
Self-Assembled Amphiphilic Fluorinated Random Copolymers for the Encapsulation and Release of the Hydrophobic Combretastatin A-4 Drug


 , , , ,
, , , ,  and
and 
Abstract
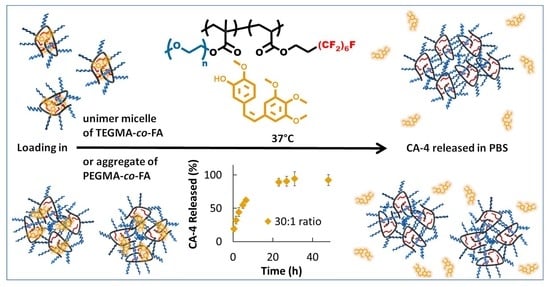
:
1. Introduction
2. Results and Discussion
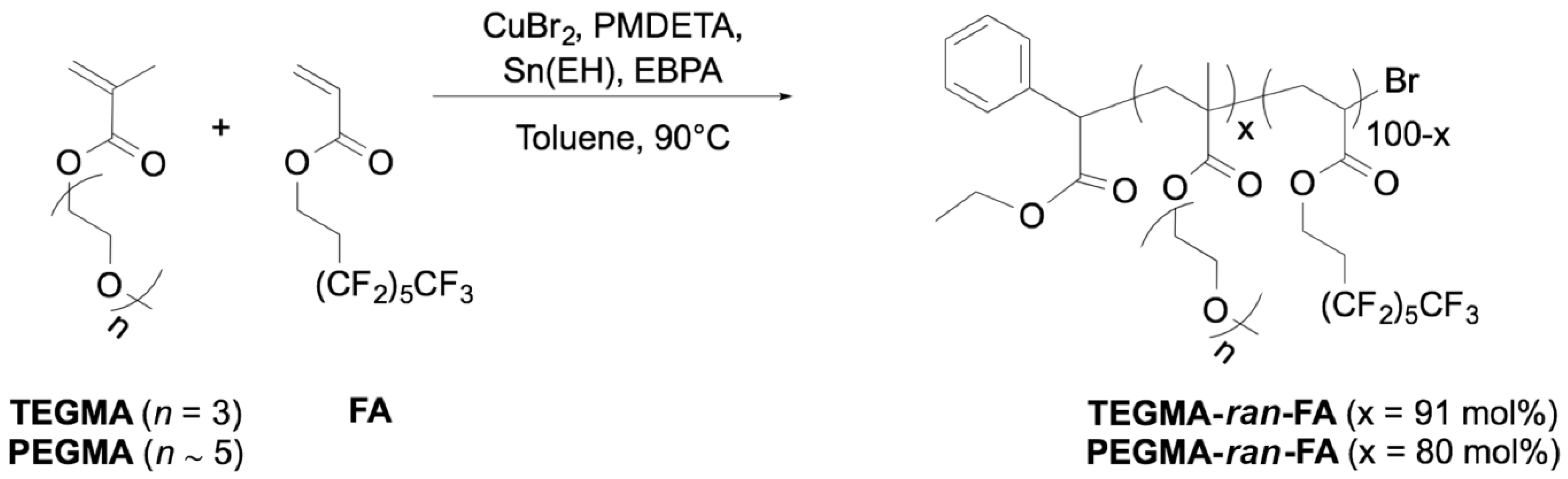
2.1. Syntheses of the Copolymers
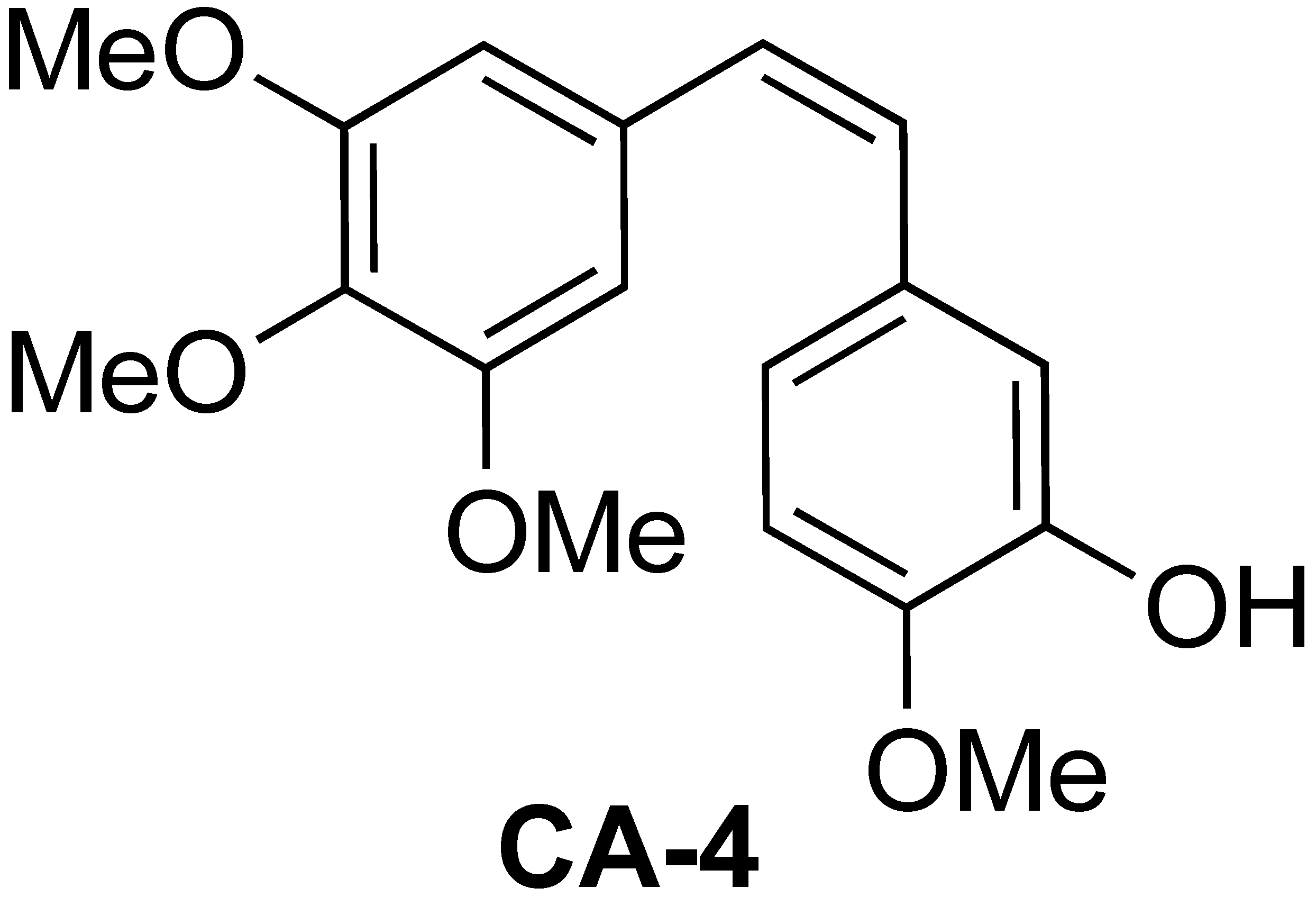
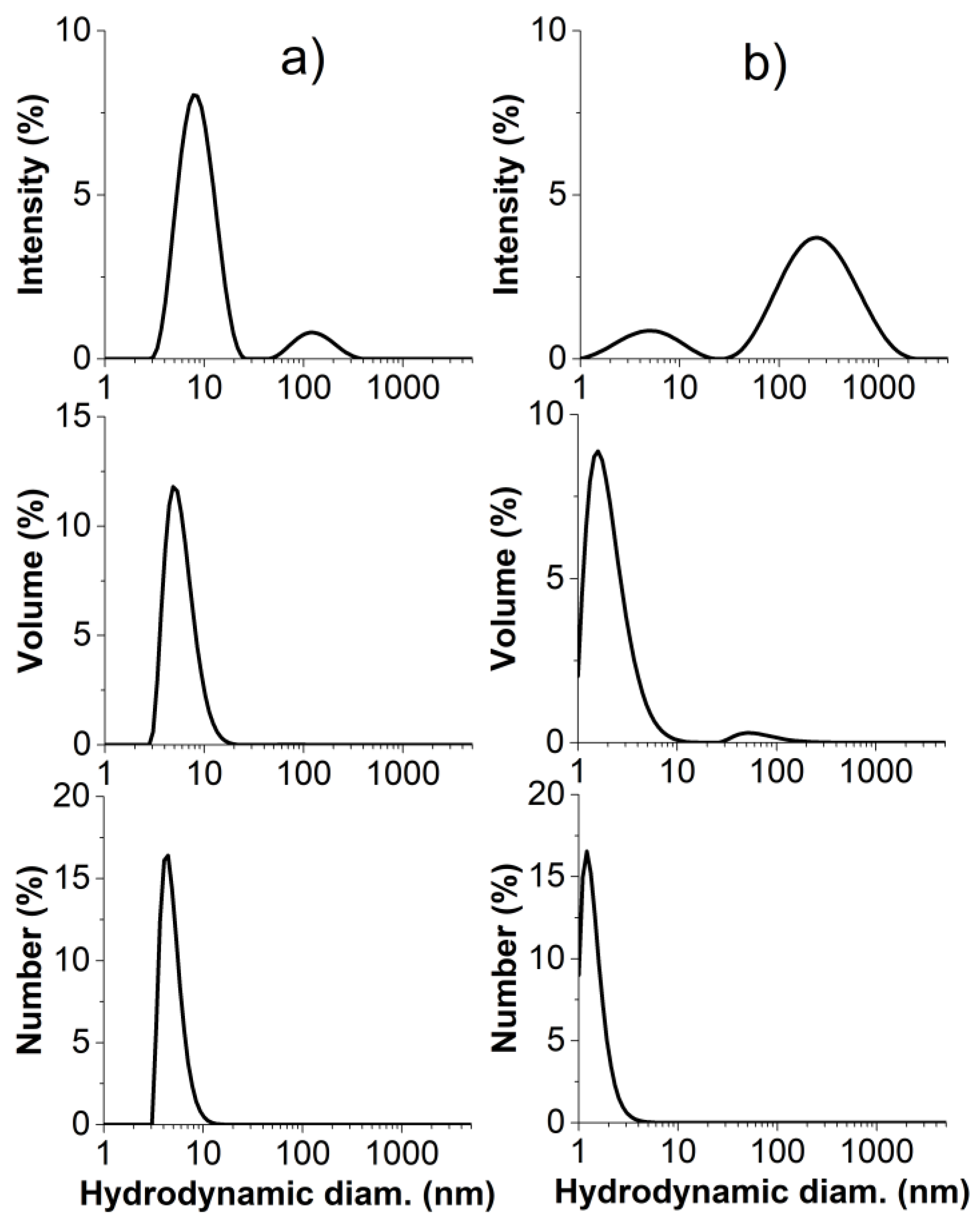
2.2. Self-Assembling Behavior of Amphiphilic Copolymers
2.3. Drug Encapsulation
2.4. Drug Release
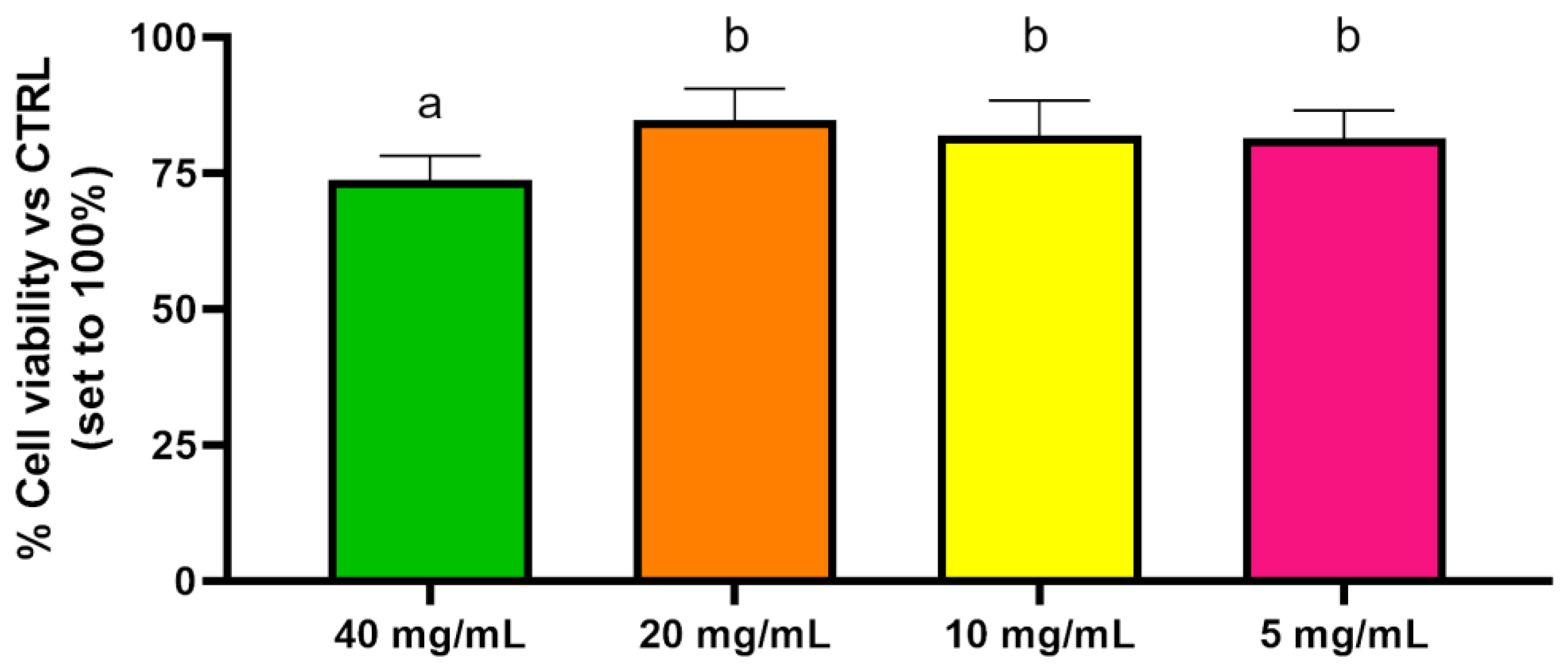
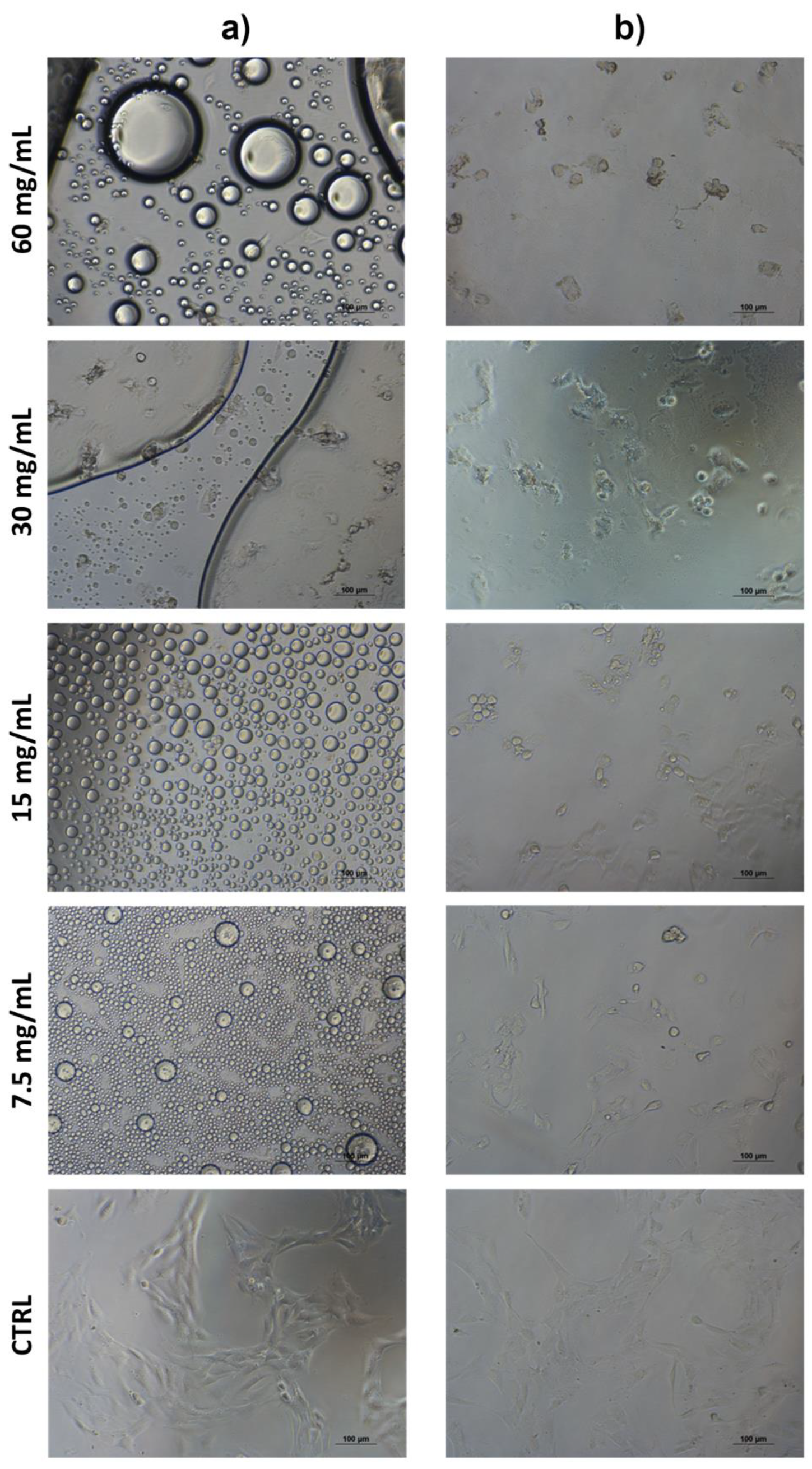
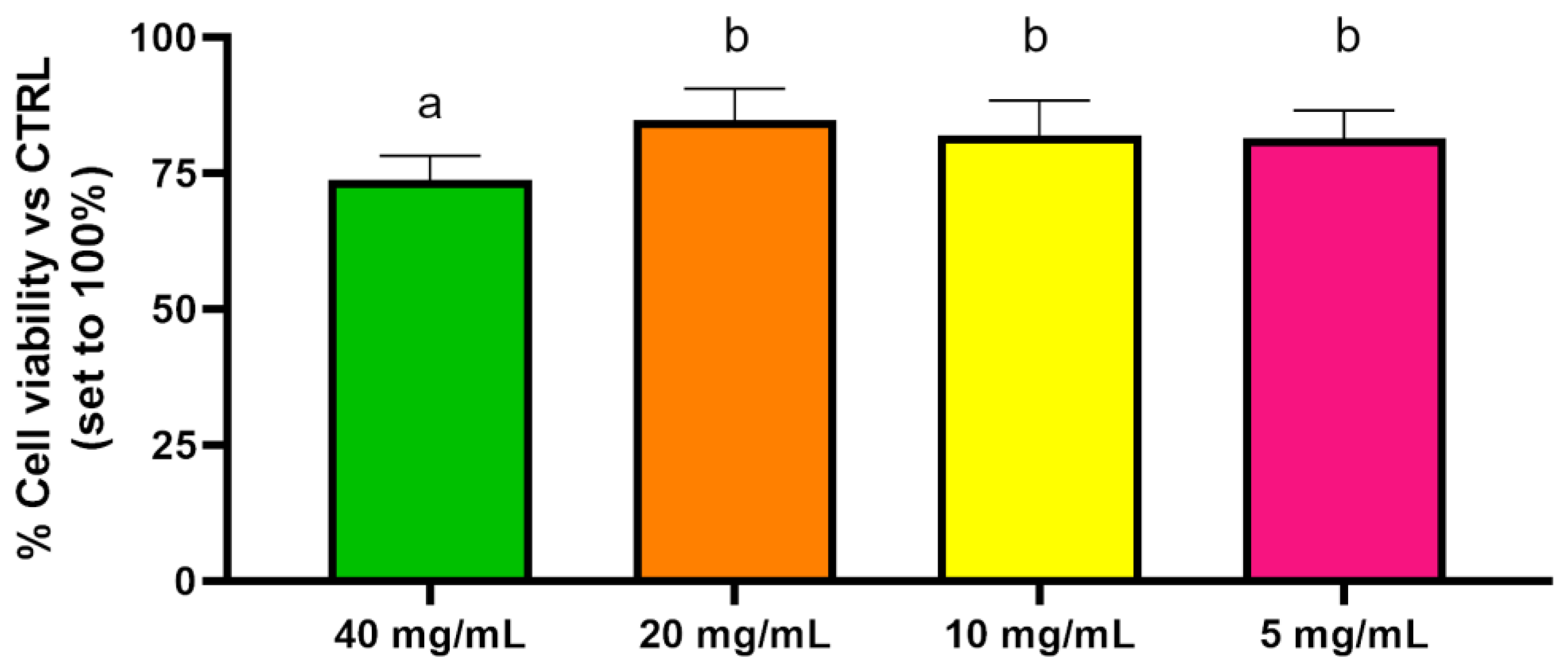
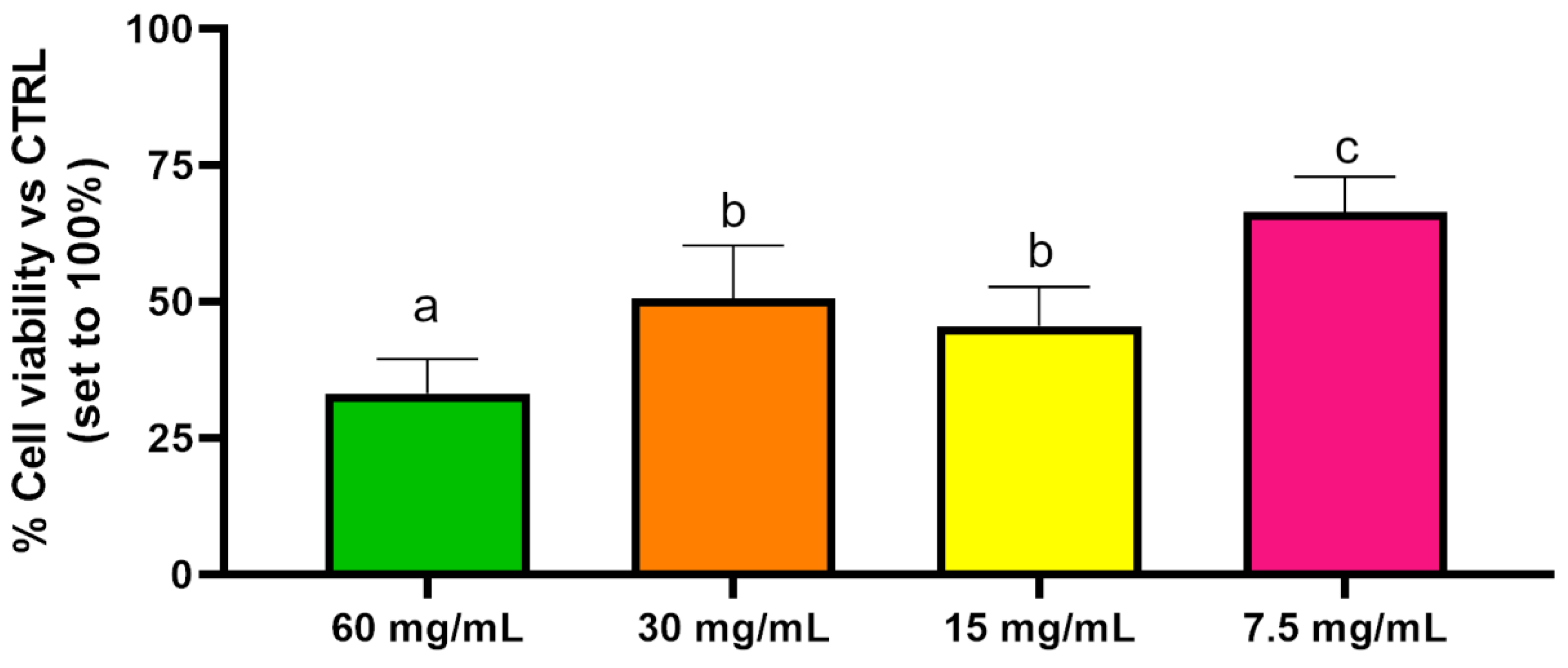
2.5. Cytotoxicity Assay
3. Materials and Methods
3.1. Polymer Synthesis
3.1.1. Materials
3.1.2. Synthesis of Copolymer TEGMA-ran-FA
3.1.3. Synthesis of Copolymer PEGMA-ran-FA
3.2. Characterization
3.3. Solubility Assay for Combretastatin A-4 in Water
3.4. Preparation of Combretastatin A-4 Loaded Micelles
3.5. In Vitro Drug Release
3.6. Cytotoxicity Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Burgos, M.; Latorre-Sanchez, A.; Pomposo, J.A. Advances in Single Chain Technology. Chem. Soc. Rev. 2015, 44, 6122–6142. [Google Scholar] [CrossRef] [Green Version]
- Lyon, C.K.; Prasher, A.; Hanlon, A.M.; Tuten, B.T.; Tooley, C.A.; Frank, P.G.; Berda, E.B. A Brief User’s Guide to Single-Chain Nanoparticles. Polym. Chem. 2014, 6, 181–197. [Google Scholar] [CrossRef]
- Altintas, O.; Fischer, T.S.; Barner-Kowollik, C. Synthetic Methods Toward Single-Chain Polymer Nanoparticles. In Single-Chain Polymer Nanoparticles; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–45. ISBN 978-3-527-80638-6. [Google Scholar]
- Alqarni, M.A.M.; Waldron, C.; Yilmaz, G.; Becer, C.R. Synthetic Routes to Single Chain Polymer Nanoparticles (SCNPs): Current Status and Perspectives. Macromol. Rapid Commun. 2021, 42, 2100035. [Google Scholar] [CrossRef]
- Altintas, O.; Barner-Kowollik, C. Single Chain Folding of Synthetic Polymers by Covalent and Non-Covalent Interactions: Current Status and Future Perspectives. Macromol Rapid Commun 2012, 33, 958–971. [Google Scholar] [CrossRef]
- Seo, M.; Beck, B.J.; Paulusse, J.M.J.; Hawker, C.J.; Kim, S.Y. Polymeric Nanoparticles via Noncovalent Cross-Linking of Linear Chains. Macromolecules 2008, 41, 6413–6418. [Google Scholar] [CrossRef]
- Terashima, T. Controlled Self-Assembly of Amphiphilic Random Copolymers into Folded Micelles and Nanostructure Materials. J. Oleo Sci. 2020. [Google Scholar] [CrossRef]
- Terashima, T.; Sawamoto, M. Single-Chain Nanoparticles via Self-Folding Amphiphilic Copolymers in Water. In Single-Chain Polymer Nanoparticles; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 313–339. ISBN 978-3-527-80638-6. [Google Scholar]
- Morishima, Y.; Nomura, S.; Ikeda, T.; Seki, M.; Kamachi, M. Characterization of Unimolecular Micelles of Random Copolymers of Sodium 2-(Acrylamido)-2-Methylpropanesulfonate and Methacrylamides Bearing Bulky Hydrophobic Substituents. Macromolecules 1995, 28, 2874–2881. [Google Scholar] [CrossRef]
- Pomposo, J.A.; Perez-Baena, I.; Lo Verso, F.; Moreno, A.J.; Arbe, A.; Colmenero, J. How Far Are Single-Chain Polymer Nanoparticles in Solution from the Globular State? ACS Macro Lett. 2014, 3, 767–772. [Google Scholar] [CrossRef]
- Kröger, A.P.P.; Paulusse, J.M.J. Single-Chain Polymer Nanoparticles in Controlled Drug Delivery and Targeted Imaging. J. Control. Release 2018, 286, 326–347. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Koda, Y.; Terashima, T.; Sawamoto, M.; Maynard, H.D. Amphiphilic/Fluorous Random Copolymers as a New Class of Non-Cytotoxic Polymeric Materials for Protein Conjugation. Polym. Chem. 2014, 6, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Koda, Y.; Terashima, T.; Sawamoto, M. Multimode Self-Folding Polymers via Reversible and Thermoresponsive Self-Assembly of Amphiphilic/Fluorous Random Copolymers. Macromolecules 2016, 49, 4534–4543. [Google Scholar] [CrossRef]
- Durmaz, Y.Y.; Sahkulubey, E.L.; Yagci, Y.; Martinelli, E.; Galli, G. A Novel Poly(p -Phenylene) Containing Alternating Poly(Perfluorooctylethyl Acrylate- Co -Methyl Methacrylate) and Polystyrene Grafts by Combination of Atom Transfer Radical Polymerization and Suzuki Coupling Processes. J. Polym. Sci. A Polym. Chem. 2012, 50, 4911–4919. [Google Scholar] [CrossRef]
- Galli, G.; Martinelli, E.; Chiellini, E.; Ober, C.K.; Glisenti, A. Low Surface Energy Characteristics of Mesophase-Forming ABC and ACB Triblock Copolymers with Fluorinated B Blocks. Molecular Crystals and Liquid Crystals 2005, 441, 211–226. [Google Scholar] [CrossRef]
- Martinelli, E.; Glisenti, A.; Gallot, B.; Galli, G. Surface Properties of Mesophase-Forming Fluorinated Bicycloacrylate/Polysiloxane Methacrylate Copolymers. Macromol. Chem. Phys. 2009, 210, 1746–1753. [Google Scholar] [CrossRef]
- Guazzelli, E.; Masotti, E.; Calosi, M.; Kriechbaum, M.; Uhlig, F.; Galli, G.; Martinelli, E. Single-Chain Folding and Self-Assembling of Amphiphilic Polyethyleneglycol-Modified Fluorinated Styrene Homopolymers in Water Solution. Polymer 2021, 231, 124107. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Galli, G.; Telling, M.T.F.; Poggetto, G.D.; Immirzi, B.; Domenici, F.; Paradossi, G. Prolate and Temperature-Responsive Self-Assemblies of Amphiphilic Random Copolymers with Perfluoroalkyl and Polyoxyethylene Side Chains in Solution. Macromol. Chem. Phys. 2018, 219, 1800210. [Google Scholar] [CrossRef]
- Guazzelli, E.; Martinelli, E.; Galli, G.; Cupellini, L.; Jurinovich, S.; Mennucci, B. Single-Chain Self-Folding in an Amphiphilic Copolymer: An Integrated Experimental and Computational Study. Polymer 2019, 161, 33–40. [Google Scholar] [CrossRef]
- Guazzelli, E.; Masotti, E.; Biver, T.; Pucci, A.; Martinelli, E.; Galli, G. The Self-Assembly over Nano- to Submicro-Length Scales in Water of a Fluorescent Julolidine-Labeled Amphiphilic Random Terpolymer. J. Polym. Sci. Part. A Polym. Chem. 2018, 56, 797–804. [Google Scholar] [CrossRef]
- Domenici, F.; Guazzelli, E.; Masotti, E.; Mahmoudi, N.; Gabrielli, S.; Telling, M.T.F.; Martinelli, E.; Galli, G.; Paradossi, G. Understanding the Temperature-Responsive Self-Assemblies of Amphiphilic Random Copolymers by SANS in D 2 O Solution. Macromol. Chem. Phys. 2021, 222, 2000447. [Google Scholar] [CrossRef]
- Chen, J.; Li, K.; Shon, J.S.L.; Zimmerman, S.C. Single-Chain Nanoparticle Delivers a Partner Enzyme for Concurrent and Tandem Catalysis in Cells. J. Am. Chem. Soc. 2020, 142, 4565–4569. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Jia, D.; Ma, X.; Liang, M.; Xue, P.; Kang, Y.; Xu, Z. Cylindrical Polymer Brushes-Anisotropic Unimolecular Micelle Drug Delivery System for Enhancing the Effectiveness of Chemotherapy. Bioact. Mater. 2021, 6, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Verde-Sesto, E.; Arbe, A.; Moreno, A.J.; Cangialosi, D.; Alegría, A.; Colmenero, J.; Pomposo, J.A. Single-Chain Nanoparticles: Opportunities Provided by Internal and External Confinement. Mater. Horiz. 2020, 7, 2292–2313. [Google Scholar] [CrossRef]
- Kröger, A.P.P.; Hamelmann, N.M.; Juan, A.; Lindhoud, S.; Paulusse, J.M.J. Biocompatible Single-Chain Polymer Nanoparticles for Drug Delivery—A Dual Approach. ACS Appl. Mater. Interfaces 2018, 10, 30946–30951. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Xing, H.; Vincil, G.A.; Lee, J.; Henderson, E.J.; Lu, Y.; Lemcoff, N.G.; Zimmerman, S.C. Practical Synthesis of Water-Soluble Organic Nanoparticles with a Single Reactive Group and a Functional Carrier Scaffold. Chem. Sci. 2014, 5, 2862–2868. [Google Scholar] [CrossRef]
- Bai, Y.; Xing, H.; Wu, P.; Feng, X.; Hwang, K.; Lee, J.M.; Phang, X.Y.; Lu, Y.; Zimmerman, S.C. Chemical Control over Cellular Uptake of Organic Nanoparticles by Fine Tuning Surface Functional Groups. ACS Nano 2015, 9, 10227–10236. [Google Scholar] [CrossRef]
- Liu, Y.; Pujals, S.; Stals, P.J.M.; Paulöhrl, T.; Presolski, S.I.; Meijer, E.W.; Albertazzi, L.; Palmans, A.R.A. Catalytically Active Single-Chain Polymeric Nanoparticles: Exploring Their Functions in Complex Biological Media. J. Am. Chem. Soc. 2018, 140, 3423–3433. [Google Scholar] [CrossRef] [Green Version]
- Hamelmann, N.M.; Paats, J.-W.D.; Paulusse, J.M.J. Cytosolic Delivery of Single-Chain Polymer Nanoparticles. ACS Macro Lett. 2021, 10, 1443–1449. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Akbari, S.; Etxeberria, A.; Arbe, A.; Gasser, U.; Moreno, A.J.; Colmenero, J.; Pomposo, J.A. “Michael” Nanocarriers Mimicking Transient-Binding Disordered Proteins. ACS Macro Lett. 2013, 2, 491–495. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Akbari, S.; Moreno, A.J.; Verso, F.L.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Design and Preparation of Single-Chain Nanocarriers Mimicking Disordered Proteins for Combined Delivery of Dermal Bioactive Cargos. Macromolecular Rapid Communications 2013, 34, 1681–1686. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Huang, S.-Y.; Fan, W.-L.; Lee, A.-W.; Chiu, C.-W.; Lee, D.-J.; Lai, J.-Y. Water-Soluble Single-Chain Polymeric Nanoparticles for Highly Selective Cancer Chemotherapy. ACS Appl. Polym. Mater. 2021, 3, 474–484. [Google Scholar] [CrossRef]
- Bartolini, A.; Tempesti, P.; Resta, C.; Berti, D.; Smets, J.; Aouad, Y.G.; Baglioni, P. Poly(Ethylene Glycol)-Graft-Poly(Vinyl Acetate) Single-Chain Nanoparticles for the Encapsulation of Small Molecules. Phys. Chem. Chem. Phys. 2017, 19, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Bhattacharya, A.; Terashima, T.; Sawamoto, M.; Maynard, H.D. Amphiphilic Fluorous Random Copolymer Self-Assembly for Encapsulation of a Fluorinated Agrochemical. J. Polym. Sci. Part. A Polym. Chem. 2019, 57, 352–359. [Google Scholar] [CrossRef]
- Martinelli, E.; Annunziata, L.; Guazzelli, E.; Pucci, A.; Biver, T.; Galli, G. The Temperature-Responsive Nanoassemblies of Amphiphilic Random Copolymers Carrying Poly(Siloxane) and Poly(Oxyethylene) Pendant Chains. Macromol. Chem. Phys. 2018, 219, 1800082. [Google Scholar] [CrossRef]
- Bordat, A.; Boissenot, T.; Nicolas, J.; Tsapis, N. Thermoresponsive Polymer Nanocarriers for Biomedical Applications. Adv. Drug Deliv. Rev. 2019, 138, 167–192. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Lee, D.-J.; Liao, Z.-S.; Huang, J.-J. Stimuli-Responsive Single-Chain Polymeric Nanoparticles towards the Development of Efficient Drug Delivery Systems. Polym. Chem. 2016, 7, 6164–6169. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garcia-Kendal, D. Isolation and Structure of the Strong Cell Growth and Tubulin Inhibitor Combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef]
- Griggs, J.; Metcalfe, J.C.; Hesketh, R. Targeting Tumour Vasculature: The Development of Combretastatin A4. Lancet Oncol. 2001, 2, 82–87. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Lin, C.M.; Ho, H.H.; Pettit, G.R.; Hamel, E. Antimitotic Natural Products Combretastatin A-4 and Combretastatin A-2: Studies on the Mechanism of Their Inhibition of the Binding of Colchicine to Tubulin. Biochemistry 1989, 28, 6984–6991. [Google Scholar] [CrossRef]
- Chaplin, D.J.; Horsman, M.R.; Siemann, D.W. Current Development Status of Small-Molecule Vascular Disrupting Agents. Curr. Opin. Investig. Drugs 2006, 7, 522–528. [Google Scholar] [PubMed]
- Hinnen, P.; Eskens, F.A.L.M. Vascular Disrupting Agents in Clinical Development. Br. J. Cancer 2007, 96, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippert, J.W. Vascular Disrupting Agents. Bioorg. Med. Chem. 2007, 15, 605–615. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.L.; Price, P. Combretastatin A4 Phosphate. Anti-Cancer Drugs 2004, 15, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.M.; Rustin, G.J.S. Vascular Damaging Agents. Clin. Oncol. 2007, 19, 443–456. [Google Scholar] [CrossRef]
- Nagaiah, G.; Remick, S.C. Combretastatin A4 Phosphate: A Novel Vascular Disrupting Agent. Future Oncol. 2010, 6, 1219–1228. [Google Scholar] [CrossRef]
- Nallamothu, R.; Wood, G.C.; Kiani, M.F.; Moore, B.M.; Horton, F.P.; Thoma, L.A. A Targeted Liposome Delivery System for Combretastatin A4: Formulation Optimization through Drug Loading and in Vitro Release Studies. PDA J. Pharm. Sci. Technol. 2006, 60, 144–155. [Google Scholar]
- Liu, Z.; Shen, N.; Tang, Z.; Zhang, D.; Ma, L.; Yang, C.; Chen, X. An Eximious and Affordable GSH Stimulus-Responsive Poly(α-Lipoic Acid) Nanocarrier Bonding Combretastatin A4 for Tumor Therapy. Biomater. Sci. 2019, 7, 2803–2811. [Google Scholar] [CrossRef]
- Zaid, A.N.; Hassan, M.; Jaradat, N.; Assali, M.; Al-Abbassi, R.; Alkilany, A.; Abulateefeh, S.R. Formulation and Characterization of Combretastatin A4 Loaded PLGA Nanoparticles. Mater. Res. Express 2020, 6, 1250d7. [Google Scholar] [CrossRef]
- Zheng, Y.; Cheng, Y.; Chen, J.; Ding, J.; Li, M.; Li, C.; Wang, J.; Chen, X. Injectable Hydrogel–Microsphere Construct with Sequential Degradation for Locally Synergistic Chemotherapy. ACS Appl. Mater. Interfaces 2017, 9, 3487–3496. [Google Scholar] [CrossRef]
- Gallego-Yerga, L.; de la Torre, C.; Sansone, F.; Casnati, A.; Mellet, C.O.; García Fernández, J.M.; Ceña, V. Synthesis, Self-Assembly and Anticancer Drug Encapsulation and Delivery Properties of Cyclodextrin-Based Giant Amphiphiles. Carbohydr. Polym. 2021, 252, 117135. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-C.; Alani, A.W.G.; Rao, D.A.; Rockich, N.C.; Kwon, G.S. Multi-Drug Loaded Polymeric Micelles for Simultaneous Delivery of Poorly Soluble Anticancer Drugs. J. Control. Release 2009, 140, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Bey, E.A.; Dong, Y.; Weinberg, B.D.; Sutton, D.M.; Boothman, D.A.; Gao, J. β-Lapachone-Containing PEG-PLA Polymer Micelles as Novel Nanotherapeutics against NQO1-Overexpressing Tumor Cells. J. Control. Release 2007, 122, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heald, C.R.; Stolnik, S.; Kujawinski, K.S.; De Matteis, C.; Garnett, M.C.; Illum, L.; Davis, S.S.; Purkiss, S.C.; Barlow, R.J.; Gellert, P.R. Poly(Lactic Acid)−Poly(Ethylene Oxide) (PLA−PEG) Nanoparticles: NMR Studies of the Central Solidlike PLA Core and the Liquid PEG Corona. Langmuir 2002, 18, 3669–3675. [Google Scholar] [CrossRef]
- Hrkach, J.S.; Peracchia, M.T.; Bomb, A.; Lotan, N.; Langer, R. Nanotechnology for Biomaterials Engineering: Structural Characterization of Amphiphilic Polymeric Nanoparticles by 1H NMR Spectroscopy. Biomaterials 1997, 18, 27–30. [Google Scholar] [CrossRef]
- Sun, X.-L.; Tsai, P.-C.; Bhat, R.; Bonder, E.M.; Michniak-Kohn, B.; Pietrangelo, A. Thermoresponsive Block Copolymer Micelles with Tunable Pyrrolidone-Based Polymer Cores: Structure/Property Correlations and Application as Drug Carriers. J. Mater. Chem. B 2015, 3, 814–823. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Chang, F.-C.; Kao, W.-Y.; Hwang, S.-M.; Liao, L.-C.; Chang, Y.-J.; Liang, M.-C.; Chen, J.-K.; Lee, D.-J. Highly Efficient Drug Delivery Systems Based on Functional Supramolecular Polymers: In Vitro Evaluation. Acta Biomater. 2016, 33, 194–202. [Google Scholar] [CrossRef]
- Miller, M.A.; Zachary, J.F. Mechanisms and Morphology of Cellular Injury, Adaptation, and Death. In Pathologic Basis of Veterinary Disease; Elsevier: Amsterdam, The Netherlands, 2017; pp. 2–43.e19. ISBN 978-0-323-35775-3. [Google Scholar]
- Gaukroger, K.; Hadfield, J.A.; Hepworth, L.A.; Lawrence, N.J.; McGown, A.T. Novel Syntheses of Cis and Trans Isomers of Combretastatin A-4. J. Org. Chem. 2001, 66, 8135–8138. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Comonomer Feed Ratio (mol/mol) 1 | Monomers/Initiator Feed Ratio (mol/mol) | Time (h) | Co-Unit Ratioin Copolymer (mol/mol) 1,2 | Mn2 (g/mol) | Mn3 (g/mol) | Đ3 |
|---|---|---|---|---|---|---|---|
| TEGMA-ran-FA | 93/7 | 60/1 | 26 | 91/9 | 16,700 | 13,200 | 1.29 |
| PEGMA-ran-FA | 80/20 | 50/1 | 18 | 80/20 | 17,800 | 13,600 | 1.22 |
| Copolymer | Dh (Peak 1) (nm) | Dh (Peak 2) (nm) | Dh above Tcp (nm) |
|---|---|---|---|
| TEGMA-ran-FA | 8 ± 2 | 120 ± 30 | 2500 ± 400 a |
| PEGMA-ran-FA | 6 ± 1 | 400 ± 100 | 600 ± 200 b |
| Copolymer | Copolymer/CA-4 Ratio (w/w) | EE (%) | DL (%) |
|---|---|---|---|
| TEGMA-ran-FA | |||
| 30/1 | 89 ± 9 | 3.0 ± 0.3 | |
| 17/1 | 81 ± 7 | 4.7 ± 0.4 | |
| PEGMA-ran-FA | |||
| 30/1 | 86 ± 10 | 2.9 ± 0.3 | |
| 19/1 | 88 ± 9 | 4.8 ± 0.7 | |
| 15/1 | 82 ± 14 | 5.5 ± 0.9 | |
| 10/1 | 82 ± 15 | 9.0 ± 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calosi, M.; Guazzelli, E.; Braccini, S.; Lessi, M.; Bellina, F.; Galli, G.; Martinelli, E. Self-Assembled Amphiphilic Fluorinated Random Copolymers for the Encapsulation and Release of the Hydrophobic Combretastatin A-4 Drug. Polymers 2022, 14, 774. https://doi.org/10.3390/polym14040774
Calosi M, Guazzelli E, Braccini S, Lessi M, Bellina F, Galli G, Martinelli E. Self-Assembled Amphiphilic Fluorinated Random Copolymers for the Encapsulation and Release of the Hydrophobic Combretastatin A-4 Drug. Polymers. 2022; 14(4):774. https://doi.org/10.3390/polym14040774
Chicago/Turabian StyleCalosi, Matteo, Elisa Guazzelli, Simona Braccini, Marco Lessi, Fabio Bellina, Giancarlo Galli, and Elisa Martinelli. 2022. "Self-Assembled Amphiphilic Fluorinated Random Copolymers for the Encapsulation and Release of the Hydrophobic Combretastatin A-4 Drug" Polymers 14, no. 4: 774. https://doi.org/10.3390/polym14040774
APA StyleCalosi, M., Guazzelli, E., Braccini, S., Lessi, M., Bellina, F., Galli, G., & Martinelli, E. (2022). Self-Assembled Amphiphilic Fluorinated Random Copolymers for the Encapsulation and Release of the Hydrophobic Combretastatin A-4 Drug. Polymers, 14(4), 774. https://doi.org/10.3390/polym14040774