
Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications


Abstract
1. Introduction
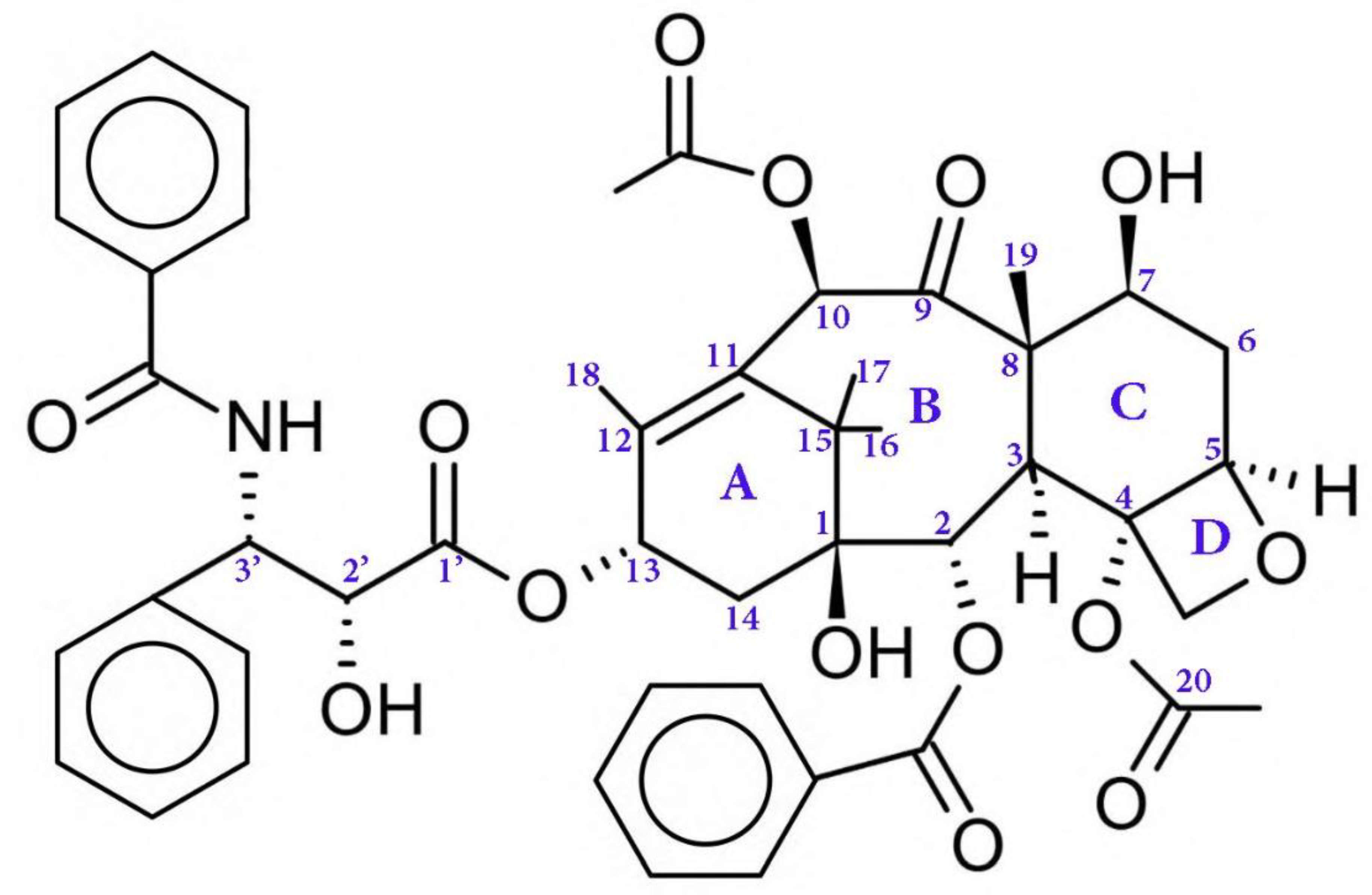
2. Paclitaxel
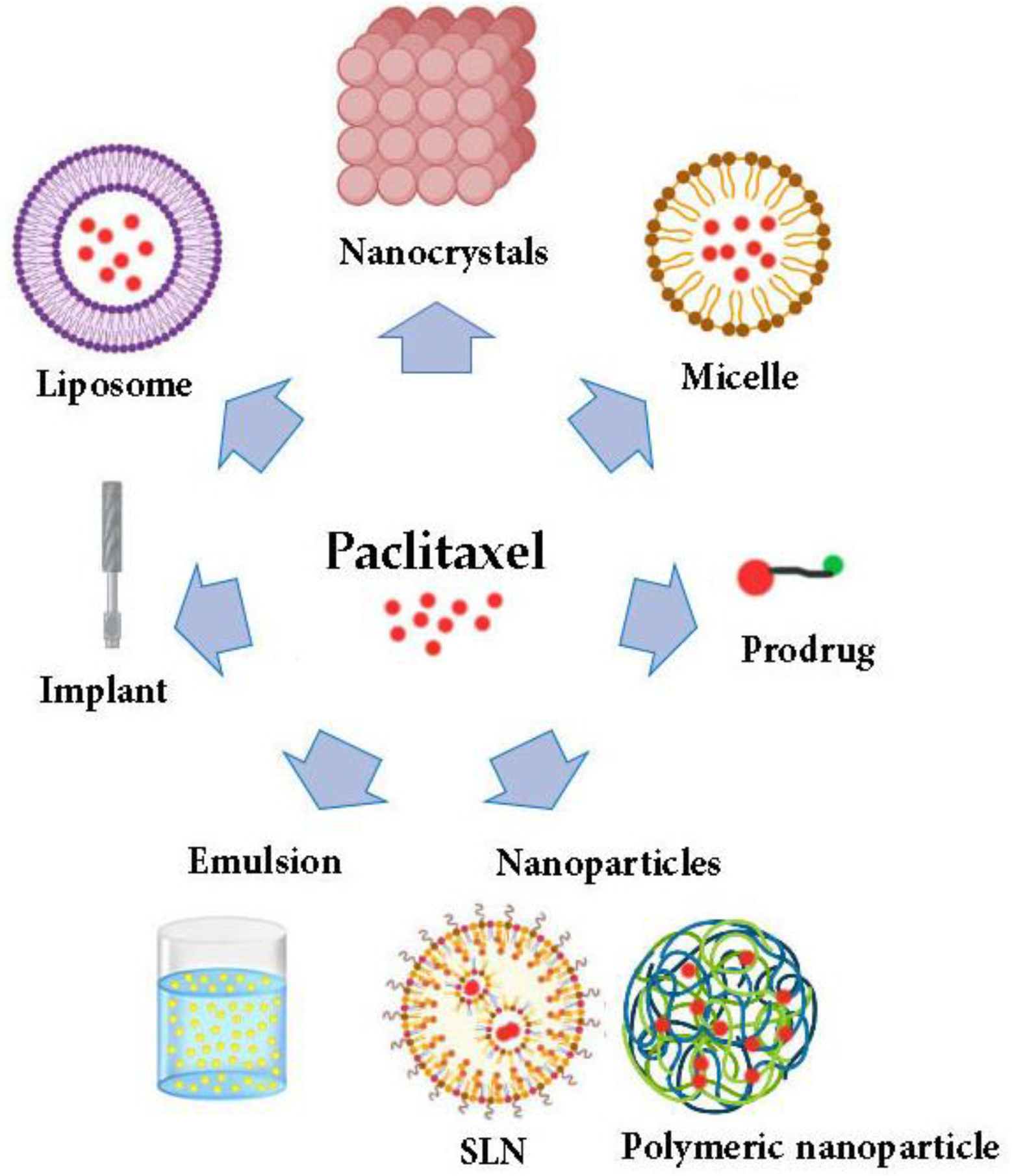
3. PTX Formulations
4. Drug Delivery of PTX
4.1. Micelles
4.2. Liposomes
4.3. Nanoparticles
4.3.1. Solid Lipid Nanoparticles
4.3.2. Polymeric Nanoparticles
Poly Lactic-co-Glycolic Acid (PLGA)
Chitosan
4.4. Prodrug Approach
4.5. Emulsions
4.6. Implants
4.7. Nanocrystals
5. PTX Nanocrystals
6. Future Aspects
7. Conclusions and Remarks
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Cancer-Fact Fheets. Available online: www.who.int/mediacentre/factsheets/fs297/en/12/9/20182/3/2020 (accessed on 2 March 2020).
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 5 March 2021).
- Patterson, R.; Fischman, V.G.; Wasserman, I.; Siu, J.; Shrime, M.G.; Fagan, J.J.; Koch, W.; Alkire, B.C. Global Burden of Head and Neck Cancer: Economic Consequences, Health, and the Role of Surgery. Otolaryngol. Neck Surg. 2020, 162, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Lopez-Olivo, M.; Advani, P.G.; Ning, M.S.; Geng, Y.; Giordano, S.H.; Volk, R.J. Financial Burdens of Cancer Treatment: A Systematic Review of Risk Factors and Outcomes. J. Natl. Compr. Cancer Netw. 2019, 17, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.-H.; Gertsch, J. Anticancer drugs from nature—Natural products as a unique source of new microtubule-stabilizing agents. Nat. Prod. Rep. 2007, 24, 327–357. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K. Signal events: Cell signal transduction and its inhibition in cancer. Oncologist 2003, 8, 5–17. [Google Scholar] [CrossRef]
- Yue, Q.-X.; Liu, X.; Guo, D.-A. Microtubule-Binding Natural Products for Cancer Therapy. Planta Med. 2010, 76, 1037–1043. [Google Scholar] [CrossRef]
- Hollis, C.P.; Li, T. Hybrid Nanocrystal as a Versatile Platform for Cancer Theranostics. In Biomaterials for Cancer Thera-peutics: Diagnosis, Prevention and Therapy; Woodhead Publishinged: Cambridge, UK, 2013. [Google Scholar]
- Chaturvedi, V.K.; Singh, A.; Singh, V.K.; Singh, M.P. Cancer Nanotechnology: A New Revolution for Cancer Diagnosis and Therapy. Curr. Drug Metab. 2019, 20, 416–429. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, Y.; A Gemeinhart, R.; Wu, W.; Li, T. Developing nanocrystals for cancer treatment. Nanomedicine 2015, 10, 2537–2552. [Google Scholar] [CrossRef]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009, 4, 99. [Google Scholar]
- Heinig, U.; Scholz, S.; Jennewein, S. Getting to the bottom of Taxol biosynthesis by fungi. Fungal Divers. 2013, 60, 161–170. [Google Scholar] [CrossRef]
- Markman, M. Managing taxane toxicities. Support. Care Cancer 2003, 11, 144–147. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Cazenave, L.A.; Donehower, R.C. Taxol: A Novel Investigational Antimicrotubule Agent. JNCI J. Natl. Cancer Inst. 1990, 82, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Yoncheva, K.; Calleja, P.; Agüeros, M.; Petrov, P.; Miladinova, I.; Tsvetanov, C.; Irache, J.M. Stabilized micelles as delivery vehicles for paclitaxel. Int. J. Pharm. 2012, 436, 258–264. [Google Scholar] [CrossRef]
- Deepa, G.; Ashwanikumar, N.; Pillai, J.J.; Kumar, G.S.V. Polymer nanoparticles-a novel strategy for administration of paclitaxel in cancer chemotherapy. Curr. Med. Chem. 2012, 19, 6207–6213. [Google Scholar] [CrossRef]
- Ezrahi, S.; Aserin, A.; Garti, N. Basic principles of drug delivery systems—The case of paclitaxel. Adv. Colloid Interface Sci. 2019, 263, 95–130. [Google Scholar] [CrossRef]
- Zhang, Z.; Mei, L.; Feng, S.-S. Paclitaxel drug delivery systems. Expert Opin. Drug Deliv. 2013, 10, 325–340. [Google Scholar] [CrossRef]
- Schiff, P.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Stinchcombe, T.E. Nanoparticle albumin-bound paclitaxel: A novel Cremphor-EL®-free formulation of paclitaxel. Nanomedicine 2007, 2, 415–423. [Google Scholar] [CrossRef]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Malingré, M.M.; Beijnen, J.H.; Schellens, J.H. Oral delivery of taxanes. Investig. New Drugs 2001, 19, 155–162. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.C.; Acharya, G.; Chang, C.; Park, K. Hydrotropic Solubilization of Paclitaxel: Analysis of Chemical Structures for Hydrotropic Property. Pharm. Res. 2003, 20, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.H.; Bhattachar, S.; Hitchingham, L.; Zocharski, P.; Naath, M.; Surendran, N.; Stoner, C.L.; El-Kattan, A. The road map to oral bioavailability: An industrial perspective. Expert Opin. Drug Metab. Toxicol. 2006, 2, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.D.; Paulus, R.; Komaki, R.; Masters, G.; Blumenschein, G.; Schild, S.; Bogart, J.; Hu, C.; Forster, K.; Magliocco, A.; et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): A randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015, 16, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tang, Z.; Li, M.; Lv, S.; Sun, H.; Deng, M.; Liu, H.; Chen, X. Polypeptide-based combination of paclitaxel and cisplatin for enhanced chemotherapy efficacy and reduced side-effects. Acta Biomater. 2014, 10, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Brotto, L.; Brundage, M.; Hoskins, P.; Vergote, I.; Cervantes, A.; Casado, H.A.; Poveda, A.; Eisenhauer, E.; Tu, N. Randomized study of sequential cisplatin-topotecan/carboplatin-paclitaxel versus carboplatin-paclitaxel: Effects on quality of life. Support Care Cancer 2016, 24, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, J.; Sima, M.; Zhou, Y.; Kopeček, J. Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl)methacrylamide copolymer paclitaxel and gemcitabine conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 12181–12186. [Google Scholar] [CrossRef]
- Panchagnula, R. Pharmaceutical aspects of paclitaxel. Int. J. Pharm. 1998, 172, 1–15. [Google Scholar] [CrossRef]
- Marupudi, N.; E Han, J.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef]
- Green, R.M.; Manikhas, M.G.; Orlov, S.; Afanasyev, B.; Makhson, M.A.; Bhar, P.; Hawkins, J.M. Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2006, 17, 1263–1268. [Google Scholar] [CrossRef]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef]
- Paál, K.; Müller, J.; Hegedûs, L. High affinity binding of paclitaxel to human serum albumin. JBIC J. Biol. Inorg. Chem. 2001, 268, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dash, A.K. Paclitaxel in cancer treatment: Perspectives and prospects of its delivery challenges. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 333–372. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III Trial of Nanoparticle Albumin-Bound Paclitaxel Compared with Polyethylated Castor Oil–Based Paclitaxel in Women with Breast Cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, Q.-Z.; Liu, J.; Li, L.-Q.; Zhao, W.-H.; Wang, Y.-J.; Zhou, Q.-H.; Li, L. Multi-center prospective randomized trial on paclitaxel liposome and traditional taxol in the treatment of breast cancer and non-small-cell lung cancer. Zhonghua Zhong Liu Za Zhi Chinese J. Oncol. 2003, 25, 190–192. [Google Scholar]
- Xu, X.; Wang, L.; Xu, Q.H.; Huang, E.X.; Qian, D.Y.; Xiang, J. Clinical comparison between paclitaxel liposome (Lipusu®) and paclitaxel for treatment of patients with meta-static gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2591–2594. [Google Scholar] [CrossRef]
- Kim, T.-Y.; Kim, D.-W.; Chung, J.-Y.; Shin, S.G.; Kim, S.-C.; Heo, D.S.; Kim, N.K.; Bang, Y.-J. Phase I and Pharmacokinetic Study of Genexol-PM, a Cremophor-Free, Polymeric Micelle-Formulated Paclitaxel, in Patients with Advanced Malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [Google Scholar] [CrossRef]
- Lim, W.T.; Tan, E.H.; Toh, C.K.; Hee, S.W.; Leong, S.S.; Ang, P.C.S.; Wong, N.S.; Chowbay, B. Phase I pharmacokinetic study of a weekly liposomal paclitaxel formulation (Genexol®-PM) in patients with solid tumors. Ann. Oncol. 2009, 21, 382–388. [Google Scholar] [CrossRef]
- Saif, M.W.; Podoltsev, N.A.; Rubin, M.S.; Figueroa, J.A.; Lee, M.Y.; Kwon, J.; Rowen, E.; Yu, J.; Kerr, R.O. Phase II Clinical Trial of Paclitaxel Loaded Polymeric Micelle in Patients with Advanced Pancreatic Cancer. Cancer Investig. 2010, 28, 186–194. [Google Scholar] [CrossRef]
- Sartori, S.; Caporale, A.; Rechichi, A.; Cufari, D.; Cristallini, C.; Barbani, N.; Giusti, P.; Ciardelli, G. Biodegradable paclitaxel-loaded microparticles prepared from novel block copolymers: Influence of polymer composition on drug encapsulation and release. J. Pept. Sci. 2013, 19, 205–213. [Google Scholar] [CrossRef]
- He, H.; Chen, S.; Zhou, J.; Dou, Y.; Song, L.; Che, L.; Zhou, X.; Chen, X.; Jia, Y.; Zhang, J.; et al. Cyclodextrin-derived pH-responsive nanoparticles for delivery of paclitaxel. Biomaterials 2013, 34, 5344–5358. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, G.; Du, Y.; Ye, L.; Chen, W.; Zhang, L.; Wang, T.; Tian, J.; Fu, F. Hypersensitivity reaction studies of a polyethoxylated castor oil-free, liposome-based alternative paclitaxel formulation. Mol. Med. Rep. 2013, 7, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-J.; Guo, R.-F.; Liu, Y.-L.; Zhang, P.-X.; Zhou, C.-P.; Jin, D.-J.; Wang, R.-Y. Formulation, Characterization and Hypersensitivity Evaluation of an Intravenous Emulsion Loaded with a Paclitaxel-Cholesterol Complex. Chem. Pharm. Bull. 2011, 59, 321–326. [Google Scholar] [CrossRef]
- Torchilin, V.P. Micellar Nanocarriers: Pharmaceutical Perspectives. Pharm. Res. 2007, 24, 1. [Google Scholar] [CrossRef] [PubMed]
- May, S.; Ben-Shaul, A. Molecular Theory of Lipid-Protein Interaction and the Lα-HII Transition. Biophys. J. 1999, 76, 751–767. [Google Scholar] [CrossRef]
- Lukyanov, A.N.; Torchilin, V.P. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Deliv. Rev. 2004, 56, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.J.; Ke, X.Y.; Huang, Y.; Chen, X.M.; Zhao, X.; Zhao, B.X.; Lu, W.L.; Lou, J.N.; Zhang, X.; Zhang, Q. The antiangiogenic efficacy of NGR-modified PEG–DSPE micelles containing paclitaxel (NGR-M-PTX) for the treatment of glioma in rats. J. Drug Target. 2011, 19, 382–390. [Google Scholar] [CrossRef]
- Chen, T.; Tu, L.; Wang, G.; Qi, N.; Wu, W.; Zhang, W.; Feng, J. Multi-functional chitosan polymeric micelles as oral paclitaxel delivery systems for enhanced bioavailability and anti-tumor efficacy. Int. J. Pharm. 2020, 578, 119105. [Google Scholar] [CrossRef]
- Mutlu-Agardan, N.B.; Sarisozen, C.; Torchilin, V. Cytotoxicity of Novel Redox Sensitive PEG 2000-SS-PTX Micelles against Drug-Resistant Ovarian and Breast Cancer Cells. Pharm. Res. 2020, 37, 65. [Google Scholar] [CrossRef]
- Feng, L.; Mumper, R.J. A critical review of lipid-based nanoparticles for taxane delivery. Cancer Lett. 2013, 334, 157–175. [Google Scholar] [CrossRef]
- Fetterly, G.J.; Grasela, T.H.; Sherman, J.W.; Dul, J.L.; Grahn, A.; LeComte, D.; Fiedler-Kelly, J.; Damjanov, N.; Fishman, M.; Kane, M.P.; et al. Pharmacokinetic/Pharmacodynamic Modeling and Simulation of Neutropenia during Phase I Development of Liposome-Entrapped Paclitaxel. Clin. Cancer Res. 2008, 14, 5856–5863. [Google Scholar] [CrossRef]
- Crosasso, P.; Ceruti, M.; Brusa, P.; Arpicco, S.; Dosio, F.; Cattel, L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J. Control. Release 2000, 63, 19–30. [Google Scholar] [CrossRef]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Yoshizawa, Y.; Kono, Y.; Ogawara, K.-I.; Kimura, T.; Higaki, K. PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int. J. Pharm. 2011, 412, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Abu Lila, A.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ran, R.; Chen, J.; Kuang, Q.; Tang, J.; Mei, L.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 2014, 35, 4835–4847. [Google Scholar] [CrossRef]
- Luo, L.-M.; Huang, Y.; Zhao, B.-X.; Zhao, X.; Duan, Y.; Du, R.; Yu, K.-F.; Song, P.; Zhao, Y.; Zhang, X.; et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials 2013, 34, 1102–1114. [Google Scholar] [CrossRef]
- Qin, L.I.; Wang, C.Z.; Fan, H.J.; Zhang, C.J.; Zhang, H.W.; Lv, M.H.; Cui, S.D. A dual-targeting liposome conjugated with transferrin and arginine-glycine-aspartic acid peptide for glio-ma-targeting therapy. Oncol. Lett. 2014, 8, 2000–2006. [Google Scholar] [CrossRef]
- Büyükköroğlu, G.; Şenel, B.; Başaran, E.; Gezgin, S. Development of paclitaxel-loaded liposomal systems with anti-her2 antibody for targeted therapy. Trop. J. Pharm. Res. 2016, 15, 895. [Google Scholar] [CrossRef]
- Chen, D.; Jiang, X.; Liu, J.; Jin, X.; Zhang, C.; Ping, Q. In vivo evaluation of novel pH-sensitive mPEG-Hz-Chol conjugate in liposomes: Pharmacokinetics, tissue distribution, efficacy assessment. Artif. Cells Blood Substit. Biotechnol. 2010, 38, 136–142. [Google Scholar] [CrossRef]
- Monteiro, L.O.; Malachias, A.; Pound-Lana, G.; Magalhaes-Paniago, R.; Mosqueira, V.C.; Oliveira, M.C.; de Barros, A.L.B.; Leite, E.A. Paclitaxel-loaded pH-sensitive liposome: New insights on structural and physicochemical characterization. Langmuir 2018, 34, 5728–5737. [Google Scholar] [CrossRef]
- Qi, J.; Lu, Y.; Wu, W. Absorption, Disposition and Pharmacokinetics of Solid Lipid Nanoparticles. Curr. Drug Metab. 2012, 13, 418–428. [Google Scholar] [CrossRef]
- Shahgaldian, P.; Da Silva, E.; Coleman, A.W.; Rather, B.; Zaworotko, M.J. Para-acyl-calix-arene based solid lipid nanoparticles (SLNs): A detailed study of preparation and stability parameters. Int. J. Pharm. 2003, 253, 23–38. [Google Scholar] [CrossRef]
- Yegin, A.B.; Benoît, J.-P.; Lamprecht, A. Paclitaxel-loaded lipid nanoparticles prepared by solvent injection or ultra-sound emulsification. Drug Dev. Ind. Pharm. 2006, 32, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Miao, J.; Du, Y.-Z.; You, J.; Hu, F.-Q.; Zeng, S. Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int. J. Pharm. 2008, 348, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Bae, E.J.; Lee, M.-K. Enhanced anticancer activity and intracellular uptake of paclitaxel-containing solid lipid nanoparticles in multidrug-resistant breast cancer cells. Int. J. Nanomed. 2018, 13, 7549–7563. [Google Scholar] [CrossRef] [PubMed]
- Valsalakumari, R.; Yadava, S.K.; Szwed, M.; Pandya, A.D.; Mælandsmo, G.M.; Torgersen, M.L.; Iversen, T.-G.; Skotland, T.; Sandvig, K.; Giri, J. Mechanism of cellular uptake and cytotoxicity of paclitaxel loaded lipid nanocapsules in breast cancer cells. Int. J. Pharm. 2021, 597, 120217. [Google Scholar] [CrossRef]
- Dong, X.; Mattingly, C.A.; Tseng, M.T.; Cho, M.J.; Liu, Y.; Adams, V.R.; Mumper, R.J. Doxorubicin and Paclitaxel-Loaded Lipid-Based Nanoparticles Overcome Multidrug Resistance by Inhibiting P-Glycoprotein and Depleting ATP. Cancer Res. 2009, 69, 3918–3926. [Google Scholar] [CrossRef]
- Tammam, S.N. Lipid Based Nanoparticles as Inherent Reversing Agents of Multidrug Resistance in Cancer. Curr. Pharm. Des. 2017, 23, 6714–6729. [Google Scholar] [CrossRef]
- Pandey, A.; Jain, D.S.; Chakraborty, S. Poly Lactic-Co-Glycolic Acid (PLGA) Copolymer and Its Pharmaceutical Application. Handb. Polym. Pharm. Technol. 2015, 2, 151–172. [Google Scholar]
- Astete, C.E.; Sabliov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef] [PubMed]
- Berthet, M.; Gauthier, Y.; Lacroix, C.; Verrier, B.; Monge, C. Nanoparticle-based dressing: The future of wound treatment? Trends Biotechnol. 2017, 35, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef]
- Jin, C.; Wu, H.; Liu, J.; Bai, L.; Guo, G. The effect of paclitaxel-loaded nanoparticles with radiation on hypoxic MCF-7 cells. J. Clin. Pharm. Ther. 2007, 32, 41–47. [Google Scholar] [CrossRef]
- Dinarvand, R.; Sepehri, N.; Manouchehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [PubMed]
- Mirakabad, F.S.T.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-Based Nanoparticles as Cancer Drug Delivery Systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef]
- Fonseca, C.; Simões, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control. Release 2002, 83, 273–286. [Google Scholar] [CrossRef]
- Danhier, F.; Lecouturier, N.; Vroman, B.; Jérôme, C.; Marchand-Brynaert, J.; Feron, O.; Préat, V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J. Control. Release 2009, 133, 11–17. [Google Scholar] [CrossRef]
- Mo, Y.; Lim, L.-Y. Paclitaxel-loaded PLGA nanoparticles: Potentiation of anticancer activity by surface conjugation with wheat germ agglutinin. J. Control. Release 2005, 108, 244–262. [Google Scholar] [CrossRef]
- Esfandyari-Manesh, M.; Mostafavi, S.H.; Majidi, R.F.; Koopaei, M.N.; Ravari, N.S.; Amini, M.; Darvishi, B.; Ostad, S.N.; Atyabi, F.; Dinarvand, R. Improved anticancer delivery of paclitaxel by albumin surface modification of PLGA nano-particles DARU. J. Pharm. Sci. 2015, 23, 28. [Google Scholar]
- Cerqueira, B.B.S.; Lasham, A.; Shelling, A.N.; Al-Kassas, R. Development of biodegradable PLGA nanoparticles surface engineered with hyaluronic acid for targeted delivery of paclitaxel to triple negative breast cancer cells. Mater. Sci. Eng. C 2017, 76, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Godara, S.; Lather, V.; Kirthanashri, S.V.; Awasthi, R.; Pandita, D. Lipid-PLGA hybrid nanoparticles of paclitaxel: Preparation, characterization, in vitro and in vivo evaluation. Mater. Sci. Eng. C 2020, 109, 110576. [Google Scholar] [CrossRef]
- Kim, C.; Lee, A.S.C.; Kang, S.W.; Kwon, I.C.; Kim, A.Y.-H.; Jeong, S.Y. Synthesis and the Micellar Characteristics of Poly(ethylene oxide)−Deoxycholic Acid Conjugates1. Langmuir 2000, 16, 4792–4797. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.C.; Kwon, I.C.; Chung, H.; Jeong, S.Y. Complexation of Poly (2-ethyl-2-oxazoline)-b lock-poly (ε-caprolactone) Micelles with Multifunctional Car-boxylic Acids. Macromolecules 2002, 35, 193–200. [Google Scholar] [CrossRef]
- Patel, N.K.; Sinha, V.K. Synthesis, Characterization and Optimization of Water-Soluble Chitosan Derivatives. Int. J. Polym. Mater. Polym. Biomater. 2009, 58, 548–560. [Google Scholar] [CrossRef]
- Chen, X.-G.; Park, H.-J. Chemical characteristics of O-carboxymethyl chitosans related to the preparation conditions. Carbohydr. Polym. 2003, 53, 355–359. [Google Scholar] [CrossRef]
- Cao, J.; Zhou, N. Progress in antitumor studies of chitosan. Chin. J. Biochem. Pharm. 2005, 26, 127. [Google Scholar]
- Gupta, U.; Sharma, S.; Khan, I.; Gothwal, A.; Sharma, A.K.; Singh, Y.; Chourasia, M.K.; Kumar, V. Enhanced apoptotic and anticancer potential of paclitaxel loaded biodegradable nanoparticles based on chitosan. Int. J. Biol. Macromol. 2017, 98, 810–819. [Google Scholar] [CrossRef]
- Lv, P.-P.; Ma, Y.-F.; Yu, R.; Yue, H.; Ni, D.-Z.; Wei, W.; Ma, G.-H. Targeted Delivery of Insoluble Cargo (Paclitaxel) by PEGylated Chitosan Nanoparticles Grafted with Arg-Gly-Asp (RGD). Mol. Pharm. 2012, 9, 1736–1747. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohamadi, N.; Zarrabi, A.; Abasi, S.; Dehghannoudeh, G.; Tamaddondoust, R.N.; Khanbabaei, H.; Mohammadinejad, R.; Thakur, V.K. Chitosan-based advanced materials for docetaxel and paclitaxel delivery: Recent advances and future directions in cancer theranostics. Int. J. Biol. Macromol. 2020, 145, 282–300. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, G.; Sun, Y.; Wu, X.; Yao, Z.; Guo, Q.; Ding, Q.; Yuan, S.; Shen, Z.; Ping, Q.; et al. Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials 2008, 29, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, H.; Zhang, H.; Li, F.; Gu, C.H.; Yang, Q. Antitumor drug Paclitaxel-loaded pH-sensitive nanoparticles targeting tumor extracellular pH. Carbohydr. Polym. 2009, 77, 773–778. [Google Scholar] [CrossRef]
- Nag, M.; Gajbhiye, V.; Kesharwani, P.; Jain, N.K. Transferrin functionalized chitosan-PEG nanoparticles for targeted delivery of paclitaxel to cancer cells. Colloids Surfaces B Biointerfaces 2016, 148, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Ursachi, V.C.; Dodi, G.; Rusu, A.G.; Mihai, C.T.; Verestiuc, L.; Balan, V. Paclitaxel-Loaded Magnetic Nanoparticles Based on Biotinylated N-Palmitoyl Chitosan: Synthesis, Characterization and Preliminary In Vitro Studies. Molecules 2021, 26, 3467. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Ettmayer, P.; Amidon, G.L.; Clement, A.B.; Testa, B. Lessons Learned from Marketed and Investigational Prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Hayashi, Y.; Kiso, Y. Paclitaxel Prodrugs: Toward Smarter Delivery of Anticancer Agents. J. Med. Chem. 2006, 49, 7253–7269. [Google Scholar] [CrossRef]
- Li, C.; Yu, D.; Inoue, T.; Yang, D.J.; Milas, L.; Hunter, N.R.; Kim, E.E.; Wallace, S. Synthesis and evaluation of water-soluble polyethylene glycol-paclitaxel conjugate as a paclitaxel prodrug. Anticancer Drugs 1996, 7, 642–648. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, C.-K.; Law, W.-C.; Mok, J.; Zou, J.; Prasad, P.N.; Cheng, C. Well-Defined Degradable Brush Polymer–Drug Conjugates for Sustained Delivery of Paclitaxel. Mol. Pharm. 2013, 10, 867–874. [Google Scholar] [CrossRef]
- Yu, Y.; Zou, J.; Yu, L.; Ji, W.; Li, Y.; Law, W.C.; Cheng, C. Functional polylactide-g-paclitaxel–poly (ethylene glycol) by azide–alkyne click chemistry. Macromolecules 2011, 44, 4793–4800. [Google Scholar] [CrossRef]
- Tong, R.; Cheng, J. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew. Chem. Int. Ed. 2008, 47, 4830–4834. [Google Scholar] [CrossRef]
- Satsangi, A.; Roy, S.S.; Satsangi, R.K.; Vadlamudi, R.K.; Ong, J.L. Design of a Paclitaxel Prodrug Conjugate for Active Targeting of an Enzyme Upregulated in Breast Cancer Cells. Mol. Pharm. 2014, 11, 1906–1918. [Google Scholar] [CrossRef] [PubMed]
- Erez, R.; Segal, E.; Miller, K.; Satchi-Fainaro, R.; Shabat, D. Enhanced cytotoxicity of a polymer–drug conjugate with triple payload of paclitaxel. Bioorg. Med. Chem. 2009, 17, 4327–4335. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.W. Paclitaxel poliglumex (XYOTAX™, CT-2103): A macromolecular taxane. J. Control. Release 2005, 109, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Karmali, P.P.; Kotamraju, V.R.; Kastantin, M.; Black, M.; Missirlis, D.; Tirrell, M.; Ruoslahti, E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Shan, X.; Zhang, T.; Zhai, K.; Gao, G.; Chen, X.; Gu, Y. Transferrin-conjugated paclitaxel prodrugs for targeted cancer therapy. RSC Adv. 2016, 6, 77987–77998. [Google Scholar] [CrossRef]
- Zhang, P.; Cheetham, A.G.; Lin, Y.-A.; Cui, H. Self-Assembled Tat Nanofibers as Effective Drug Carrier and Transporter. ACS Nano 2013, 7, 5965–5977. [Google Scholar] [CrossRef]
- Tian, R.; Wang, H.; Niu, R.; Ding, D. Drug delivery with nanospherical supramolecular cell penetrating peptide–taxol conjugates containing a high drug loading. J. Colloid Interface Sci. 2015, 453, 15–20. [Google Scholar] [CrossRef]
- Bhattacharyya, J.; Bellucci, J.J.; Weitzhandler, I.; McDaniel, J.; Spasojevic, I.; Li, X.; Lin, C.-C.; Chi, J.-T.; Chilkoti, A. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat. Commun. 2015, 6, 7939. [Google Scholar] [CrossRef]
- Bradley, M.; Swindell, C.; Anthony, F.; Witman, P.; Devanesan, P.; Webb, N.; Baker, S.; Wolff, A.; Donehower, R. Tumor targeting by conjugation of DHA to paclitaxel. J. Control. Release 2001, 74, 233–236. [Google Scholar] [CrossRef]
- Ke, X.-Y.; Zhao, B.-J.; Zhao, X.; Wang, Y.; Huang, Y.; Chen, X.-M.; Zhao, B.-X.; Zhao, S.-S.; Zhang, X.; Zhang, Q. The therapeutic efficacy of conjugated linoleic acid—Paclitaxel on glioma in the rat. Biomaterials 2010, 31, 5855–5864. [Google Scholar] [CrossRef] [PubMed]
- Tam, T.Y.; Gao, J.; Kwon, G.S. Oligo (lactic acid) n-paclitaxel prodrugs for poly (ethylene glycol)-block-poly (lactic acid) micelles: Loading, release, and backbiting conversion for anticancer activity. J. Am. Chem. Soc. 2016, 138, 8674–8677. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Koo, J.M.; Cui, H. One-component nanomedicine. J. Control. Release 2015, 219, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Ajaj, K.A.; Biniossek, M.L.; Kratz, F. Development of protein-binding bifunctional linkers for a new generation of dual-acting prodrugs. Bioconjug. Chem. 2009, 20, 390–396. [Google Scholar] [CrossRef]
- Cheetham, A.G.; Zhang, P.; Lin, Y.-A.; Lin, R.; Cui, H. Synthesis and self-assembly of a mikto-arm star dual drug amphiphile containing both paclitaxel and camptothecin. J. Mater. Chem. B 2014, 2, 7316–7326. [Google Scholar] [CrossRef]
- Tan, X.; Lu, X.; Jia, F.; Liu, X.; Sun, Y.; Logan, J.K.; Zhang, K. Blurring the Role of Oligonucleotides: Spherical Nucleic Acids as a Drug Delivery Vehicle. J. Am. Chem. Soc. 2016, 138, 10834–10837. [Google Scholar] [CrossRef]
- Berg, J. An Introduction to Interfaces of Colloids: The Bridge to Nanoscience; World Scientific Publishing Co.: Singapore, 2009. [Google Scholar]
- Paul, B.K.; Moulik, S.P. Microemulsions: An overview. J. Dispers. Sci. Technol. 1997, 18, 301–367. [Google Scholar] [CrossRef]
- Lawrence, M.J.; Warisnoicharoen, W. Recent Advances in Microemulsions as Drug Delivery Vehicles. In Nanoparticulates Drug Carr; World Scientific: Singapore, 2006; pp. 125–171. [Google Scholar]
- Forgiarini, A.M.; Esquena, J.; Gonzalez, C.; Solans, C. Formation of Nano-emulsions by Low-Energy Emulsification Methods at Constant Temperature. Langmuir 2001, 17, 2076–2083. [Google Scholar] [CrossRef]
- Kunieda, H.; Solans, C. Nano-Emulsions: Where Macro-and Microemulsions Meet; Imperial College Press: London, UK, 2001. [Google Scholar]
- Ma, P.; Mumper, R.J. Paclitaxel nano-delivery systems: A comprehensive review. J. Nanomed. Nano Technol. 2013, 4, 1000164. [Google Scholar] [CrossRef]
- Shakhwar, S.; Darwish, R.; Kamal, M.M.; Nazzal, S.; Pallerla, S.; Abu Fayyad, A. Development and evaluation of paclitaxel nanoemulsion for cancer therapy. Pharm. Dev. Technol. 2020, 25, 510–516. [Google Scholar] [CrossRef]
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997, 25, 47–58. [Google Scholar] [CrossRef]
- Veltkamp, S.A.; Thijssen, B.; Garrigue, J.S.; Lambert, G.; Lallemand, F.; Binlich, F.; Huitema, A.D.; Nuijen, B.; Nol, A.; Beijnen, J.H.; et al. A novel self-microemulsifying formulation of paclitaxel for oral administration to patients with advanced cancer. Br. J. Cancer 2006, 95, 729–734. [Google Scholar] [CrossRef]
- Meher, J.G.; Dixit, S.; Pathan, D.K.; Singh, Y.; Chandasana, H.; Pawar, V.K.; Sharma, M.; Bhatta, R.S.; Konwar, R.; Kesharwani, P.; et al. Paclitaxel-loaded TPGS enriched self-emulsifying carrier causes apoptosis by modulating survivin ex-pression and inhibits tumour growth in syngeneic mammary tumours. Artif. Cells Nanomed. Biotechnol. 2018, 46, S344–S358. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-S.; Maniar, M.; Shah, J.C. Biodegradable polyanhydride devices of cefazolin sodium, bupivacaine, and taxol for local drug delivery: Preparation, and kinetics and mechanism of in vitro release. J. Control. Release 1998, 52, 179–189. [Google Scholar] [CrossRef]
- Bode, C.; Kranz, H.; Siepmann, F.; Siepmann, J. In-situ forming PLGA implants for intraocular dexamethasone delivery. Int. J. Pharm. 2018, 548, 337–348. [Google Scholar] [CrossRef]
- Kamali, H.; Khodaverdi, E.; Hadizadeh, F.; Mohajeri, S.A. In-vitro, ex-vivo, and in-vivo evaluation of buprenorphine HCl release from an in situ forming gel of PLGA-PEG-PLGA using N-methyl-2-pyrrolidone as solvent. Mater. Sci. Eng. C 2019, 96, 561–575. [Google Scholar] [CrossRef]
- Samy, W.M.; I Ghoneim, A.; A Elgindy, N. Novel microstructured sildenafil dosage forms as wound healing promoters. Expert Opin. Drug Deliv. 2014, 11, 1525–1536. [Google Scholar] [CrossRef]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Packhaeuser, C.B.; Schnieders, J.; Oster, C.G.; Kissel, T. In situ forming parenteral drug delivery systems: An overview. Eur. J. Pharm. Biopharm. Drug Dispos. 2004, 58, 445–455. [Google Scholar] [CrossRef]
- Amini-Fazl, M.S. Biodegradation study of PLGA as an injectable in situ depot-forming implant for controlled release of paclitaxel. Polym. Bull. 2021, 3, 1–14. [Google Scholar] [CrossRef]
- Hollis, C.P.; Li, T. Nanocrystals Production, Characterization, and Application for Cancer Therapy. In Pharmaceutical Sciences Encyclopedia: Drug Discovery, Development, Manufacturing; Wiley & Sons, Inc.: New York, NY, USA, 2013; pp. 181–206. [Google Scholar]
- Wong, J.; Brugger, A.; Khare, A.; Chaubal, M.; Papadopoulos, P.; Rabinow, B.; Kipp, J.; Ning, J. Suspensions for intravenous (IV) injection: A review of development, preclinical and clinical aspects. Adv. Drug Deliv. Rev. 2008, 60, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Müller, H.R.; Gohla, S.; Keck, C.M. State of the art of nanocrystals—Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. Drug Dispos. 2011, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shegokar, R.; Müller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Zhang, H.; Hollis, C.P.; Zhang, Q.; Li, T. Preparation and antitumor study of camptothecin nanocrystals. Int. J. Pharm. 2011, 415, 293–300. [Google Scholar] [CrossRef]
- Wang, J.; Muhammad, N.; Li, T.; Wang, H.; Liu, Y.; Liu, B.; Zhan, H. Hyaluronic Acid-Coated Camptothecin Nanocrystals for Targeted Drug Delivery to Enhance Anticancer Efficacy. Mol. Pharm. 2020, 17, 2411–2425. [Google Scholar] [CrossRef]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X.; Wang, F. Application of Drug Nanocrystal Technologies on Oral Drug Delivery of Poorly Soluble Drugs. Pharm. Res. 2013, 30, 307–324. [Google Scholar] [CrossRef]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Xia, D.; Quan, P.; Piao, H.; Piao, H.; Sun, S.; Yin, Y.; Cui, F. Preparation of stable nitrendipine nanosuspensions using the precipitation–ultrasonication method for enhancement of dissolution and oral bioavailability. Eur. J. Pharm. Sci. 2010, 40, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Hollis, C.P.; Li, T. Nanocrystals production, characterization, and application for cancer therapy. Pharm. Sci. Encycl. Drug Discov. Dev. Manuf. 2010, 1–26. [Google Scholar]
- De Castro, M.L.; Priego-Capote, F. Ultrasound-assisted crystallization (sonocrystallization). Ultrason. Sonochem. 2007, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Kakran, M.; Sahoo, N.G.; Tan, I.-L.; Li, L. Preparation of nanoparticles of poorly water-soluble antioxidant curcumin by antisolvent precipitation methods. J. Nanopart. Res. 2012, 14, 757. [Google Scholar] [CrossRef]
- Lonare, A.A.; Patel, S.R. Antisolvent crystallization of poorly water soluble drugs. Int. J. Chem. Eng. Appl. 2013, 4, 337. [Google Scholar] [CrossRef]
- Pawar, N.; Agrawal, S.; Methekar, R. Modeling, Simulation, and Influence of Operational Parameters on Crystal Size and Morphology in Semibatch Antisolvent Crystallization of α-Lactose Monohydrate. Cryst. Growth Des. 2018, 18, 4511–4521. [Google Scholar] [CrossRef]
- Crisp, J.; Dann, S.; Blatchford, C. Antisolvent crystallization of pharmaceutical excipients from aqueous solutions and the use of preferred orientation in phase identification by powder X-ray diffraction. Eur. J. Pharm. Sci. 2011, 42, 568–577. [Google Scholar] [CrossRef]
- Sharma, C.; Desai, M.; Patel, S.R. Effect of surfactants and polymers on morphology and particle size of telmisartan in ultrasound-assisted anti-solvent crystallization. Chem. Pap. 2019, 73, 1685–1694. [Google Scholar] [CrossRef]
- Miao, X.; Yang, W.; Feng, T.; Lin, J.; Huang, P. Drug nanocrystals for cancer therapy. Nanomed. Nanobiotechnol. 2018, 10, e1499. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, X.; Yu, C.; Nan, X.; Chen, X.; Zhang, X.-H. Shape regulated anticancer activities and systematic toxicities of drug nanocrystals in vivo. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 181–189. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.-H.; Li, T.; McNally, H.; Park, K.; Sturek, M. Development and evaluation of transferrin-stabilized paclitaxel nanocrystal formulation. J. Control. Release 2014, 176, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tu, L.; Cheng, M.; Feng, J.; Jin, Y. Mechanisms for oral absorption enhancement of drugs by nanocrystals. J. Drug Deliv. Sci. Technol. 2020, 56, 101607. [Google Scholar] [CrossRef]
- Park, J.; Sun, B.; Yeo, Y. Albumin-coated nanocrystals for carrier-free delivery of paclitaxel. J. Control. Release 2017, 263, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, J.E.; Hedrick, V.E.; Wood, K.V.; Bonham, C.; Lee, W.; Yeo, Y. A Comparative In Vivo Study of Albumin-Coated Paclitaxel Nanocrystals and Abraxane. Small 2018, 14, 1703670. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.S.; Yoon, D.S.; Sohn, J.Y.; Park, J.S.; Choi, J.S. Development and evaluation of targeting ligands surface modified paclitaxel nanocrystals. Mater. Sci. Eng. C 2017, 72, 228–237. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, J.; Verma, A.; Teja, B.V.; Shukla, R.P.; Singh, S.K.; Sharma, V.; Konwar, R.; Mishra, P.R. Hyaluronic acid anchored paclitaxel nanocrystals improves chemotherapeutic efficacy and inhibits lung metastasis in tumor-bearing rat model. RSC Adv. 2016, 6, 73083–73095. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, H.; Zhang, H.; Dai, W.; Wang, X.; Wang, X.; Zhang, Q. Effects of PEGylated paclitaxel nanocrystals on breast cancer and its lung metastasis. Nanoscale 2015, 7, 10790–10800. [Google Scholar] [CrossRef]
- Polomska, A.; Gauthier, M.A.; Leroux, J.C. In Vitro and In Vivo Evaluation of PEGylated Layer-by-Layer Polyelectro-lyte-Coated Paclitaxel Nanocrystals. Small 2017, 13, 1602066. [Google Scholar] [CrossRef]
- Zhao, J.; Du, J.; Wang, J.; An, N.; Zhou, K.; Hu, X.; Dong, Z.; Liu, Y. Folic Acid and Poly(ethylene glycol) Decorated Paclitaxel Nanocrystals Exhibit Enhanced Stability and Breast Cancer-Targeting Capability. ACS Appl. Mater. Interfaces 2021, 13, 14577–14586. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Zhao, G.; Zhuang, J.; Wu, W. Improving systemic circulation of paclitaxel nanocrystals by surface hybridization of DSPE-PEG2000. Colloids Surfaces B Biointerfaces 2019, 182, 110337. [Google Scholar] [CrossRef]
- Guo, F.; Shang, J.; Zhao, H.; Lai, K.; Li, Y.; Fan, Z.; Hou, Z.; Su, G. Cube-shaped theranostic paclitaxel prodrug nanocrystals with surface functionalization of SPC and MPEG-DSPE for imaging and chemotherapy. Colloids Surfaces B Biointerfaces 2017, 160, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Park, J.-S. Effects of paclitaxel nanocrystals surface charge on cell internalization. Eur. J. Pharm. Sci. 2016, 93, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; Liu, D.; Fallon, J.K.; Liu, F. The Effect of Surfactant on Paclitaxel Nanocrystals: An In Vitro and In Vivo Study. J. Biomed. Nanotechnol. 2016, 12, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.-K.; Naeem, M.; Cao, J.; Lee, E.H.; Kim, M.-S.; Jung, Y.; Yoo, J.-W. Herceptin-functionalized pure paclitaxel nanocrystals for enhanced delivery to HER2-postive breast cancer cells. Int. J. Pharm. 2016, 513, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chen, Y.; Thompson, D.H.; Park, K.; Li, T. Impact of surfactant treatment of paclitaxel nanocrystals on biodistribution and tumor accumulation in tumor-bearing mice. J. Control. Release 2016, 237, 168–176. [Google Scholar] [CrossRef]
- Han, X.; Su, R.; Huang, X.; Wang, Y.; Kuang, X.; Zhou, S.; Liu, H. Triphenylphosphonium-modified mitochondria-targeted paclitaxel nanocrystals for overcoming multidrug resistance. Asian J. Pharm. Sci. 2019, 14, 569–580. [Google Scholar] [CrossRef]
- Mei, D.; Gong, L.; Zou, Y.; Yang, D.; Liu, H.; Liang, Y.; Sun, N.; Zhao, L.; Zhang, Q.; Lin, Z. Platelet membrane-cloaked paclitaxel-nanocrystals augment postoperative chemotherapeutical efficacy. J. Control. Release 2020, 324, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-G.; Lv, F.-M.; Wang, J.; Cao, S.-J.; Liu, Z.-P.; Liu, Y.; Lu, W.-Y. RGD-modified PEGylated paclitaxel nanocrystals with enhanced stability and tumor-targeting capability. Int. J. Pharm. 2019, 556, 217–225. [Google Scholar] [CrossRef]
- Wang, J.; Lv, F.M.; Wang, D.L.; Du, J.L.; Guo, H.Y.; Chen, H.N.; Zhao, S.J.; Liu, Z.P.; Liu, Y. Synergistic Antitumor Effects on Drug-Resistant Breast Cancer of Paclitaxel/Lapatinib Composite Nano-crystals. Molecules 2020, 25, 604. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Pandey, G.; Mittapelly, N.; Mishra, P.R. Investigating the role of Pluronic-g-Cationic polyelectrolyte as functional stabilizer for nanocrystals: Impact on Paclitaxel oral bioavailability and tumor growth. Acta Biomater. 2015, 26, 169–183. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Teja, B.V.; Shukla, P.; Mishra, P.R. Development of stabilized Paclitaxel nanocrystals: In-vitro and in-vivo efficacy studies. Eur. J. Pharm. Sci. 2015, 69, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Li, X.; Zhou, C.; Hu, X.; Zhou, Y.; Jin, Y.; Liu, Q.; Wang, L.; Li, X.; Liu, Y. Further Enhancement in Intestinal Absorption of Paclitaxel by Using Transferrin-Modified Paclitaxel Nano-crystals. ACS Appl. Bio Mater. 2020, 3, 4684–4695. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chang, Y.-N.; Xing, G.; Li, M.; Zhao, Y. Study on orally delivered paclitaxel nanocrystals: Modification, characterization and activity in the gastrointestinal tract. R. Soc. Open Sci. 2017, 4, 170753. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Xu, S.; Gao, W.; Hu, H.; Chen, M.; Wang, Y.; He, B.; Dai, W.; Zhang, H.; Wang, X.; et al. A comparative investigation between paclitaxel nanoparticle- and nanocrystal-loaded thermosensitive PECT hydrogels for peri-tumoural administration. Nanoscale 2016, 8, 18782–18791. [Google Scholar] [CrossRef]
- Tiet, P.; Li, J.; Abidi, W.; Mooney, R.; Flores, L.; Aramburo, S.; Batalla-Covello, J.; Gonzaga, J.; Tsaturyan, L.; Kang, Y.; et al. Silica Coated Paclitaxel Nanocrystals Enable Neural Stem Cell Loading for Treatment of Ovarian Cancer. Bioconjug. Chem. 2019, 30, 1415–1424. [Google Scholar] [CrossRef]
- Huang, X.; Shi, Q.; Du, S.; Lu, Y.; Han, N. Poly-tannic acid coated paclitaxel nanocrystals for combinational photothermal-chemotherapy. Colloids Surfaces B Biointerfaces 2021, 197, 111377. [Google Scholar] [CrossRef]
- Lin, Z.; Mei, D.; Chen, M.; Wang, Y.; Chen, X.; Wang, Z.; He, B.; Zhang, H.; Wang, X.; Dai, W.; et al. A comparative study of thermo-sensitive hydrogels with water-insoluble paclitaxel in molecule, nanocrystal and microcrystal dispersions. Nanoscale 2015, 7, 14838–14847. [Google Scholar] [CrossRef]
- Sun, B.; Taha, M.S.; Ramsey, B.; Torregrosa-Allen, S.; Elzey, B.D.; Yeo, Y. Intraperitoneal chemotherapy of ovarian cancer by hydrogel depot of paclitaxel nanocrystals. J. Control. Release 2016, 235, 91–98. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, C.; Fu, Q.; Lv, H. Combined chemotherapy for triple negative breast cancer treatment by paclitaxel and niclosamide nano-crystals loaded thermosensitive hydrogel. Eur. J. Pharm. Sci. 2021, 167, 105992. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PTX NC | Method of Preparation | The Models Used and the Reference or Control Formula | Benefits, Aims, and Other Notes | Refs. |
|---|---|---|---|---|
| Albumin-coated PTX-NC (Alb-PTX NCs) | NC crystallized in the medium containing Pluronic F-127 and then coated with albumin “Cim-F-alb” | The new formula was compared to Abraxane and solvent-dissolved PTX In vitro models including Biolayer interferometry analysisCell culture models: J774A.1 macrophages and SPARC+ B16F10 melanoma cells In vivo model: mouse model of B16F10 melanoma | High drug loading (90%) and serum stability Equivalent cytotoxicity. More stability in undiluted serum. Less interaction with serum proteins. In cell culture studies, demonstrated suitable cell interaction profiles (depressed uptake by macrophages and great uptake by melanoma cells). In the in vivo studies, exhibited prolonged plasma t1/2 and superior accumulation in tumors by about 1.5 and 4.6 times, respectively. Exhibited superior antitumor efficacy. | [160,161] |
| Surface modified PTX-NCs with apo-transferrin (Tf) or hyaluronic acid (HA) | PTX NCs were prepared by the nanoprecipitation Method, and then the surface was modified by grafting with Tf or HA | The new formula was compared to PTX-NC and pure PTX drug In vitro models: drug release in PBS with or without tween 80 Cell culture models: HaCaT normal cells and MCF-7 cancer cells | PTX release was faster. Improve the cellular uptake, permeability, and cell growth inhibition (60%) against the cancer cells. The effect on the normal cells was inferior. Provide targeted delivery to cancer cells. | [162] |
| Hyaluronic acid (HA) coated PTX NCs | The NCs were prepared by the top-down method using homogenization | The new formula was compared to Taxol® and heparin-coated PTX NCs In vitro models: 2D monolayer and 3D spheroids Cell culture models: MDA-MB 231 cells In vivo model: LA-7 tumor-bearing rat model | Exhibited superior in vitro efficacy. HA-PTX NCs incur receptor-mediated endocytosis by binding to CD44 receptors. The in vivo studies indicated significantly prolonged blood circulation time of PTX. Exhibited superior efficacy with reduced lung metastasis and toxicity. | [163] |
| PEGylated PTX NCs | The NCs were prepared by the antisolvent precipitation method combined with probe sonication | The new formula was compared to PTX NCs and Taxol® In vivo model: breast cancer xenografted mice model and a model of lung tumor metastasis quantified by the luciferase activity | Superior stability under both storage and physiological conditions. In vivo studies showed significant improvement of the antitumor activity in facing in situ or metastatic tumors. | [164] |
| PEGylated polyelectrolyte multilayer-coated PTX NCs | The layer-by-layer method was used to coat PTX NCs with alternating layers of oppositely charged polyelectrolytes, utilizing a PEGylated copolymer as the upper layer, and PTX NCs were prepared by a wet milling approach | The new formula was compared to Abraxane and PTX NCs In vitro models: physiologically relevant media and human RBC hemolysis Cell culture models: HT-29 cells In vivo model: NMRI-nu mice bearing HT-29 subcutaneous xenografts | Slowed down the dissolution. Offered colloidal stability in physiologically simulated media. Showed no innate effect on cell viability using HT-29 cells. No hemolytic activity was detected. Quickly eliminated from the bloodstream and accumulated in the liver and spleen (mononuclear phagocyte organs). Poor tumor accumulation. | [165] |
| PTX NCs modified with PEG and folic acid (FA)(PTX NCs-PEG-FA) | PTX NCs were prepared by thin-film hydration method, which is a bottom-up method, and then modified with both PEG and FA derivatives using thin-film hydration technique | The new formula was compared to Taxol®, PTX NCs, and PTX NCs-PEG In vitro models: plasma Cell culture models: 4T1 breast cancer cells In vivo model: PK rat model and 4T1 orthotopic breast cancer-bearing nude mice | More size stability in plasma. Improved cellular uptake and growth inhibition in cells. An in vivo pharmacokinetic study showed a significant increase in the circulation of PTX. In vivo cancer model showed that it significantly enhanced the accumulation of PTX in the tumor and effectively inhibited tumor growth. | [166] |
| Surface hybridization of PTX NCs by DSPE-PEG 2000 | PTX NCs were prepared by anti-solvent method, and DSPE-PEG 2000 was incorporated by hybridization | The new formula was compared to PTX solution and PTX NCs In vitro models: in vitro release study In vivo model: PK rats’ model | Similar size with an increased negative charge. The in vitro study showed that the release of PTX was significantly slower. The pharmacokinetics studies showed a greater area under the curve (AUC) and a lower clearance rate. | [167] |
| Cube-shaped PTX NC prodrug with surface functionalization of SPC and MPEG-DSPE | PTX was labeled with fluorophore conjugate 4-chloro-7-nitro-1, 2, 3-benzoxadiazole (NBD-Cl) (PTX-NBD), which was synthesized by a nucleophilic substitution reaction of PTX with NBD-Cl in high yield. The PTX-NBD NCs were prepared by the anti-solvent method followed by surface functionalization of SPC and MPEG-DSPE. | The new formula was compared to free PTX-NBD and the sphere-shaped PTX-NBD nanocrystals with surface functionalization of SPC and MPEG-DSPE (PTX-NBD@PC-PEG NSs) Cell culture models: HeLa cells | The cube-shaped PTX-NBD@PC-PEG NCs exhibited better drug loading and stability properties. It showed a remarkable decrease in burst release, efficiently enhanced cellular uptake, and had a better ability to kill cancer cells in vitro using HeLa cells. These NCs can be useful for cell imaging and chemotherapy. | [168] |
| Surface-modified PTX with positively charged poly(allylamine hydrochloride) (PAH) | Nano-precipitation method (bottom-up approach) was employed to prepare PTX NCs, and the surface-modified NCs were obtained by an absorption method with the positively charged polymer | The new formula was compared to pure PTX, PTX NCs, and negatively charged poly (sodium 4-styrene sulfonate) PSS PTX NCs In vitro models: PBS (pH 7.4) containing 0.5% (w/v) tween 80 and bovine serum albumin (BSA) Cell culture models: A549 cells | Higher drug release. Stronger interaction with bovine serum albumin. Greater cellular internalization, uptake, and cytotoxicity. | [169] |
| A non-covalent transferrin-stabilized PTX NCs | The NCs were prepared by the antisolvent precipitation method augmented by sonication | The new formula was compared to PTX solution, PTX NCs, and Taxol® Cell culture models: human KB epidermal carcinoma cells and SKOV-3 ovarian cancer cells In vivo model: mice inoculated with KB cells | The in vivo efficacy studies on KB-bearing mice showed a significantly superior tumor inhibition rate compared with PTX NCs and less efficacy than Taxol, but with a better toxicity profile. However, in cellular models, it showed similar efficacy 72 h after treatment. | [158] |
| PTX NCs stabilized by D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) | The NCs were prepared by three-phase nanoparticle engineering technology (3PNET) | The new formula was compared to Taxol® and PTX/Pluronic F127 (F127) NCs Cell culture models: P-glycoprotein-overexpressing PTX-resistant (H460/TaxR) cancer cells In vivo model: PK using CD-1 mice | The greater the amount of TPGS in the formula, the greater cytotoxicity and cellular internalization. TPGS PTX NCs demonstrated a significantly sustained and prolonged in vitro release pattern. PK studies indicated more rapid clearance. However, they were more effective in promoting the accumulation of PTX in drug-resistant tumors. | [170] |
| Herceptin (HCT)-functionalized PTX NCs | PTX NCs were prepared by sono-precipitation approach, and then HCT was coated, applying a facile non-covalent technique | The new formula was compared to PTX NCs and PTX powder In vitro models: release study Cell culture models: HER2-positive breast cancer cell lines | Exhibited a sustained release pattern comparable to PTX NCs. Demonstrated a higher binding affinity, greater cell-specific internalization, and inhibition of growth to HER2-positive breast cancer cell lines. | [171] |
| PTX-NCs coated with Pluronic® F68 (PEG-PPG-PEG block polymer) | The NCs were prepared by the anti-solvent method | The new formula was compared to Taxol® and PTX NCs In vivo model: tumor-bearing (HT-29 and KB cells) mice and female nude outbred mice | These NCs exhibited similar or better antitumor efficacy and lower toxicity in comparison with Taxol. The in vivo study showed a significant enhancement in the blood circulation of PTX and accumulation in tumor tissue. However, the definite amount that reached the tumor was still minimal for the administered dose. The maximum amount of the coated NCs was significantly obtained in the liver compared with the other organs relative to the uncoated PTX NCs. | [172] |
| Triphenylphosphonium (TPP+)-stabilized PTX NCs (TPP+ PTX NCs) | Precipitation-resuspending method | The new formula was compared to free PTX and unmodified PTX NCs In vitro cell culture models: 2D monolayer and 3D multicellular spheroids (MCs) of MCF-7 cells and MCF-7/ADR cells | A mitochondria-targeted system was developed. Showed the strongest cytotoxicity that was associated with a reduction in mitochondrial membrane potential. Showed greater penetration and superior growth inhibition. | [173] |
| Platelet membrane-coated or cloaked PEG-PTX NCs (PPNCs) | The modified emulsion-lyophilized crystallization method | The new formula was compared to PTX NCs Platelet aggregation was examined using a spectrophotometric method In vitro drug releasee Cell culture models: 4T1 breast cancer cells In vivo model: BALB/c mice injected with 4T1 cells model | Minor risk of thrombus formation after injection was observed. Higher cellular uptake and greater cytotoxicity. In vivo studies showed the ability to deliver a higher dose of the drug and target the site of the coagulation (surgery or vascular disrupting), which improved the antitumor efficacy and decreased toxicities. | [174] |
| RGD peptide -PEGylated PTX NCs coated by polydopamine (PDA) (NC@PDA-PEG-RGD) | The NCs were prepared using modified antisolvent–sonication method | The new formula was compared to free PTX, PTX NCs, PTX NCs-PEG, and PTX NCs-PDA-PEG In vitro models: plasma for size stabilityCell culture models: A549 lung cancer cell line In vivo model: nude mice A549 bearing cancer model | More size stability in plasma. Showed superior cellular uptake, growth inhibition, and cytotoxicity on A549 lung cancer cell line. In vivo demonstrated significantly greater accumulation in the tumor and slower tumor growth. | [175] |
| PTX and lapatinib (LAPA) composite nanocrystals with PDA and PEG modification(cNC@PDA-PEG) | PEG coat was introduced into the cNC via PDA) coat to get PEGylated composite NCs (cNC@PDA-PEG). The NCs were prepared using the bottom-up method or precipitation-resuspending method. | The new formula was compared to free PTX and unmodified PTX NCs In vitro models: plasma and blood Cell culture models: MCF-7/ADR cancer cells | cNC@PDA-PEG had optimum size and stability. The in vitro release study showed that both PTX and LAPA were released completely from cNC@PDA-PEG in 3 days, while only 30% of the drug was released from bulk drugs or unmodified NCs. Showed negligible hemocytolysis and improved therapeutic effect on MCF-7/ADR through endocytosis of whole NCs. | [176] |
| PTX NC | Method of Preparation | The Models Used and the Reference or Control Formula | Benefits, Aims, and Other Notes | Ref. |
|---|---|---|---|---|
| Pluronic-grafted chitosan as a stabilizer for PTX NC (Pl-g-CH PTX NCs) | A novel Pluronic-grafted chitosan copolymer was established and then utilized as a functional stabilizer for PTX NCs. Generally, the NCs were prepared using a high-pressure homogenizer. | The new formula was compared to Taxol® Cell culture models: Caco-2 cells and B16 F10 murine melanoma cells In vivo model for oral PK evaluation: Wistar ratsIn vivo model for efficacy study: healthy Balb/C mice injected with B16 F10 murine melanoma model | Improving intra-cellular accumulation. Improving the absorption by the transcellular and paracellular routes. Showed a P-gp inhibitory property. The in vivo model demonstrated more anti-tumor efficacy and growth reduction after oral delivery, and this was related to the enhancement in the systemic circulation as both the absorption and bioavailability were improved significantly. | [177] |
| PTX NCs stabilized by tween 80 or low molecular weight synthetic polymer sodium polystyrene sulfonate (PSS) | The top-down method was performed using a microfluidizer as a high-pressure homogenizer that was used to prepare the NCs without using any organic solvent | The new formula was compared to formulas stabilized with high molecular weight polymers glycol chitosan (GC) and sodium alginate (SA), as well as with PTX solution and PTX-NCs Cell culture models: MCF7 and MDA-MB breast cancer cell lines In vivo model: PK in male Wistar rat model | The prepared NCs were more suitable, efficient, and exhibited a considerable increase in the dissolution rate, which indicated an enhancement in its bioavailability. The in vitro cell culture study showed more efficiency and potency in killing and inhibiting the growth of the cancer cells. In vivo pharmacokinetic studies demonstrated a considerable increase in AUC0–t, Cmax, and MRT and a decrease in Tmax. | [178] |
| Transferrin (TF)-modified PTX NCs | PTX NCs were prepared using the precipitation–resuspension method | The new formula was compared to Taxol® and unmodified PTX NCs In vitro models: in situ intestinal perfusion study Cell culture models: Caco-2 cells and MCF-7 cancer cells In vivo model: PK Sprague Dawley rat model | Showed an enhancement of cellular monolayer penetration. Had superior suppression in MCF-7 cell growth. Showed an enhancement of intestinal absorption. The pharmacokinetic studies also demonstrated greater Cmax and AUC than both PTX NCs and Taxol® while having the lowest tmax. | [179] |
| Poly(sodium pstyrenesulfonate) (PSS)-modified PTX NCs | Not mentioned | In vitro models: interactions with biomolecules in oral delivery pathways Cell culture models: Caco-2 cell lines | Suitable mono-dispersion and stability in the gastrointestinal tract (GIT) environments for at least 24 h. No substantial interactions with pepsin or trypsin enzymes were detected in the GIT environments. PSS-modified PTX NCs passed through the mimical intestinal epithelial cell (Caco-2 cell lines) with about 25% transmittance. However, the concentration of the NCs should be controlled to avoid toxic effects on the cells. | [180] |
| PTX NC | Route of Administration | Method of Preparation | The Models Used and the Reference or Control Formula | Benefits, Aims, and Other Notes | Ref. |
|---|---|---|---|---|---|
| PTX NC-loaded PECT hydrogels | Local delivery and peritumoral administration | PTX NCs were prepared by three-phase nanoparticle engineering technology (3PNET), while PTX-NC-based PECT (PTX-NC-PECT) gel was prepared based on the “cold” method | The new formula was compared to a nanoparticle-based system (PTX-NP-PECT) and controlled hydrogel of Pluronic® F127 In vitro models: release study In vivo model: MCF-7 tumor-bearing mouse models | High loading capacity of the drug. In vitro release was more effective and homogeneous. In vivo near-infrared fluorescence (NIRF) imaging indicated the ability to maintain the payloads of 1,1-dioctadecyltetramethyl indotricarbocyanine iodide (DiR) at a peri-tumoral site for about 21 days. Exhibited the most complete release system with the greatest anti-tumor efficacy and apoptosis effect. | [181] |
| Silica-coated PTX NCs Si | Intra-peritoneal (IP) | Precipitation–resuspending method | The new formula was compared to uncoated PTX-NC or Abraxane Cell culture model: neural stem cells and OVCAR-8 cells In vivo model: athymic nude mice which inoculated with 2 M OVCAR-8.eGFP.ffluc human ovarian cancer cells | More effective in loading neural stem cells (NSCs). In vivo studies showed that loaded NSCs preserved their migratory ability and, for low PTX dose, were more effective against ovarian tumors. | [182] |
| Poly-tannic acid-coated PTX NCs (PTA-PTX NCs) | Intertumoral injection | The NCs were prepared using the thin-film hydration method followed by probe sonication | The new formula was compared with or without laser irradiation to PTX Cell culture models: 4T1, A549, and HepG2 cells In vivo model: 4T1 tumor-bearing mice | PTX NCs were prepared to act as a chemo-therapeutic agent and poly-tannic acid (pTA)-coated PTX NCs in the presence of Fe3+ acting as a potential agent for photothermal therapy (PTT). The cellular uptake was significantly improved. A synergistic effect with laser irradiation was observed. Demonstrated mild photothermal effect in vivo and the greatest effect in tumor inhibition upon laser irradiation. | [183] |
| PTX NC with F127 hydrogel | Intertumoral injection | Precipitation–resuspending method. The cold method was used for hydrogel preparation. | The new formula was compared to PTX or PTX microcrystal-based hydrogels In vitro erosion of the hydrogels and drug release In vivo model: 4T1 tumor-bearing BALB/c mice | PTX NCs gel offered optimum properties with high drug loading combined with moderate drug release and erosion profiles. Superior anti-tumor efficacy in 4T1 tumor-bearing BALB/c mice. | [184] |
| In situ cross-linkable hydrogel depot containing PTX NCs | Intraperitoneal (IP) | Anti-solvent and temperature-induced crystallization method | The new formula was compared to Taxol® and microparticulate PTX precipitates (PPT) Cell culture models: SKOV3 cells In vivo model: healthy Balb/c mice for toxicity studies and Balb/c mice (SKOV3-Luc) cell-bearing mice for the efficacy study | Superior killing efficiency and more toxicity in SKOV3 cell line. The in vivo study indicated improved dissolution, cellular uptake, and lower maximum tolerated dose. It also showed that a single IP dose was sufficient in extending the survival of tumor-bearing mice. | [185] |
| PTX-NCs combined with niclosamide (NLM) NLM-NCs co-loaded PLGA-PEG-PLGA thermosensitive hydrogel (PN-NCs-Ts) | Intratumoral injection | PTX-NCs were prepared by the “3PNET” method | The new formula was compared to PTX-NCs, PTX-NCs-Ts Gel, NLM-NCs, NLM-NCs-Ts gel, and PN–NCs-Ts gel In vitro drug release Cell culture models: MDA-MB-231 cells In vivo model: BALB/c nude mice inoculated with MDA-MB-231 cells | Sustained and significantly delayed drug release both in vitro and in vivo. The combination with NLM improved PTX cellular uptake, apoptosis, and provided inhibition of cell migration. The in vivo studies showed significant inhibition of tumor growth with acceptable safety and effectively overcoming it. Triple-negative breast cancer (TNBC) progress and drastically prevented breast cancer stem cells (BCSCs). | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haddad, R.; Alrabadi, N.; Altaani, B.; Li, T. Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications. Polymers 2022, 14, 658. https://doi.org/10.3390/polym14040658
Haddad R, Alrabadi N, Altaani B, Li T. Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications. Polymers. 2022; 14(4):658. https://doi.org/10.3390/polym14040658
Chicago/Turabian StyleHaddad, Razan, Nasr Alrabadi, Bashar Altaani, and Tonglei Li. 2022. "Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications" Polymers 14, no. 4: 658. https://doi.org/10.3390/polym14040658
APA StyleHaddad, R., Alrabadi, N., Altaani, B., & Li, T. (2022). Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications. Polymers, 14(4), 658. https://doi.org/10.3390/polym14040658