
Evaluation of Lapatinib-Loaded Microfibers Prepared by Centrifugal Spinning





Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solution Preparation
2.3. Centrifugal Spinning
2.4. Scanning Electron Microscopy (SEM)
2.5. Measurement of Lap Content in the Fiber Mats
2.6. In Vitro Dissolution Study
2.7. Differential Scanning Calorimetry (DSC)
3. Results and Discussion
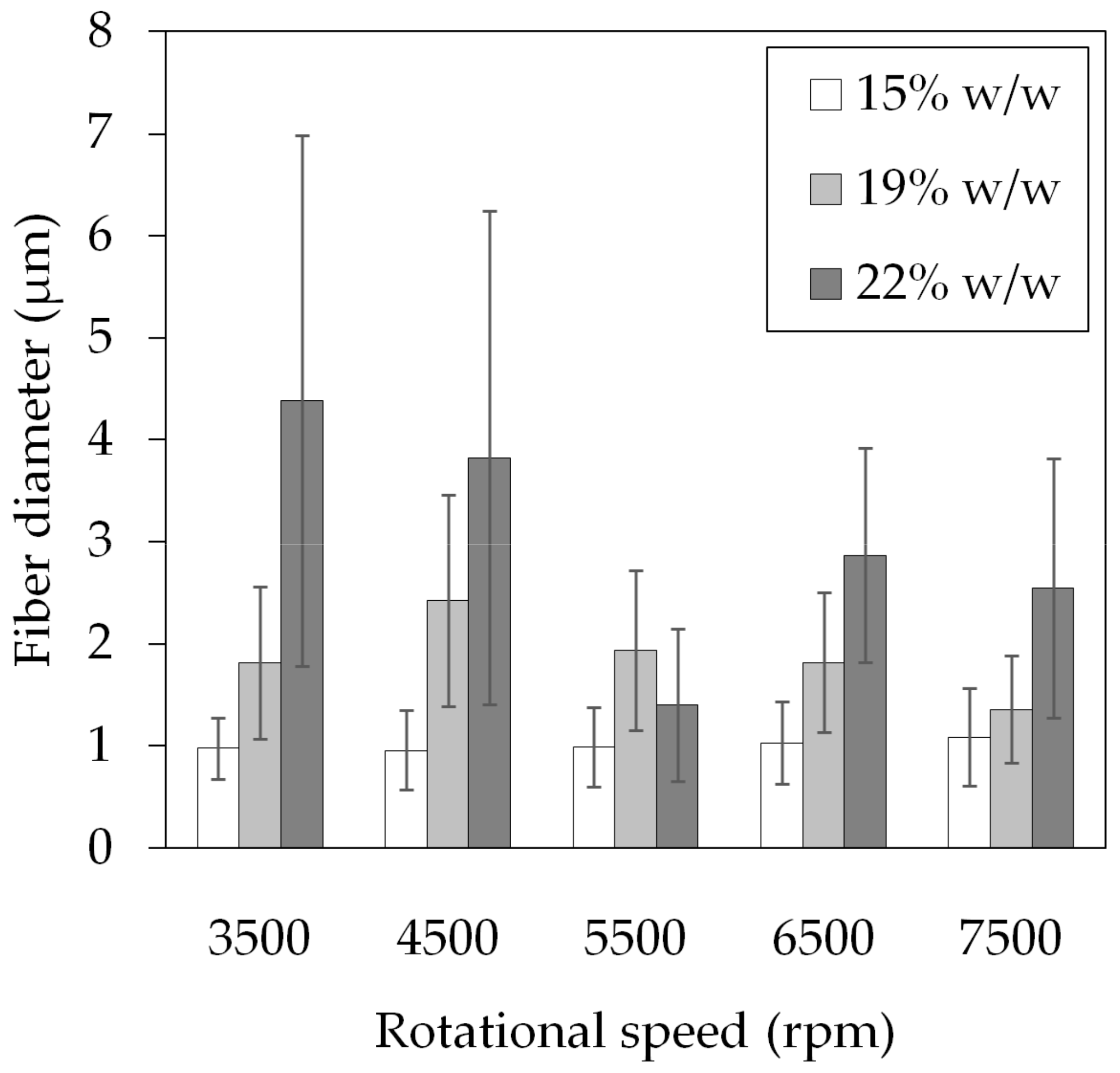
3.1. Polymer Fiber Production
3.2. Analytical Studies
3.3. Lap Dissolution
3.4. General Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Oh, D.-Y.; Bang, Y.-J. HER2-Targeted Therapies—A Role beyond Breast Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Jánoska, Á.; Völgyi, G.; Szabó, Z.-I.; Orgován, G.; Mirzahosseini, A.; Noszál, B. Physicochemical Characterization and Cyclodextrin Complexation of the Anticancer Drug Lapatinib. J. Chem. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, M.; Lonati, V.; Tomasello, G.; Borgonovo, K.; Ghilardi, M.; Cabiddu, M.; Barni, S. The Efficacy of Lapatinib and Capecitabine in HER-2 Positive Breast Cancer with Brain Metastases: A Systematic Review and Pooled Analysis. Eur. J. Cancer 2017, 84, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Budha, N.R.; Frymoyer, A.; Smelick, G.S.; Jin, J.Y.; Yago, M.R.; Dresser, M.J.; Holden, S.N.; Benet, L.Z.; Ware, J.A. Drug Absorption Interactions Between Oral Targeted Anticancer Agents and PPIs: Is PH-Dependent Solubility the Achilles Heel of Targeted Therapy? Clin. Pharmacol. Ther. 2012, 92, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Devriese, L.A.; Koch, K.M.; Mergui-Roelvink, M.; Matthys, G.M.; Ma, W.W.; Robidoux, A.; Stephenson, J.J.; Chu, Q.S.C.; Orford, K.W.; Cartee, L.; et al. Effects of Low-Fat and High-Fat Meals on Steady-State Pharmacokinetics of Lapatinib in Patients with Advanced Solid Tumours. Investig. New Drugs 2014, 32, 481–488. [Google Scholar] [CrossRef]
- Burris, H.A.; Taylor, C.W.; Jones, S.F.; Koch, K.M.; Versola, M.J.; Arya, N.; Fleming, R.A.; Smith, D.A.; Pandite, L.; Spector, N.; et al. A Phase I and Pharmacokinetic Study of Oral Lapatinib Administered Once or Twice Daily in Patients with Solid Malignancies. Clin. Cancer Res. 2009, 15, 6702–6708. [Google Scholar] [CrossRef]
- Koch, K.M.; Im, Y.-H.; Kim, S.-B.; Ribate, A.U.; Stephenson, J.; Botbyl, J.; Cartee, L.; Holshouser, J.; Ridgway, D. Effects of Esomeprazole on the Pharmacokinetics of Lapatinib in Breast Cancer Patients: Clinical Pharmacology in Drug Development. CPDD 2013, 2, 336–341. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing Solubility of Poorly Aqueous Soluble Drugs: Critical Appraisal of Techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Khan, K.U.; Minhas, M.U.; Badshah, S.F.; Suhail, M.; Ahmad, A.; Ijaz, S. Overview of Nanoparticulate Strategies for Solubility Enhancement of Poorly Soluble Drugs. Life Sci. 2022, 291, 120301. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, J.; Xie, Y. Improvement Strategies for the Oral Bioavailability of Poorly Water-Soluble Flavonoids: An Overview. Int. J. Pharm. 2019, 570, 118642. [Google Scholar] [CrossRef]
- Yu, H.; Ma, Y.; Zhang, Y.; Zhang, H.; Zuo, L.; Hao, C.; Yu, W.; Lin, X.; Zhang, Y.; Qi, X.; et al. Tailored Supersaturable Immediate Release Behaviors of Hypotensive Supersaturating Drug-Delivery Systems Combined with Hot-Melt Extrusion Technique and Self-Micellizing Polymer. Polymers 2022, 14, 4800. [Google Scholar] [CrossRef]
- Yu, L. Amorphous Pharmaceutical Solids: Preparation, Characterization and Stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- De Araujo, G.L.B.; Benmore, C.J.; Byrn, S.R. Local Structure of Ion Pair Interaction in Lapatinib Amorphous Dispersions Characterized by Synchrotron X-Ray Diffraction and Pair Distribution Function Analysis. Sci. Rep. 2017, 7, 46367. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Chen, X.; Nie, H.; Byrn, S.; Lubach, J.W. Investigation of Drug–Excipient Interactions in Lapatinib Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy. Mol. Pharm. 2015, 12, 857–866. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Lou, H.; Hageman, M.J. Preparation of Lapatinib Ditosylate Solid Dispersions Using Solvent Rotary Evaporation and Hot Melt Extrusion for Solubility and Dissolution Enhancement. Int. J. Pharm. 2018, 552, 154–163. [Google Scholar] [CrossRef]
- Prabhu, P.P.; Prathvi; Gujaran, T.V.; Mehta, C.H.; Suresh, A.; Koteshwara, K.B.; Pai, K.G.; Nayak, U.Y. Development of Lapatinib Nanosponges for Enhancing Bioavailability. J. Drug Deliv. Sci. Technol. 2021, 65, 102684. [Google Scholar] [CrossRef]
- Yu, D.-G.; Li, J.-J.; Williams, G.R.; Zhao, M. Electrospun Amorphous Solid Dispersions of Poorly Water-Soluble Drugs: A Review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef]
- Nagy, Z.K.; Balogh, A.; Démuth, B.; Pataki, H.; Vigh, T.; Szabó, B.; Molnár, K.; Schmidt, B.T.; Horák, P.; Marosi, G.; et al. High Speed Electrospinning for Scaled-up Production of Amorphous Solid Dispersion of Itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef]
- Reneker, D.H.; Yarin, A.L. Electrospinning Jets and Polymer Nanofibers. Polymer 2008, 49, 2387–2425. [Google Scholar] [CrossRef]
- Bitay, E.; Gergely, A.L.; Balint, I.; Molnar, K.; Fulop, I.; Fogarasi, E.; Szabo, Z.I. Preparation and Characterization of Lapatinib-Loaded PVP Nanofiber Amorphous Solid Dispersion by Electrospinning. Express Polym. Lett. 2021, 15, 1041–1050. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, J.; Zhou, L.; Xu, Z.; Xing, J.; Feng, Q. Recent Progress of the Needleless Electrospinning for High Throughput of Nanofibers. NANOTEC 2020, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Bitay, E.; Szabo, Z.-I.; Kantor, J.; Molnar, K.; Gergely, A.L. Scale-up and Optimization of Fenofibrate-Loaded Fibers Electrospun by Corona-Electrospinning. Express Polym. Lett. 2021, 15, 375–387. [Google Scholar] [CrossRef]
- Molnar, K.; Nagy, Z.K. Corona-Electrospinning: Needleless Method for High-Throughput Continuous Nanofiber Production. Eur. Polym. J. 2016, 74, 279–286. [Google Scholar] [CrossRef]
- Li, T.-T.; Yan, M.; Xu, W.; Shiu, B.-C.; Lou, C.-W.; Lin, J.-H. Mass-Production and Characterizations of Polyvinyl Alcohol/Sodium Alginate/Graphene Porous Nanofiber Membranes Using Needleless Dynamic Linear Electrospinning. Polymers 2018, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, J.J.; Bastiaansen, C.W.M.; Peijs, T. Rotary Jet Spinning Review—A Potential High Yield Future for Polymer Nanofibers. Nanocomposites 2017, 3, 97–121. [Google Scholar] [CrossRef]
- Jindal, A.; Puskas, J.E.; McClain, A.; Nedic, K.; Luebbers, M.T.; Baker, J.R.; Dos Santos, B.P.; Camassola, M.; Jennings, W.; Einsporn, R.L.; et al. Encapsulation and Release of Zafirlukast from Electrospun Polyisobutylene-Based Thermoplastic Elastomeric Fiber Mat. Eur. Polym. J. 2018, 98, 254–261. [Google Scholar] [CrossRef]
- Farkas, B.; Balogh, A.; Cselkó, R.; Molnár, K.; Farkas, A.; Borbás, E.; Marosi, G.; Nagy, Z.K. Corona Alternating Current Electrospinning: A Combined Approach for Increasing the Productivity of Electrospinning. Int. J. Pharm. 2019, 561, 219–227. [Google Scholar] [CrossRef]
- Padron, S.; Fuentes, A.; Caruntu, D.; Lozano, K. Experimental Study of Nanofiber Production through Forcespinning. J. Appl. Phys. 2013, 113, 024318. [Google Scholar] [CrossRef]
- Doan, H.N.; Nguyen, D.K.; Vo, P.P.; Hayashi, K.; Kinashi, K.; Sakai, W.; Tsutsumi, N.; Huynh, D.P. Facile and Scalable Fabrication of Porous Polystyrene Fibers for Oil Removal by Centrifugal Spinning. ACS Omega 2019, 4, 15992–16000. [Google Scholar] [CrossRef]
- Niu, H.; Wang, X.; Lin, T. Needleless Electrospinning: Influences of Fibre Generator Geometry. J. Text. Inst. 2012, 103, 787–794. [Google Scholar] [CrossRef]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Craig, D.Q.M. Development of Micro-Fibrous Solid Dispersions of Poorly Water-Soluble Drugs in Sucrose Using Temperature-Controlled Centrifugal Spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. [Google Scholar] [CrossRef]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Aliev, A.E.; Craig, D.Q.M. Microfibrous Solid Dispersions of Poorly Water-Soluble Drugs Produced via Centrifugal Spinning: Unexpected Dissolution Behavior on Recrystallization. Mol. Pharm. 2017, 14, 1666–1680. [Google Scholar] [CrossRef]
- Sebe, I.; Kállai-Szabó, B.; Oldal, I.; Zsidai, L.; Zelkó, R. Development of Laboratory-Scale High-Speed Rotary Devices for a Potential Pharmaceutical Microfibre Drug Delivery Platform. Int. J. Pharm. 2020, 588, 119740. [Google Scholar] [CrossRef]
- Fong, H.; Chun, I.; Reneker, D.H. Beaded Nanofibers Formed during Electrospinning. Polymer 1999, 40, 4585–4592. [Google Scholar] [CrossRef]
- Turner, D.T.; Schwartz, A. The Glass Transition Temperature of Poly(N-Vinyl Pyrrolidone) by Differential Scanning Calorimetry. Polymer 1985, 26, 757–762. [Google Scholar] [CrossRef]
- Zetina-Rocha, C.; Cammisa, E.G.; Weeratunga, G. Polymorphic Forms of Lapatinib Ditosylate and Processes for Their Preparation. US8993579 B2, 31 March 2015. [Google Scholar]
- Amponsah-Efah, K.K.; Mistry, P.; Eisenhart, R.; Suryanarayanan, R. The Influence of the Strength of Drug–Polymer Interactions on the Dissolution of Amorphous Solid Dispersions. Mol. Pharm. 2021, 18, 174–186. [Google Scholar] [CrossRef]
- Schmied, F.-P.; Bernhardt, A.; Moers, C.; Meier, C.; Endres, T.; Klein, S. A Novel Aminomethacrylate-Based Copolymer for Solubility Enhancement—From Radical Polymer Synthesis to Manufacture and Characterization of Amorphous Solid Dispersions. Polymers 2022, 14, 1281. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Speed (rpm) | Fiber Diameter (μm) | Beading |
|---|---|---|
| 3500 | 4.38 ± 2.60 | yes |
| 4500 | 3.82 ± 2.42 | yes |
| 5500 | 1.40 ± 0.75 | no |
| 6500 | 2.87 ± 1.05 | no |
| 7500 | 2.55 ± 1.27 | no |
| Sample | Measured LB Content (%) | Theoretical LB Content (%) | Relative Content (%) | Average Relative Content (%) | Standard Deviation |
|---|---|---|---|---|---|
| Lap-PVP_1 | 12.07 | 102.59 | |||
| Lap-PVP_2 | 11.68 | 11.76 | 99.27 | 100.99 | 1.66 |
| Lap-PVP_3 | 11.89 | 101.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitay, E.; Gergely, A.L.; Kántor, J.; Szabó, Z.-I. Evaluation of Lapatinib-Loaded Microfibers Prepared by Centrifugal Spinning. Polymers 2022, 14, 5557. https://doi.org/10.3390/polym14245557
Bitay E, Gergely AL, Kántor J, Szabó Z-I. Evaluation of Lapatinib-Loaded Microfibers Prepared by Centrifugal Spinning. Polymers. 2022; 14(24):5557. https://doi.org/10.3390/polym14245557
Chicago/Turabian StyleBitay, Enikő, Attila Levente Gergely, József Kántor, and Zoltán-István Szabó. 2022. "Evaluation of Lapatinib-Loaded Microfibers Prepared by Centrifugal Spinning" Polymers 14, no. 24: 5557. https://doi.org/10.3390/polym14245557
APA StyleBitay, E., Gergely, A. L., Kántor, J., & Szabó, Z.-I. (2022). Evaluation of Lapatinib-Loaded Microfibers Prepared by Centrifugal Spinning. Polymers, 14(24), 5557. https://doi.org/10.3390/polym14245557