
Comparison of Synthetic vs. Biogenic Polymeric Process-Directing Agents for Intrafibrillar Mineralization of Collagen

 ,
,
Abstract
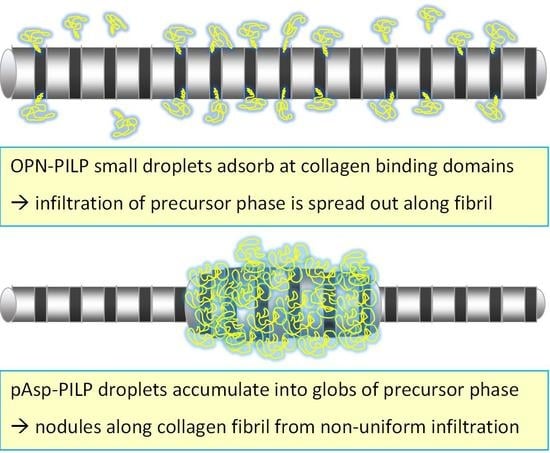
:
1. Introduction
2. Materials and Methods
2.1. Mineralization
2.2. X-ray Diffraction (XRD)
2.3. Thermogravimetric Analysis (TGA)
2.4. Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDS)
2.5. Fibril Diameter Analysis
2.6. Transmission Electron Microscopy (TEM) and Electron Diffraction
2.7. Fluorescent Tagging and Imaging
2.8. Nanoparticle Tracking Analysis
3. Results
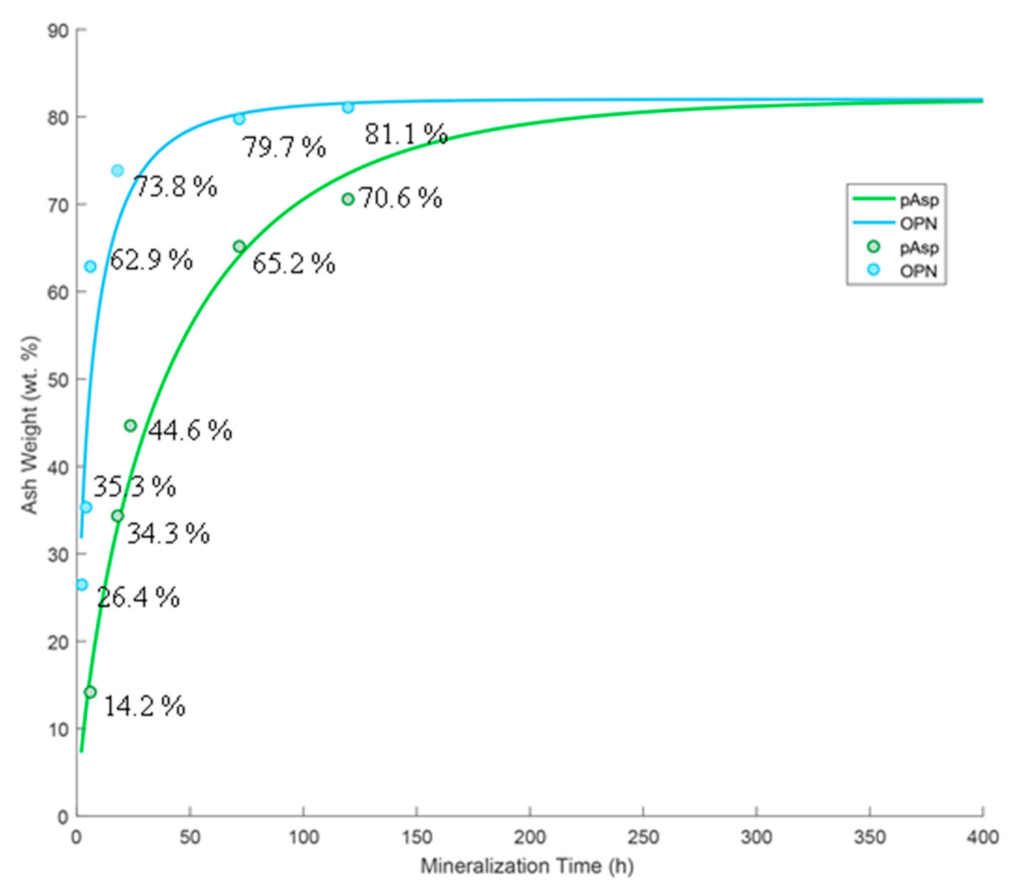
3.1. XRD and TGA Analysis of Mineralization Kinetics
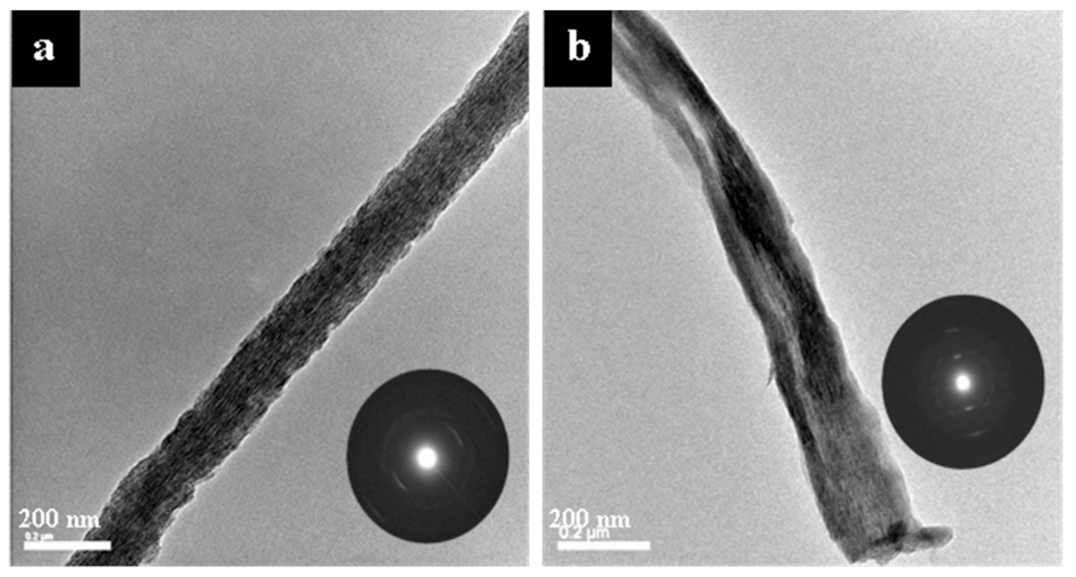
3.2. Microscopy Analysis of Mineralized Fibril Morphologies and Textures
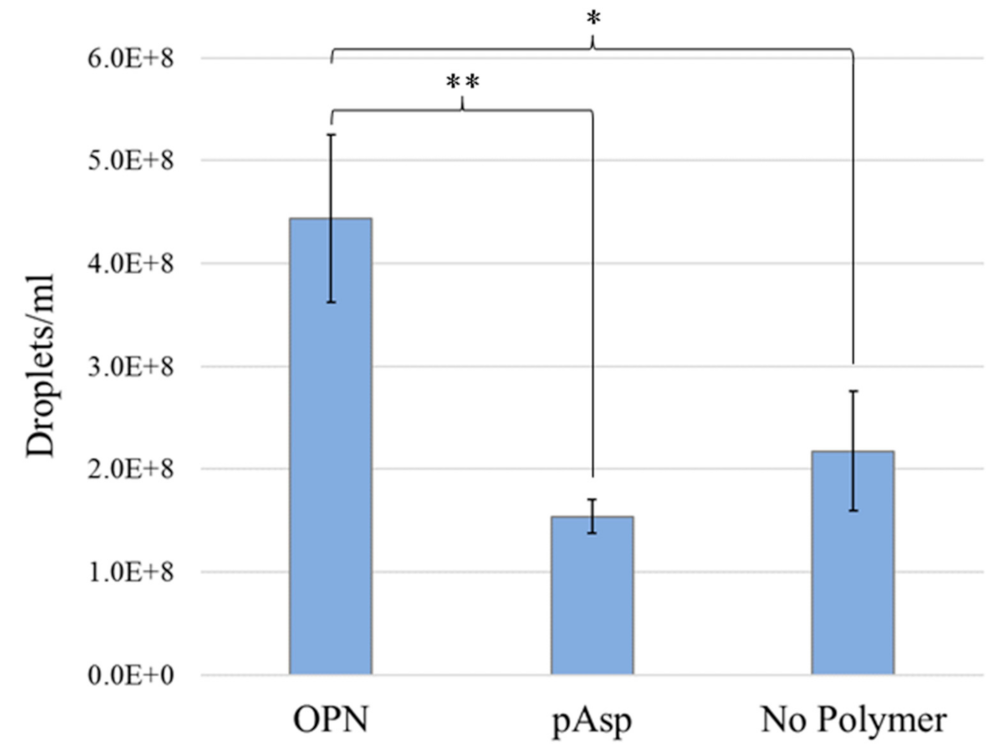
3.3. NanoSight Nanoparticle Tracking Analysis
3.4. Fluorescence Analysis of Polymer–Collagen Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone Structure and Formation: A New Perspective. Mater. Sci. Eng. R-Rep. 2007, 58, 77–116. [Google Scholar]
- Jee, S.S.; Kasinath, R.K.; DiMasi, E.; Kim, Y.Y.; Gower, L. Oriented Hydroxyapatite in Turkey Tendon Mineralized Via the Polymer-Induced Liquid-Precursor (PILP) Process. CrystEngComm 2011, 13, 2077–2083. [Google Scholar]
- Li, Y.; Thula, T.T.; Jee, S.; Perkins, S.L.; Aparicio, C.; Douglas, E.P.; Gower, L.B. Biomimetic Mineralization of Woven Bone-Like Nanocomposites: Role of Collagen Cross-Links. Biomacromolecules 2012, 13, 49–59. [Google Scholar] [PubMed]
- Wingender, B.; Bradley, P.; Saxena, N.; Ruberti, J.W.; Gower, L. Biomimetic Organization of Collagen Matrices to Template Bone-Like Microstructures. Matrix Biol. 2016, 52–54, 384–396. [Google Scholar]
- Wingender, B.; Ni, Y.; Zhang, Y.; Taylor, C.; Gower, L. Hierarchical Characterization and Nanomechanical Assessment of Biomimetic Scaffolds Mimicking Lamellar Bone Via Atomic Force Microscopy Cantilever-Based Nanoindentation. Materials 2018, 11, 1257. [Google Scholar] [CrossRef] [Green Version]
- Jee, S.S.; Thula, T.T.; Gower, L.B. Development of Bone-Like Composites Via the Polymer-Induced Liquid-Precursor (PILP) Process. Part 1: Influence of Polymer Molecular Weight. Acta Biomater. 2010, 6, 3676–3686. [Google Scholar]
- Gower, L.B.; Odom, D.J. Deposition of Calcium Carbonate Films by a Polymer-Induced Liquid-Precursor (PILP) Process. J. Cryst. Growth 2000, 210, 719–734. [Google Scholar] [CrossRef]
- Olszta, M.J.; Douglas, E.P.; Gower, L.B. Scanning Electron Microscopic Analysis of the Mineralization of Type I Collagen Via a Polymer-Induced Liquid-Precursor (PILP) Process. Calcif. Tissue Int. 2003, 72, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Gower, L.B. Biomimetic Model Systems for Investigating the Amorphous Precursor Pathway and Its Role in Biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [PubMed] [Green Version]
- Niu, L.; Jee, S.E.; Jiao, K.; Tonggu, L.; Li, M.; Wang, L.; Yang, Y.; Bian, J.; Breschi, L.; Jang, S.S.; et al. Collagen Intrafibrillar Mineralization as a Result of the Balance between Osmotic Equilibrium and Electroneutrality. Nat. Mater. 2017, 16, 370–378. [Google Scholar] [PubMed] [Green Version]
- Burwell, A.K.; Thula-Mata, T.; Gower, L.B.; Habeliz, S.; Kurylo, M.; Ho, S.P.; Chien, Y.C.; Cheng, J.; Cheng, N.F.; Gansky, S.A.; et al. Functional Remineralization of Dentin Lesions Using Polymer-Induced Liquid-Precursor Process. PLoS ONE 2012, 7, e38852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.C.; Saeki, K.; Ho, S.P.; Habelitz, S.; Marshall, G.; Gower, L. PILP Remineralized Dentin Lesions Have Two Zones of Distinct Properties. In Proceedings of the American Association for Dental Research (AADR) Annual Meeting, Tampa, FL, USA, 21–24 March 2012. [Google Scholar]
- Saxena, N.; Cremer, M.A.; Dolling, E.S.; Nurrohman, H.; Habelitz, S.; Marshall, G.W.; Gower, L.B. Influence of Fluoride on the Mineralization of Collagen Via the Polymer-Induced Liquid-Precursor (PILP) Process. Dent. Mater. 2018, 34, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Bacino, M.; Girn, V.; Nurrohman, H.; Saeki, K.; Marshall, S.J.; Gower, L.; Saeed, E.; Stewart, R.; Le, T.; Marshall, G.W.; et al. Integrating the PILP-Mineralization Process into a Restorative Dental Treatment. Dent. Mater. 2019, 35, 53–63. [Google Scholar] [PubMed]
- Saxena, N.; Habelitz, S.; Marshall, G.W.; Gower, L.B. Remineralization of Demineralized Dentin Using a Dual Analog System. Orthod. Craniofacial Res. 2019, 22, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Noncollagenous Matrix Proteins and Their Role in Mineralization. Bone Miner. 1989, 6, 111–123. [Google Scholar] [PubMed]
- Zurick, K.M.; Qin, C.; Bernards, M.T. Mineralization Induction Effects of Osteopontin, Bone Sialoprotein, and Dentin Phosphoprotein on a Biomimetic Collagen Substrate. J. Biomed. Mater. Res. Part A 2013, 101, 1571–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Singh, K.; Mukherjee, B.B.; Sodek, J. Developmental Expression of Osteopontin (Opn) Mrna in Rat Tissues: Evidence for a Role for Opn in Bone Formation and Resorption. Matrix 1993, 13, 113–123. [Google Scholar] [PubMed]
- Giachelli, C.M.; Steitz, S. Osteopontin: A Versatile Regulator of Inflammation and Biomineralization. Matrix Biol. 2000, 19, 615–622. [Google Scholar] [PubMed]
- Sodek, J.; Ganss, B.; McKee, M.D. Osteopontin. Crit. Rev. Oral Biol. Med. 2000, 11, 279–303. [Google Scholar]
- Azuma, N.; Maeta, A.; Fukuchi, K.; Kanno, C. A Rapid Method for Purifying Osteopontin from Bovine Milk and Interaction between Osteopontin and Other Milk Proteins. Int. Dairy J. 2006, 16, 370–378. [Google Scholar] [CrossRef]
- Gericke, A.; Qin, C.; Spevak, L.; Fujimoto, Y.; Butler, W.T.; Sorensen, E.S.; Boskey, A.L. Importance of Phosphorylation for Osteopontin Regulation of Biomineralization. Calcif. Tissue Int. 2005, 77, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licini, C.; Vitale-Brovarone, C.; Mattioli-Belmonte, M. Collagen and Non-Collagenous Proteins Molecular Crosstalk in the Pathophysiology of Osteoporosis. Cytokine Growth Factor Rev. 2019, 49, 59–69. [Google Scholar] [PubMed]
- Fisher, L.W.; Torchia, D.A.; Fohr, B.; Young, M.F.; Fedarko, N.S. Flexible Structures of Sibling Proteins, Bone Sialoprotein, and Osteopontin. Biochem. Biophys. Res. Commun. 2001, 280, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Bal, B.S.; Gorski, J.P. Calcium and Collagen Binding Properties of Osteopontin, Bone Sialoprotein, and Bone Acidic Glycoprotein-75 from Bone. J. Biol. Chem. 1992, 267, 24871–24878. [Google Scholar] [CrossRef]
- Kläning, E.; Christensen, B.; Sørensen, E.S.; Vorup-Jensen, T.; Jensen, J.K. Osteopontin Binds Multiple Calcium Ions with High Affinity and Independently of Phosphorylation Status. Bone 2014, 66, 90–95. [Google Scholar] [CrossRef]
- Hunter, G.K.; O’Young, J.; Grohe, B.; Karttunen, M.; Goldberg, H.A. The Flexible Polyelectrolyte Hypothesis of Protein-Biomineral Interaction. Langmuir 2010, 26, 18639–18646. [Google Scholar] [CrossRef] [PubMed]
- Huq, N.L.; Cross, K.J.; Ung, M.; Reynolds, E.C. A Review of Protein Structure and Gene Organisation for Proteins Associated with Mineralised Tissue and Calcium Phosphate Stabilisation Encoded on Human Chromosome 4. Arch. Oral Biol. 2004, 50, 599–609. [Google Scholar]
- Hunter, G.K.; Kyle, C.L.; Goldberg, H.A. Modulation of Crystal Formation by Bone Phosphoproteins: Structural Specificity of the Osteopontin-Mediated Inhibition of Hydroxyapatite Formation. Biochem. J. 1994, 300, 723–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, D.E.; Thula-Mata, T.; Toro, E.J.; Yeh, Y.-W.; Holt, C.; Holliday, L.S.; Gower, L.B. Multifunctional Role of Osteopontin in Directing Intrafibrillar Mineralization of Collagen and Activation of Osteoclasts. Acta Biomater. 2014, 10, 494–507. [Google Scholar] [PubMed] [Green Version]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin. A Multifunctional Molecule Regulating Chronic Inflammation and Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senger, D.R.; Perruzzi, C.A.; Papadopoulos-Sergiou, A.; Water, L.V.D. Adhesive Properties of Osteopontin: Regulation by a Naturally Occurring Thrombin-Cleavage in Close Proximity to the Grgds Cell-Binding Domain. Mol. Biol. Cell. 1994, 5, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, N.M.T.; Hoac, B.; Neves, R.L.; Addison, W.N.; Assis, D.M.; Murshed, M.; Carmona, A.K.; McKee, M.D. Proteolytic Processing of Osteopontin by Phex and Accumulation of Osteopontin Fragments in Hyp Mouse Bone, the Murine Model of X-Linked Hypophosphatemia. J. Bone Miner. Res. 2013, 28, 688–699. [Google Scholar]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. 2020, 26, e919159-1. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Pilkington, M.F.; Lakkakorpi, P.T.; Lipfert, L.; Sims, S.M.; Dixon, S.J.; Rodan, G.A.; Duong, L.T. Role of Alpha(V)Beta(3) Integrin in Osteoclast Migration and Formation of the Sealing Zone. J. Cell Sci. 1999, 112, 3985–3993. [Google Scholar] [PubMed]
- Duong, L.T.; Lakkakorpi, P.I.; Nakamura, I.; Rodan, G.A. Integrins and Signaling in Osteoclast Function. Matrix Biol. 2000, 19, 97–105. [Google Scholar] [PubMed]
- Duong, L.T.; Rodan, G.A. Regulation of Osteoclast Formation and Function. Rev. Endocr. Metab. Disord. 2001, 2, 95–104. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Coupling the Activities of Bone Formation and Resorption: A Multitude of Signals within the Basic Multicellular Unit. BoneKEy Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjidakis, D.J.; Androulakis, I.I. Bone Remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, Not Osteoblasts or Lining Cells, Are the Main Source of the Rankl Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Targeting Osteoclast-Osteoblast Communication. Nat. Med. 2011, 17, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of Bone Resorption and Formation by Rankl Reverse Signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar]
- Wingender, B.; Tong, E.; Emery, J.; Gower, L.; Kröger, R. Time-Resolved in Situ Raman Spectroscopic Observations of a Biomineralization Model System. Microsc. Microanal. 2019, 25, 826–827. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.A.; Abrahamson, D.R.; John, P.L.S.; Clapp, W.L.; Williams, M.J.; Terada, N.; Hamazaki, T.; Ellison, G.W.; Batich, C.D. Mouse Stem Cells Seeded into Decellularized Rat Kidney Scaffolds Endothelialize and Remodel Basement Membranes. Organogenesis 2012, 8, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Lovett, A.C.; Khan, S.R.; Gower, L.B. Development of a Two-Stage in Vitro Model System to Investigate the Mineralization Mechanisms Involved in Idiopathic Stone Formation: Stage 1—Biomimetic Randall’s Plaque Using Decellularized Porcine Kidneys. Urolithiasis 2019, 47, 321–334. [Google Scholar]
- Mark, J.; Ngai, K.; Graessley, W.; Mandelkern, L.; Samulski, E.; Koenig, J.; Wingall, G. The Crystalline State. In Physical Properties of Polymers, 3rd ed.; Cambridge University Press: New York, NY, USA, 2004. [Google Scholar]
- Tye, C.E.; Hunter, G.K.; Goldberg, H.A. Identification of the Type I Collagen-Binding Domain of Bone Sialoprotein and Characterization of the Mechanism of Interaction. J. Biol. Chem. 2005, 280, 13487–13492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.M.; Schwartz, J.L.; Giachelli, C.M.; Ratner, B.D. Enhancing the Biological Activity of Immobilized Osteopontin Using a Type-1 Collagen Affinity Coating. J. Biomed. Mater. Res. Part A 2004, 70, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Schwartz, S.M.; Liaw, L. Molecular and Cellular Biology of Osteopontin: Potential Role in Cardiovascular Disease. Trends Cardiovasc. Med. 1995, 5, 88–95. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Choo, J.-E.; Park, H.-J.; Park, J.-B.; Lee, S.-C.; Jo, I.; Lee, S.-J.; Chung, C.-P.; Park, Y.-J. Injectable Gel with Synthetic Collagen-Binding Peptide for Enhanced Osteogenesis in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2007, 357, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Thula, T.T.; Svedlund, F.; Rodriguez, D.E.; Podschun, J.; Pendi, L.; Gower, L.B. Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents. Polymers 2011, 3, 10–35. [Google Scholar] [PubMed] [Green Version]
- Antebi, B.; Cheng, X.; Harris, J.N.; Gower, L.B.; Chen, X.-D.; Ling, J. Biomimetic Collagen-Hydroxyapatite Composite Fabricated Via a Novel Perfusion-Flow Mineralization Technique. Tissue Eng. Part C 2013, 19, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Landis, W.J.; Song, M.J.; Leith, A.; McEwen, L.; McEwen, B.F. Mineral and Organic Matrix Interaction in Normally Calcifying Tendon Visualized in Three Dimensions by High-Voltage Electron Microscopic Tomography and Graphic Image Reconstruction. J. Struct. Biol. 1993, 110, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E. Role of Collagen Fibrils in Calcification. In Calcification of Biological Systems; Bonucci, E., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 19–39. [Google Scholar]
- Liu, Y.; Kim, Y.-K.; Dai, L.; Li, N.; Khan, S.O.; Pashley, D.H.; Tay, F.R. Hierarchical and Non-Hierarchical Mineralisation of Collagen. Biomaterials 2011, 32, 1291–1300. [Google Scholar] [PubMed] [Green Version]
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; de With, G.; Sommerdijk, N.A.J.M. The Role of Collagen in Bone Apatite Formation in the Presence of Hydroxyapatite Nucleation Inhibitors. Nat. Mater. 2010, 9, 1004–1009. [Google Scholar] [PubMed] [Green Version]
- Xu, Y.; Nudelman, F.; Eren, E.D.; Wirix, M.J.M.; Cantaert, B.; Nijhuis, W.H.; Hermida-Merino, D.; Portale, G.; Bomans, P.H.H.; Ottmann, C.; et al. Intermolecular Channels Direct Crystal Orientation in Mineralized Collagen. Nat. Commun. 2020, 11, 5068. [Google Scholar] [PubMed]
- Cantaert, B.; Beniash, E.; Meldrum, F.C. The Role of Poly(Aspartic Acid) in the Precipitation of Calcium Phosphate in Confinement. J. Mater. Chem. B 2013, 1, 6586–6595. [Google Scholar]
- Orgel, J.; Irving, T.C.; Miller, A.; Wess, T.J. Microfibrillar Structure of Type I Collagen in Situ. Proc. Natl. Acad. Sci. USA 2006, 103, 9001–9005. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, N.; Qi, Y.-p.; Dai, L.; Bryan, T.E.; Mao, J.; Pashley, D.H.; Tay, F.R. Intrafibrillar Collagen Mineralization Produced by Biomimetic Hierarchical Nanoapatite Assembly. Adv. Mater. 2011, 23, 975–980. [Google Scholar]
- Dai, L.; Qi, Y.-P.; Niu, L.-N.; Liu, Y.; Pucci, C.R.; Looney, S.W.; Ling, J.-Q.; Pashley, D.H.; Tay, F.R. Inorganic–Organic Nanocomposite Assembly Using Collagen as a Template and Sodium Tripolyphosphate as a Biomimetic Analog of Matrix Phosphoprotein. Cryst. Growth Des. 2011, 11, 3504–3511. [Google Scholar]
- Gu, L.; Kim, Y.K.; Liu, Y.; Ryou, H.; Wimmer, C.E.; Dai, L.; Arola, D.D.; Looney, S.W.; Pashley, D.H.; Tay, F.R. Biomimetic Analogs for Collagen Biomineralization. J. Dent. Res. 2011, 90, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Y.; Zhao, W.; Wang, Z.; Landis, W.J.; Cui, Q.; Sahai, N. Molecular Mechanisms for Intrafibrillar Collagen Mineralization in Skeletal Tissues. Biomaterials 2015, 39, 59–66. [Google Scholar] [PubMed]
- Lenton, S.; Wang, Q.; Nylander, T.; Teixeira, S.; Holt, C. Structural Biology of Calcium Phosphate Nanoclusters Sequestered by Phosphoproteins. Crystals 2020, 10, 755. [Google Scholar]
- Ibsen, C.J.S.; Gebauer, D.; Birkedal, H. Osteopontin Stabilizes Metastable States Prior to Nucleation during Apatite Formation. Chem. Mater. 2016, 28, 8550–8555. [Google Scholar] [CrossRef]
- Bewernitz, M.A.; Gebauer, D.; Long, J.; Cölfen, H.; Gower, L.B. A Metastable Liquid Precursor Phase of Calcium Carbonate and Its Interactions with Polyaspartate. Faraday Discuss. 2012, 159, 291–312. [Google Scholar]
- Shipovskov, S.; Oliveira, C.L.; Hoffmann, S.V.; Schauser, L.; Sutherland, D.S.; Besenbacher, F.; Pedersen, J.S. Water-in-Oil Micro-Emulsion Enhances the Secondary Structure of a Protein by Confinement. Chemphyschem 2012, 13, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, E.; Liu, T.; Olszta, M.J.; Gower, L.B. Laser Light Scattering Studies of a Polymer-Induced Liquid-Precursor (PILP) Process for Mineralization. In Biological and Bio-Inspired Materials and Devices; Sandhage, K.H., Yang, S., Douglas, T., Parker, A.R., DiMasi, E., Eds.; Materials Research Society: Warrendale, PA, USA, 2005; Volume 873E, pp. 177–183. [Google Scholar]
- Toroian, D.; Lim, J.E.; Price, P.A. The Size Exclusion Characteristics of Type I Collagen—Implications for the Role of Noncollagenous Bone Constituents in Mineralization. J. Biol. Chem. 2007, 282, 22437–22447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, S.H.; Grier, D.G. The Charge of Glass and Silica Surfaces. J. Chem. Phys. 2008, 115, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gower, L.; Elias, J. Colloid Assembly and Transformation (Cat): The Relationship of PILP to Biomineralization. J. Struct. Biol. X 2022, 6, 100059. [Google Scholar] [CrossRef]
- Lönnerdal, B.; Kvistgaard, A.S.; Peerson, J.M.; Donovan, S.M.; Peng, Y. Growth, Nutrition, and Cytokine Response of Breast-Fed Infants and Infants Fed Formula with Added Bovine Osteopontin. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 650–657. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Droplet Size (nm) | Droplet Concentration (per mL) |
|---|---|---|
| OPN Mix | 81 ± 41 | 4.44 × 108 ± 0.82 × 108 |
| 27 kDa pAsp | 92 ± 50 | 1.54 × 108 ± 0.16 × 108 |
| No Polymer | 83 ± 49 | 2.17 × 108 ± 0.58 × 108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saxena, N.; Mizels, J.; Cremer, M.A.; Guarnizo, V.; Rodriguez, D.E.; Gower, L.B. Comparison of Synthetic vs. Biogenic Polymeric Process-Directing Agents for Intrafibrillar Mineralization of Collagen. Polymers 2022, 14, 775. https://doi.org/10.3390/polym14040775
Saxena N, Mizels J, Cremer MA, Guarnizo V, Rodriguez DE, Gower LB. Comparison of Synthetic vs. Biogenic Polymeric Process-Directing Agents for Intrafibrillar Mineralization of Collagen. Polymers. 2022; 14(4):775. https://doi.org/10.3390/polym14040775
Chicago/Turabian StyleSaxena, Neha, Joshua Mizels, Maegan A. Cremer, Vanessa Guarnizo, Douglas E. Rodriguez, and Laurie B. Gower. 2022. "Comparison of Synthetic vs. Biogenic Polymeric Process-Directing Agents for Intrafibrillar Mineralization of Collagen" Polymers 14, no. 4: 775. https://doi.org/10.3390/polym14040775
APA StyleSaxena, N., Mizels, J., Cremer, M. A., Guarnizo, V., Rodriguez, D. E., & Gower, L. B. (2022). Comparison of Synthetic vs. Biogenic Polymeric Process-Directing Agents for Intrafibrillar Mineralization of Collagen. Polymers, 14(4), 775. https://doi.org/10.3390/polym14040775