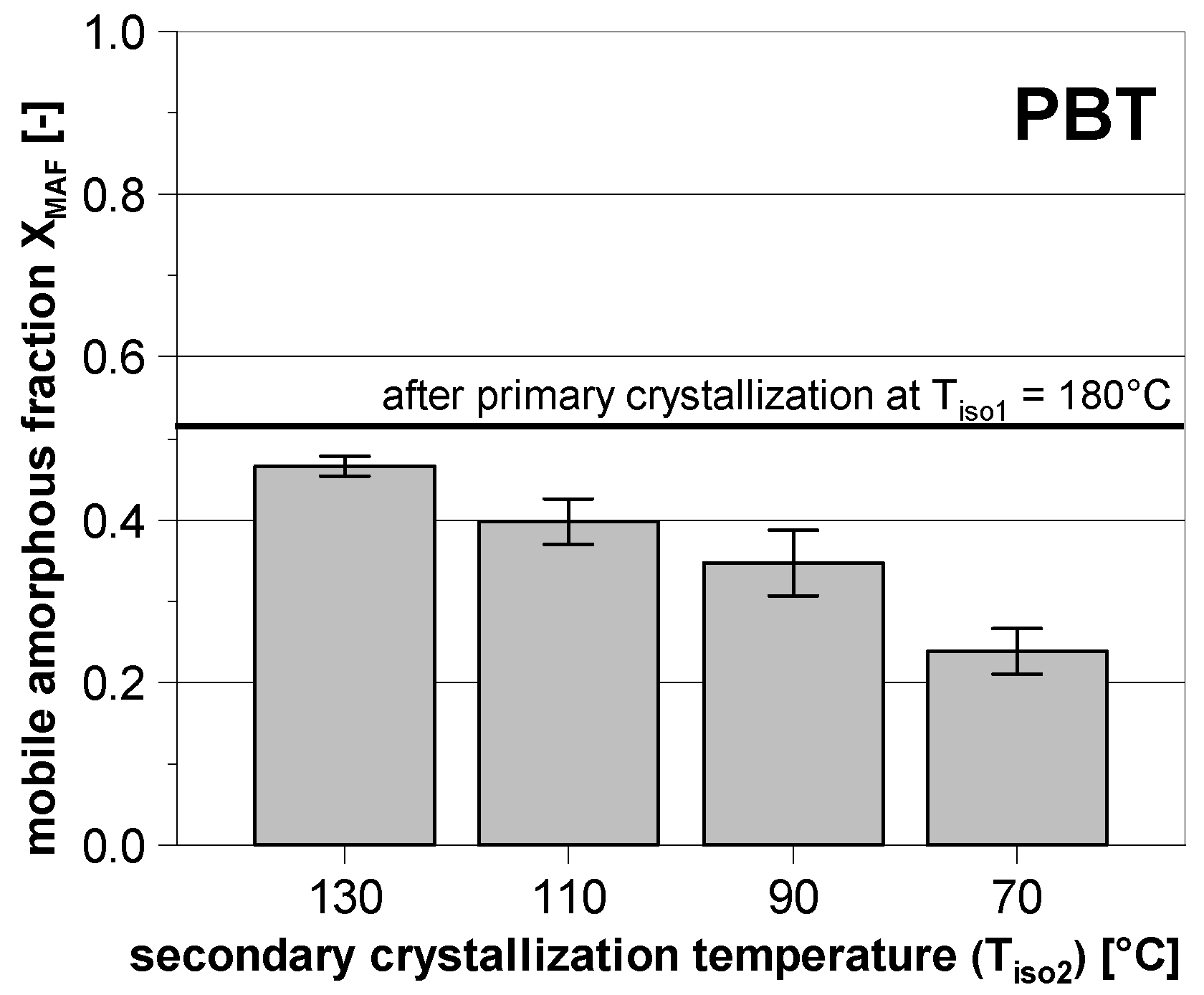
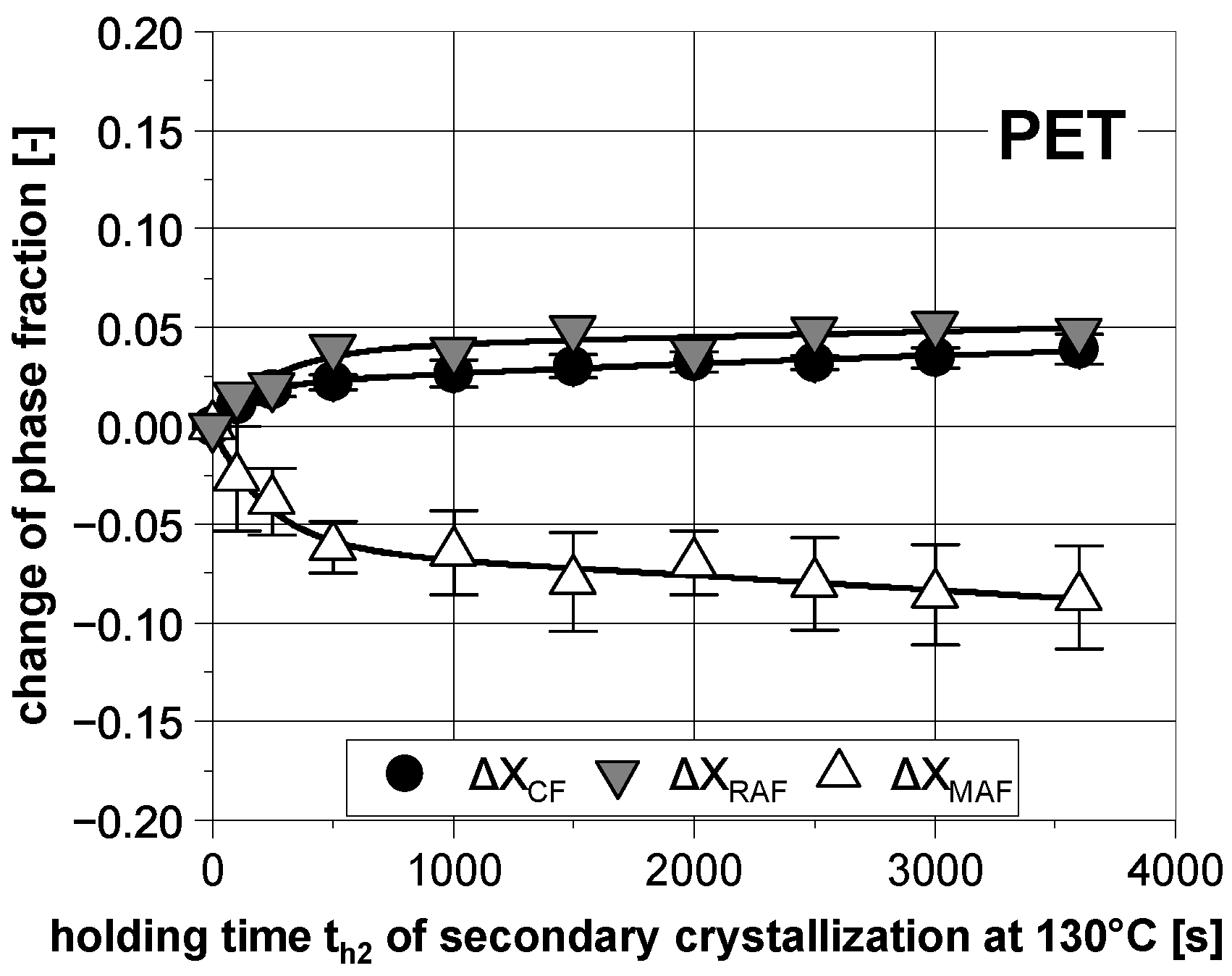
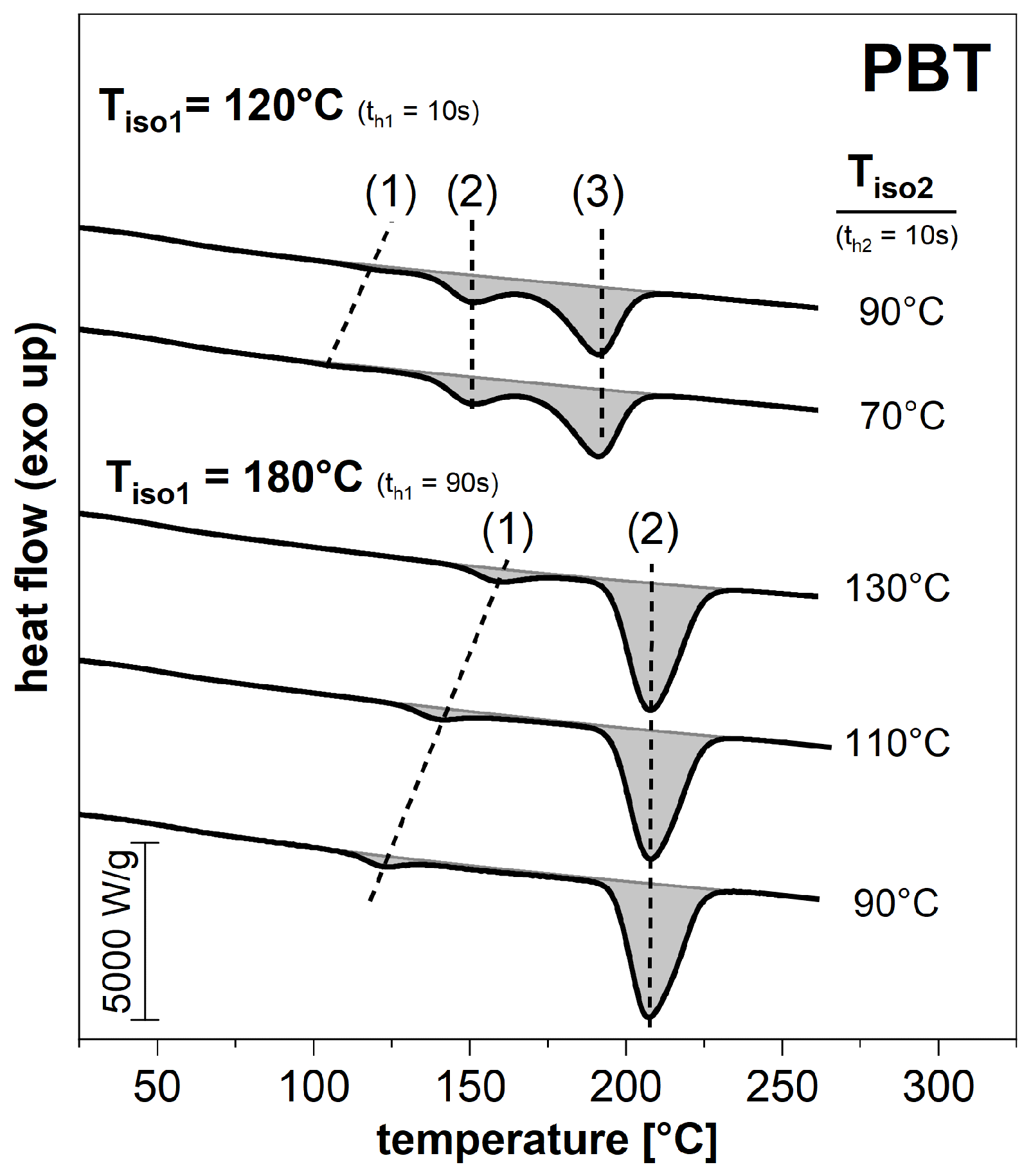
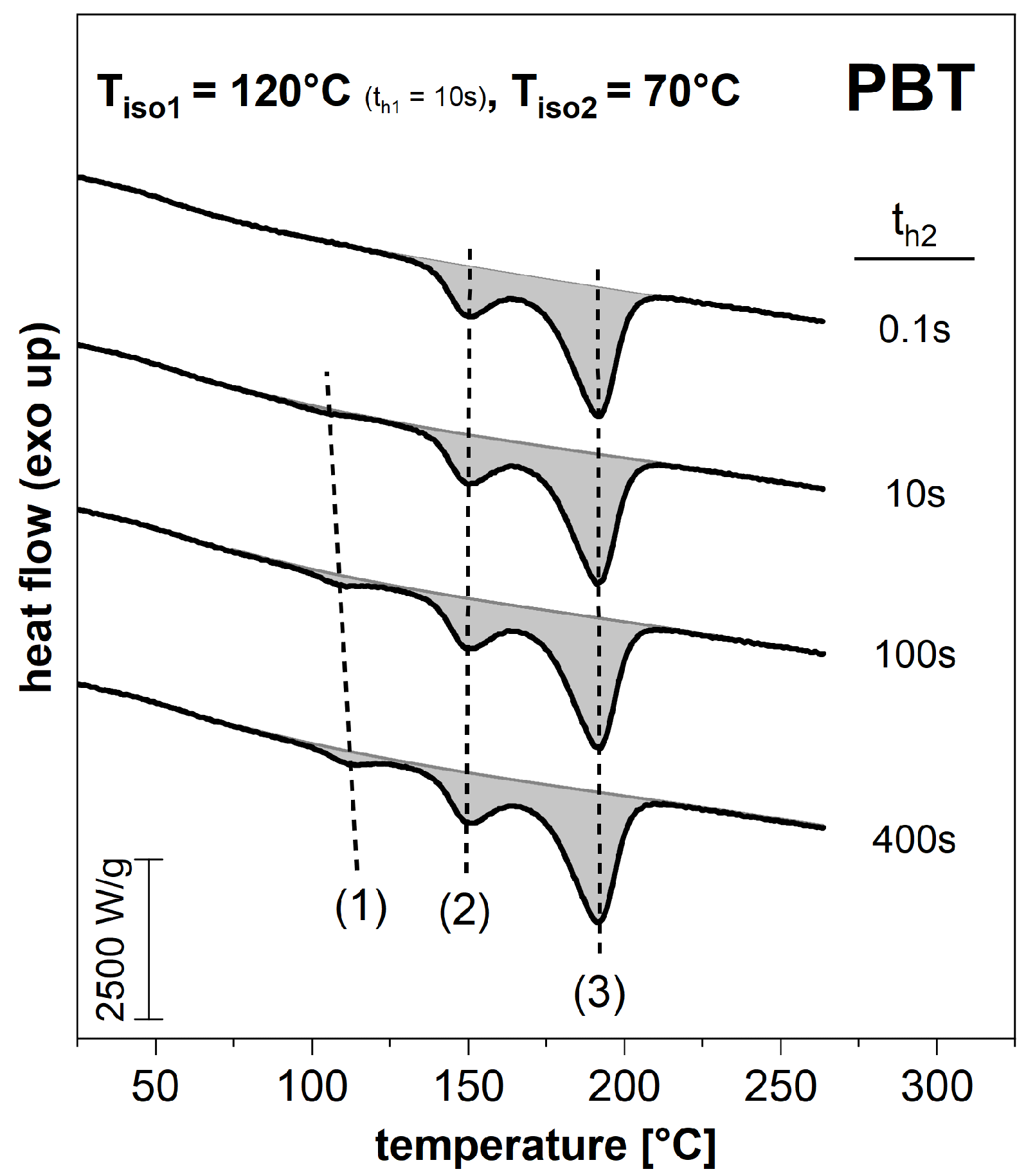
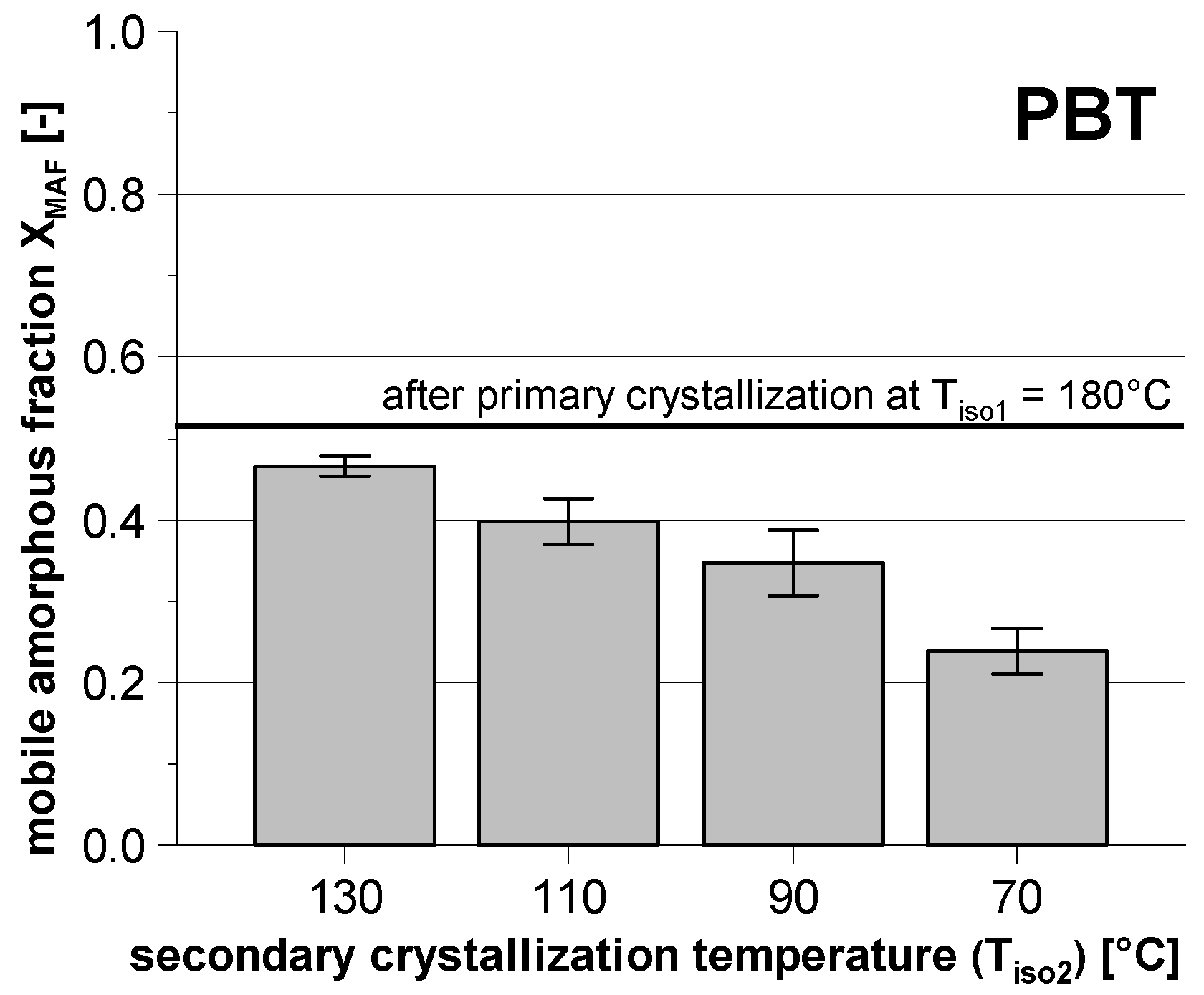
3.2. Subsequent Secondary Crystallization
3.3. Phase Composition after Non-Isothermal Crystallization
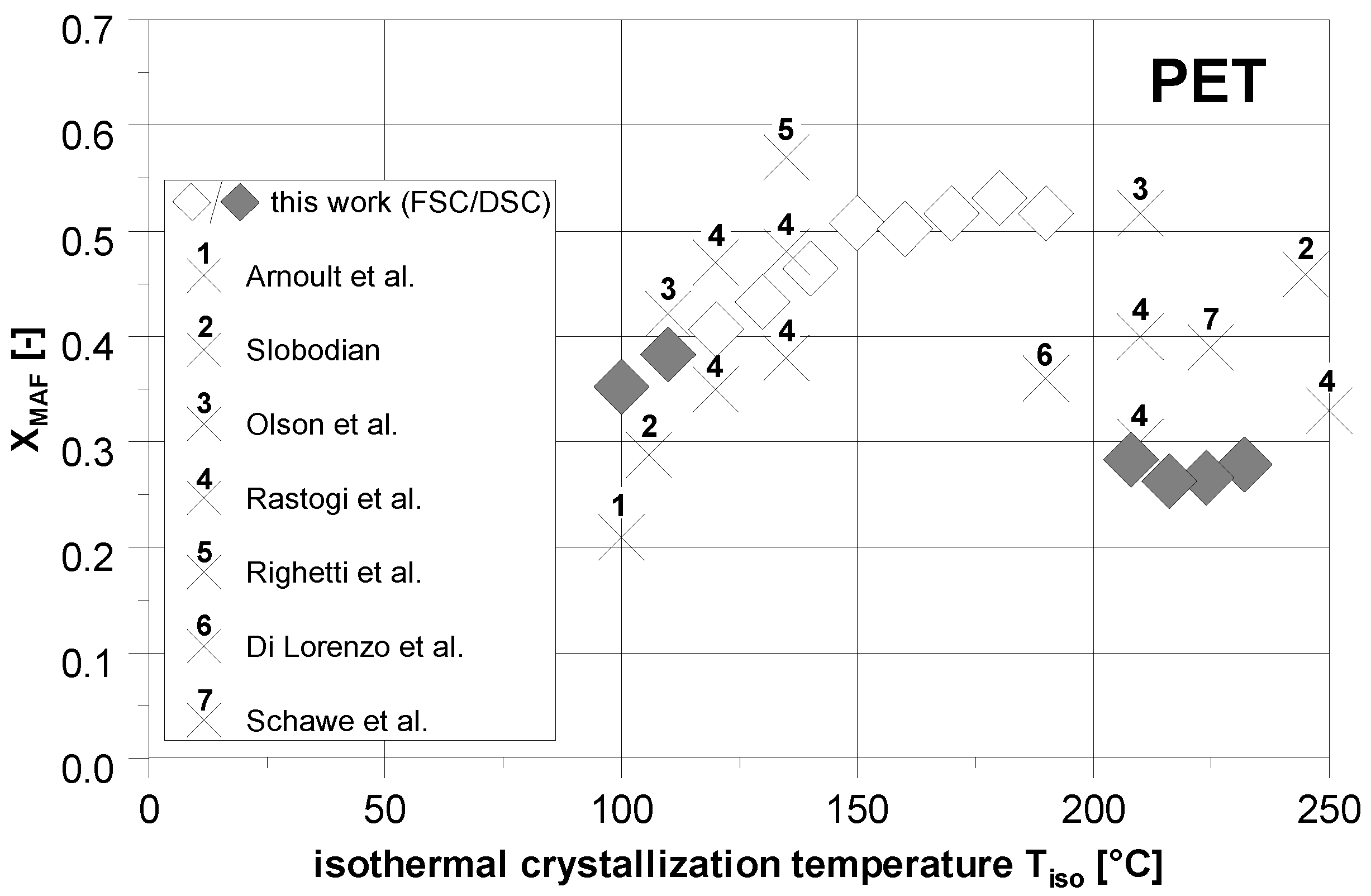
3.4. Comparison to the Literature
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Menczel, J.; Wunderlich, B. Heat capacity hysteresis of semicrystalline macromolecular glasses. J. Polym. Sci. Polym. Lett. Ed. 1981, 19, 261–264. [Google Scholar] [CrossRef]
- Reiter, G.; Strobl, G.R. Progress in Understanding of Polymer Crystallization; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Di Lorenzo, M.L.; Righetti, M.C. Crystallization-induced formation of rigid amorphous fraction. Polym. Cryst. 2018, 1, e10023. [Google Scholar] [CrossRef]
- Pyda, M.; Nowak-Pyda, E.; Mays, J.; Wunderlich, B. Heat capacity of poly(butylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4401–4411. [Google Scholar] [CrossRef]
- Rastogi, R.; Vellinga, W.; Rastogi, S.; Schick, C.; Meijer, H.E.H. The three-phase structure and mechanical properties of poly(ethylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 2092–2106. [Google Scholar] [CrossRef]
- Wissler, G.E.; Crist, B. Glass transition in semicrystalline polycarbonate. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 1257–1270. [Google Scholar] [CrossRef]
- Drieskens, M.; Peeters, R.; Mullens, J.; Franco, D.; Lemstra, P.; Hristova-Bogaerds, D. Structure Versus Properties Relationship of Poly(lactic acid)—Effect of Crystallinity on Barrier Properties. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 2247–2258. [Google Scholar] [CrossRef]
- Menczel, J.D.; Jaffe, M. How did we find the rigid amorphous phase? J. Therm. Anal. Calorim. 2007, 89, 357–362. [Google Scholar] [CrossRef]
- Chen, H.; Cebe, P. Investigation of the rigid amorphous fraction in Nylon6. J. Therm. Anal. Calorim. 2007, 89, 417–425. [Google Scholar] [CrossRef]
- Schawe, J.E.K. Mobile amorphous, rigid amorphous and crystalline fractions in isotactic polypropylene during fast cooling. J. Therm. Anal. Calorim. 2017, 127, 931–937. [Google Scholar] [CrossRef]
- Kitamaru, R.; Horii, F.; Murayama, K. Phase structure of lamellar crystalline polyethylene by solid-state high-resolution carbon-13 NMR detection of the crystalline-amorphous interphase. Macromolecules 1986, 19, 636–643. [Google Scholar] [CrossRef]
- Cheng, J.; Fone, M.; Reddy, V.N.; Schwartz, K.B.; Fisher, H.P.; Wunderlich, B. Identification and quantitative analysis of the intermediate phase in a linear high-density polyethylene. J. Polym. Sci. Part B Polym. Phys. 1994, 32, 2683–2693. [Google Scholar] [CrossRef]
- Suzuki, H.; Grebowicz, J.; Wunderlich, B. Glass transition of poly(oxymethylene). Br. Polym. J. 1985, 17, 1–3. [Google Scholar] [CrossRef]
- Parodi, E.; Govaert, L.E.; Peters, G.W.M. Glass transition temperature versus structure of polyamide 6: A flash-DSC study. Thermochim. Acta 2017, 657, 110–122. [Google Scholar] [CrossRef]
- Menczel, J.D. The rigid amorphous fraction in semicrystalline macromolecules. J. Therm. Anal. Calorim. 2011, 106, 7–24. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L. Vitrification and devitrification of the rigid amorphous fraction in poly(ethylene terephthalate). e-Polymers 2009, 9. [Google Scholar] [CrossRef]
- Righetti, M.C.; Laus, M.; Di Lorenzo, M.L. Temperature dependence of the rigid amorphous fraction in poly(ethylene terephthalate). Eur. Polym. J. 2014, 58, 60–68. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L. Rigid amorphous fraction and multiple melting behavior in poly(butylene terephthalate) and isotactic polystyrene. J. Therm. Anal. Calorim. 2016, 126, 521–530. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Pan, R.; Wunderlich, B. Thermal analysis of poly(butylene terephthalate) for heat capacity, rigid-amorphous content, and transition behavior. Die Makromol. Chem. 1988, 189, 2443–2458. [Google Scholar] [CrossRef]
- Schawe, J.E.K.; Wrana, C. Competition between Structural Relaxation and Crystallization in the Glass Transition Range of Random Copolymers. Polymers 2020, 12, 1778. [Google Scholar] [CrossRef]
- Slobodian, P. Rigid amorphous fraction in poly(ethylene terephthalate) determined by dilatometry. J. Therm. Anal. Calorim. 2008, 94, 545–551. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C.; Cocca, M.; Wunderlich, B. Coupling between Crystal Melting and Rigid Amorphous Fraction Mobilization in Poly(ethylene terephthalate). Macromolecules 2010, 43, 7689–7694. [Google Scholar] [CrossRef]
- Arnoult, M.; Dargent, E.; Mano, J.F. Mobile amorphous phase fragility in semi-crystalline polymers: Comparison of PET and PLLA. Polymer 2007, 48, 1012–1019. [Google Scholar] [CrossRef]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Vyazovkin, S.; Stone, J.; Sbirrazzuoli, N. Hoffman-Lauritzen parameters for non-isothermal crystallization of poly(ethylene terephthalate) and poly(ethylene oxide) melts. J. Therm. Anal. Calorim. 2005, 80, 177–180. [Google Scholar] [CrossRef]
- Conix, A.; Van Kerpel, R. Crystallization behavior and melting properties of m-phenylene group containing polyesters. J. Polym. Sci. 1959, 40, 521–532. [Google Scholar] [CrossRef]
- Poel, G.; Daniel, I.; Magon, A.; Mathot, V. Performance and calibration of the Flash DSC 1, a new, MEMS-based fast scanning calorimeter. J. Therm. Anal. Calorim. 2012, 110, 1533–1546. [Google Scholar] [CrossRef]
- Cheng, S.; Wunderlich, B. Heat capacities and entropies of liquid, high-melting-point polymers containing phenylene groups (PEEK, PC, and PET). J. Polym. Sci. Part B Polym. Phys. 1986, 24, 1755–1765. [Google Scholar] [CrossRef]
- Standard DIN EN ISO 11357-7:2015-12; Plastics—Differential scanning calorimetry (DSC)—Part 7: Determination of crystallization kinetics (ISO 11357-7:2015). ISO: Geneva, Switzerland, 2015.
- Heidrich, D.; Gehde, M. Calorimetric analysis of the crystallization of PBT considering the dynamic cooling condition of real processing. Polym. Test. 2020, 85, 106459. [Google Scholar] [CrossRef]
- Bornschlegl, E.; Bonart, R. Small angle X-ray scattering studies of poly(ethylene terephthalate) and poly(butylene terephthalate). Colloid Polym. Sci. 1980, 258, 319–331. [Google Scholar] [CrossRef]
- Kim, H.G.; Robertson, R.E. Multiple melting endotherms in isothermally melt-crystallized poly(butylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 1998, 36, 1757–1767. [Google Scholar]
- Yeh, J.; Runt, J. Multiple melting in annealed poly(butylene terephthalate). J. Polym. Sci. Part B 1989, 27, 1543–1550. [Google Scholar] [CrossRef]
- Furushima, Y.; Kumazawa, S.; Umetsu, H.; Toda, A.; Zhuravlev, E.; Schick, C. Melting and recrystallization kinetics of poly(butylene terephthalate). Polymer 2016, 109, 307–314. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L. Melting process of Poly(butylene terephthalate) analyzed by temperature-modulated differential scanning calorimetry. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 2191–2201. [Google Scholar] [CrossRef]
- Hay, J.N. Secondary crystallization kinetics. Polym. Cryst. 2018, 1, e10007. [Google Scholar] [CrossRef]
- Olson, B.; Lin, J.; Nazarenko, S.; Jamieson, A. Positron Annihilation Lifetime Spectroscopy of Poly(ethylene terephthalate): Contributions from Rigid and Mobile Amorphous Fractions. Macromolecules 2003, 36, 7618–7623. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heidrich, D.; Gehde, M. The 3-Phase Structure of Polyesters (PBT, PET) after Isothermal and Non-Isothermal Crystallization. Polymers 2022, 14, 793. https://doi.org/10.3390/polym14040793
Heidrich D, Gehde M. The 3-Phase Structure of Polyesters (PBT, PET) after Isothermal and Non-Isothermal Crystallization. Polymers. 2022; 14(4):793. https://doi.org/10.3390/polym14040793
Chicago/Turabian StyleHeidrich, Dario, and Michael Gehde. 2022. "The 3-Phase Structure of Polyesters (PBT, PET) after Isothermal and Non-Isothermal Crystallization" Polymers 14, no. 4: 793. https://doi.org/10.3390/polym14040793
APA StyleHeidrich, D., & Gehde, M. (2022). The 3-Phase Structure of Polyesters (PBT, PET) after Isothermal and Non-Isothermal Crystallization. Polymers, 14(4), 793. https://doi.org/10.3390/polym14040793