

Polyacrylonitrile-b-Polystyrene Block Copolymer-Derived Hierarchical Porous Carbon Materials for Supercapacitor

 ,
,  ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PS−CPDT MacroCTA
2.3. Synthesis of PS−b−PAN Copolymer
2.4. Preparation of Porous Carbon Materials
2.5. Characterization
3. Results and Discussion
3.1. Synthesis of PS−b−PAN Copolymer
3.2. Thermal Characterization
3.3. Structural Characterization
3.4. Electrochemical Characterization

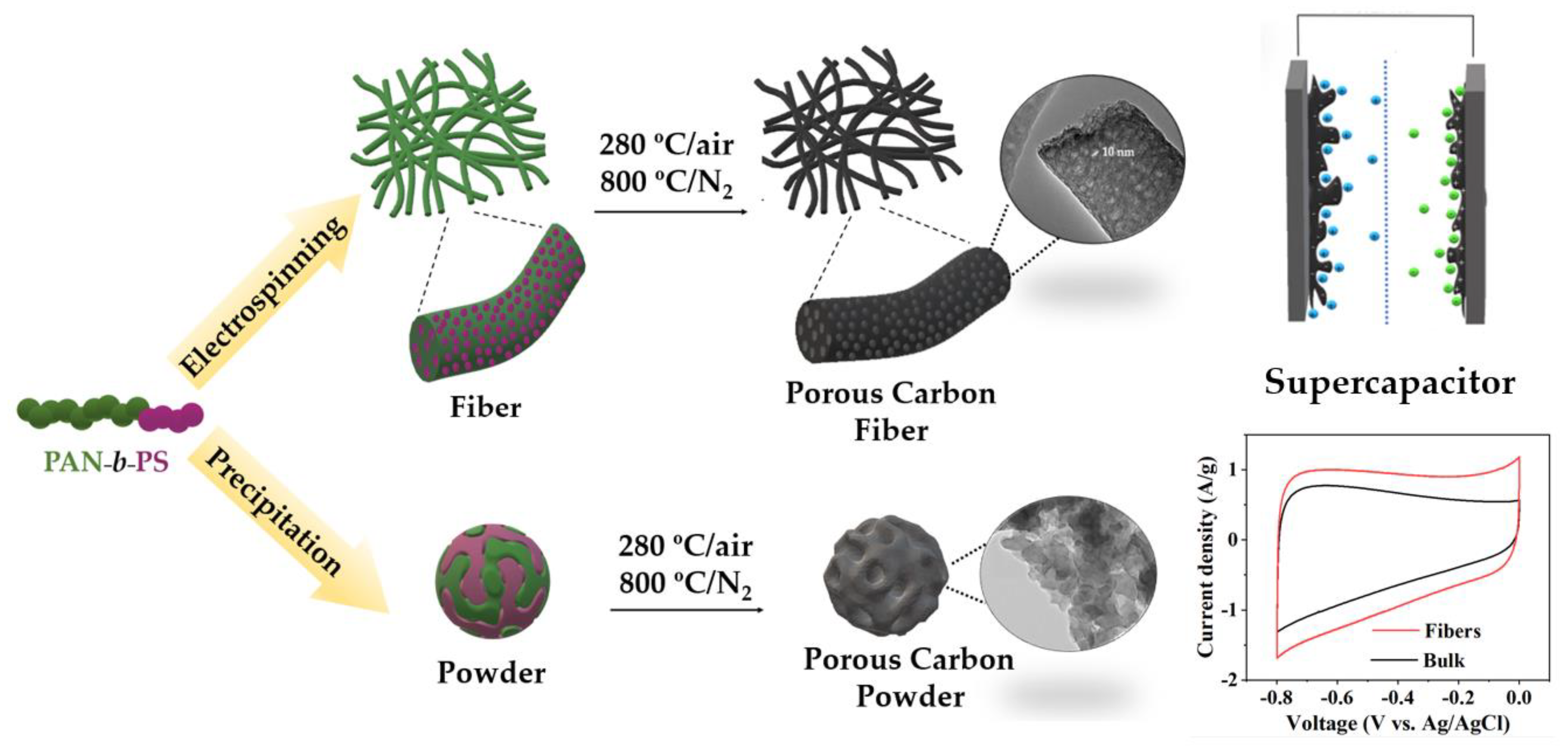
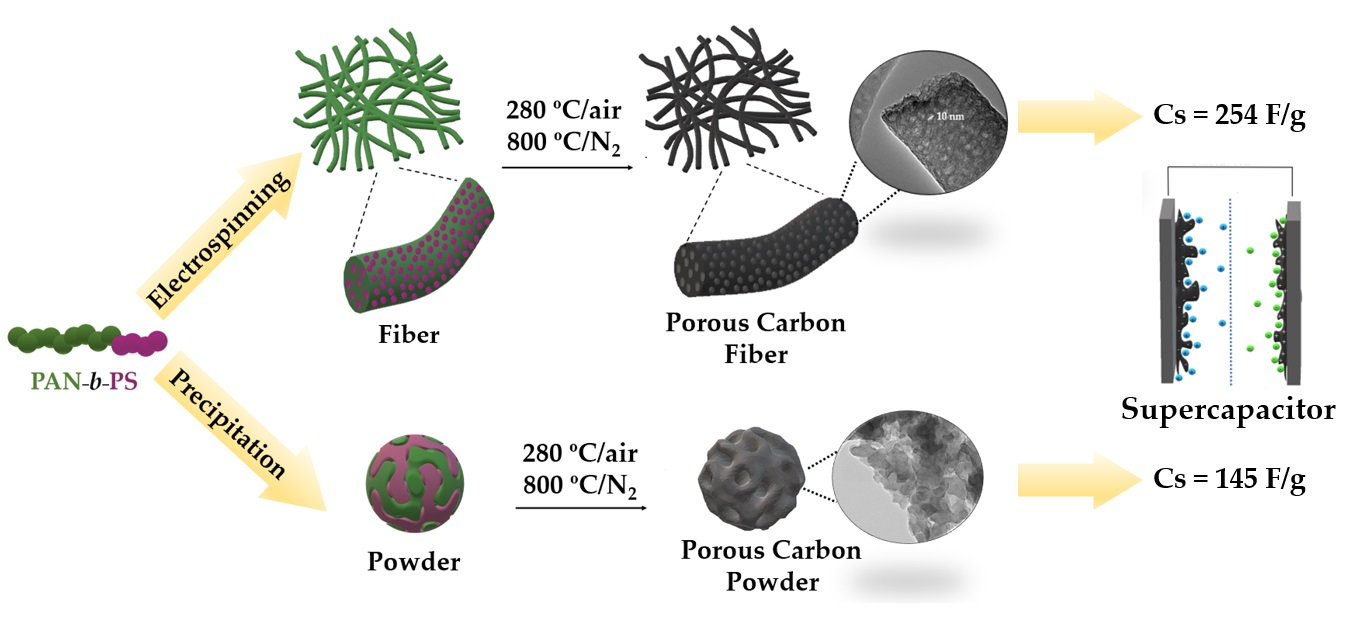{kind=link}
{kind=link}
{kind=link}
{kind=link}
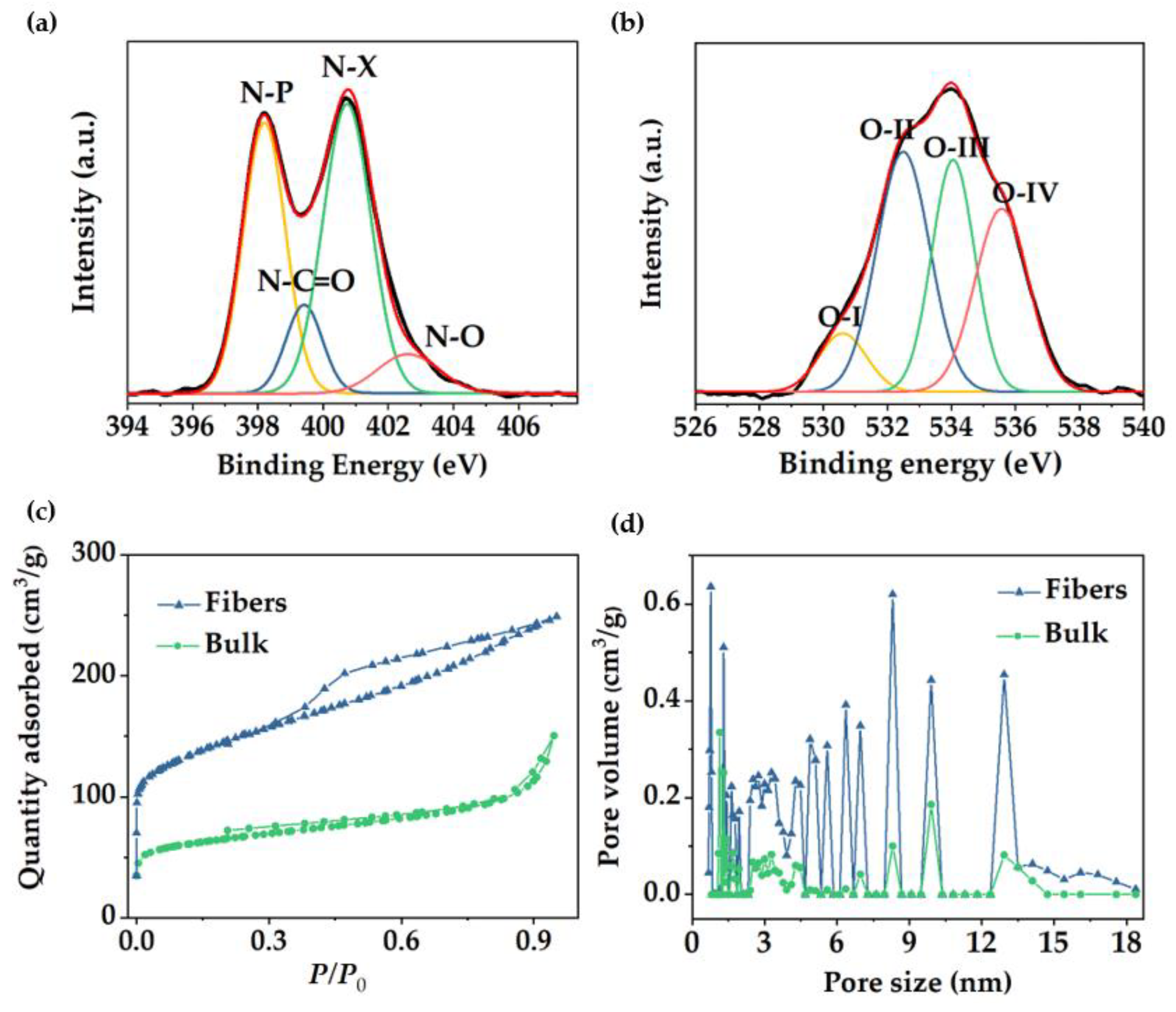{kind=link}
{kind=link}
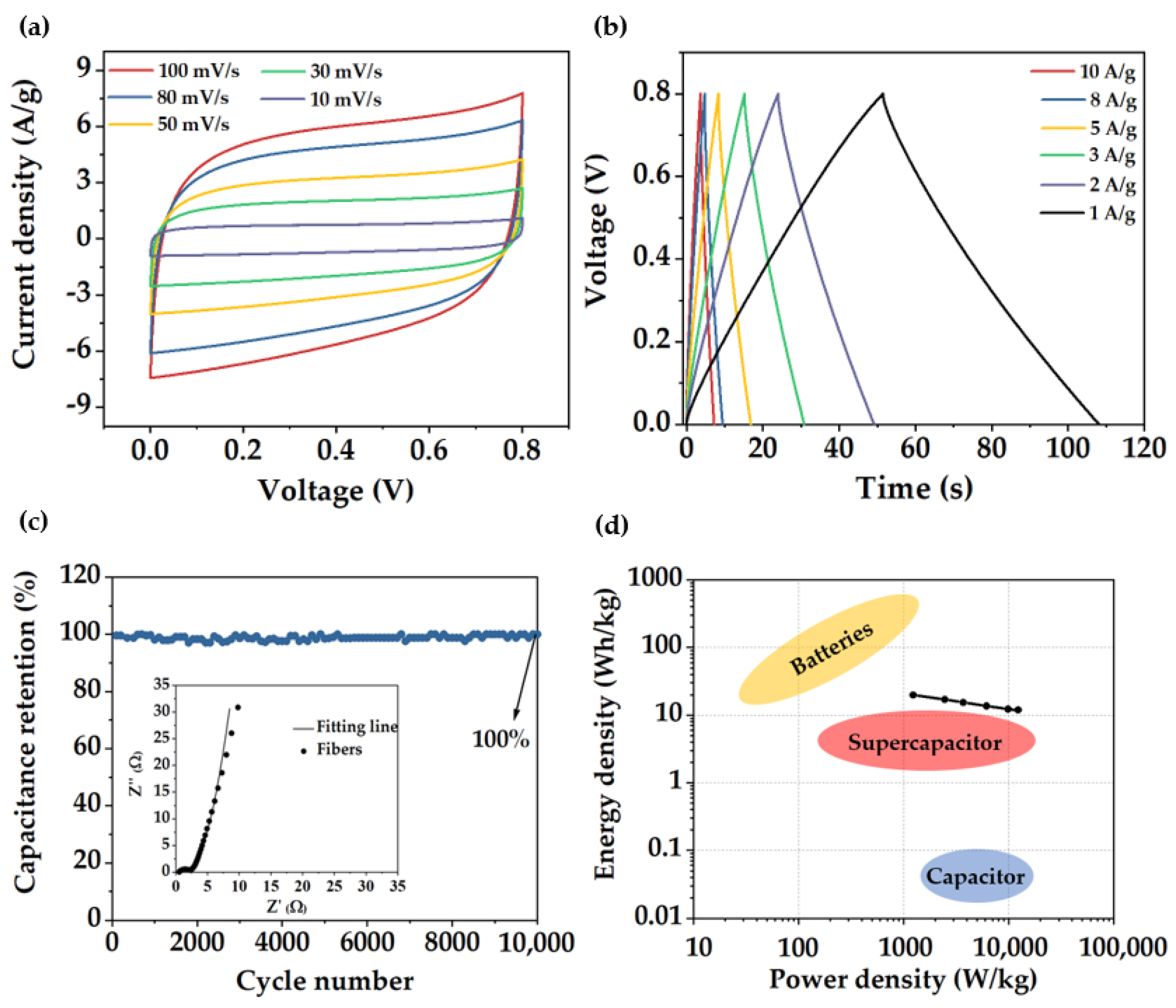{kind=link}
{kind=link}
| Material | Precursor | SSA (m2/g) | Cs (F/g) | Electrolyte | Reference |
|---|---|---|---|---|---|
| Powder carbons | |||||
| S/N-doped porous carbons | PBA−b−PAN | 478 | 236 (0.1 A/g) | KOH 6M | [58] |
| Open-ended hollow carbon spheres (HCS) | PS−b−P4VP/ KOH activation | 1583 | 249 (0.5 A/g) | KOH 6M | [59] |
| N-doped hierarchical porous carbons (NHPC) | PS−b−PAN/KOH activation | 2105 | 230 (1 A/g) | KOH 6M | [47] |
| Mesoporous carbons | PAN−b−PS−b−PAN | 954 | 185 (0.625 A/g) | KOH 2M | [21] |
| Fibers | |||||
| Linear-tube carbon nanofibers (LTCNF) | PAN/PS | 212 | 188 (0.5 A/g) | LiOH 3M | [60] |
| Multichannel PCFs | PAN/PS | 750 | 250 (1 A/g) | KOH 6M | [61] |
| N-doped multi-nano-channel PCFs | PAN/PS | 840 | 461 (0.25 A/g) | H2SO4 1M | [62] |
| Mesoporous carbon nanofibers (MCNFs) | PAN/PAA−b−PAN−b−PAA | 250 | 256 (0.5 A/g) | KOH 4M | [23] |
| PCFs | PAN/PMMMA | 683 | 140 (0.5 A/g) | KOH 6M | [63] |
| Interconnected meso-PCFs | PMMA−b−PAN | 503 | 360 (1 A/g) | KOH 6M | [34] |
| Hierarchical PCFs | PS−b−PAN | 492 | 254 (1 A/g) | KOH 6M | This work |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salanne, M.; Rotenberg, B.; Naoi, K.; Kaneko, K.; Taberna, P.L.; Grey, C.P.; Dunn, B.; Simon, P. Efficient Storage Mechanisms for Building Better Supercapacitors. Nat. Energy 2016, 1, 16070. [Google Scholar] [CrossRef]
- Patrice, S.; Yuri, G.; Bruce, D. Where Do Batteries End and Supercapacitors Begin? Science 2014, 343, 1208–1210. [Google Scholar]
- Pandolfo, A.G.; Hollenkamp, A.F. Carbon Properties and Their Role in Supercapacitors. J. Power Sources 2006, 157, 11–27. [Google Scholar] [CrossRef]
- González, A.; Goikolea, E.; Barrena, J.A.; Mysyk, R. Review on Supercapacitors: Technologies and Materials. Renew. Sustain. Energy Rev. 2016, 58, 1189–1206. [Google Scholar] [CrossRef]
- Liu, C.F.; Liu, Y.C.; Yi, T.Y.; Hu, C.C. Carbon Materials for High-Voltage Supercapacitors. Carbon N. Y. 2019, 145, 529–548. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Materials for Electrochemical Capacitors. Nat. Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef]
- Liu, N.; Shen, J.; Liu, D. Activated High Specific Surface Area Carbon Aerogels for EDLCs. Microporous Mesoporous Mater. 2013, 167, 176–181. [Google Scholar] [CrossRef]
- Menéndez-Díaz, J.A.; Martín-Gullón, I. Chapter 1 Types of Carbon Adsorbents and Their Production. Interface Sci. Technol. 2006, 7, 1–47. [Google Scholar] [CrossRef]
- Kim, Y.J.; Horie, Y.; Ozaki, S.; Matsuzawa, Y.; Suezaki, H.; Kim, C.; Miyashita, N.; Endo, M. Correlation between the Pore and Solvated Ion Size on Capacitance Uptake of PVDC-Based Carbons. Carbon N. Y. 2004, 42, 1491–1500. [Google Scholar] [CrossRef]
- Vatamanu, J.; Bedrov, D. Capacitive Energy Storage: Current and Future Challenges. J. Phys. Chem. Lett. 2015, 6, 3594–3609. [Google Scholar] [CrossRef]
- Wu, D.; Xu, F.; Sun, B.; Fu, R.; He, H.; Matyjaszewski, K. Design and Preparation of Porous Polymers. Chem. Rev. 2012, 112, 3959–4015. [Google Scholar] [CrossRef]
- Serrano, J.M.; Liu, T.; Khan, A.U.; Botset, B.; Stovall, B.J.; Xu, Z.; Guo, D.; Cao, K.; Hao, X.; Cheng, S.; et al. Composition Design of Block Copolymers for Porous Carbon Fibers. Chem. Mater. 2019, 31, 8898–8907. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Keddie, D.J. A Guide to the Synthesis of Block Copolymers Using Reversible-Addition Fragmentation Chain Transfer (RAFT) Polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tang, Z.; Ou, B.; Liu, L.; Zhou, Z.; Shen, S.; Duan, Y. Design, Preparation, and Application of Ordered Porous Polymer Materials. Mater. Chem. Phys. 2014, 144, 213–225. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Y.; Mei, S.; Lu, Y.; Zhang, M.; Sun, J.K.; Matyjaszewski, K.; Antonietti, M.; Yuan, J. Polymer-Derived Heteroatom-Doped Porous Carbon Materials. Chem. Rev. 2020, 120, 9363–9419. [Google Scholar] [CrossRef]
- McGann, J.P.; Zhong, M.; Kim, E.K.; Natesakhawat, S.; Jaroniec, M.; Whitacre, J.F.; Matyjaszewski, K.; Kowalewski, T. Block Copolymer Templating as a Path to Porous Nanostructured Carbons with Highly Accessible Nitrogens for Enhanced (Electro)Chemical Performance. Macromol. Chem. Phys. 2012, 213, 1078–1090. [Google Scholar] [CrossRef]
- Liu, T.; Liu, G. Block Copolymer-Based Porous Carbons for Supercapacitors. J. Mater. Chem. A 2019, 7, 23476–23488. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, W.; Alhebshi, N.A.; Salah, N.; Alshareef, H.N. Synthesis Strategies of Porous Carbon for Supercapacitor Applications. Small Methods 2020, 4, 1900853. [Google Scholar] [CrossRef]
- Kopeć, M.; Yuan, R.; Gottlieb, E.; Abreu, C.M.R.; Song, Y.; Wang, Z.; Coelho, J.F.J.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile-b-Poly(Butyl Acrylate) Block Copolymers as Precursors to Mesoporous Nitrogen-Doped Carbons: Synthesis and Nanostructure. Macromolecules 2017, 50, 2759–2767. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, L.B.; Li, X.M.; Ran, F.; Luo, Y.C.; Kang, L. Mesoporous Carbons for Supercapacitors Obtained by the Pyrolysis of Block Copolymers. Xinxing Tan Cailiao/New Carbon Mater. 2015, 30, 302–309. [Google Scholar] [CrossRef]
- Kruk, M.; Dufour, B.; Celer, E.B.; Kowalewski, T.; Jaroniec, M.; Matyjaszewski, K. Well-Defined Poly(Ethylene Oxide)-Polyacrylonitrile Diblock Copolymers as Templates for Mesoporous Silicas and Precursors for Mesoporous Carbons. Chem. Mater. 2006, 18, 1417–1424. [Google Scholar] [CrossRef]
- Dong, W.; Wang, Z.; Zhang, Q.; Ravi, M.; Yu, M.; Tan, Y.; Liu, Y.; Kong, L.; Kang, L.; Ran, F. Polymer/Block Copolymer Blending System as the Compatible Precursor System for Fabrication of Mesoporous Carbon Nanofibers for Supercapacitors. J. Power Sources 2019, 419, 137–147. [Google Scholar] [CrossRef]
- Nguyen, C.T.; Kim, D.P. Direct Preparation of Mesoporous Carbon by Pyrolysis of Poly(Acrylonitrile-b-Methylmethacrylate) Diblock Copolymer. J. Mater. Chem. 2011, 21, 14226–14230. [Google Scholar] [CrossRef]
- Carriazo, D.; Picó, F.; Gutiérrez, M.C.; Rubio, F.; Rojo, J.M.; Del Monte, F. Block-Copolymer Assisted Synthesis of Hierarchical Carbon Monoliths Suitable as Supercapacitor Electrodes. J. Mater. Chem. 2010, 20, 773–780. [Google Scholar] [CrossRef]
- Ran, F.; Shen, K.; Tan, Y.; Peng, B.; Chen, S.; Zhang, W.; Niu, X.; Kong, L.; Kang, L. Activated Hierarchical Porous Carbon as Electrode Membrane Accommodated with Triblock Copolymer for Supercapacitors. J. Memb. Sci. 2016, 514, 366–375. [Google Scholar] [CrossRef]
- Wen, Y.; Kok, M.D.R.; Tafoya, J.P.V.; Sobrido, A.B.J.; Bell, E.; Gostick, J.T.; Herou, S.; Schlee, P.; Titirici, M.M.; Brett, D.J.L.; et al. Electrospinning as a Route to Advanced Carbon Fibre Materials for Selected Low-Temperature Electrochemical Devices: A Review. J. Energy Chem. 2021, 59, 492–529. [Google Scholar] [CrossRef]
- Newcomb, B.A. Processing, Structure, and Properties of Carbon Fibers. Compos. Part A Appl. Sci. Manuf. 2016, 91, 262–282. [Google Scholar] [CrossRef]
- Kim, C.; Yang, K.S.; Lee, W.J. The Use of Carbon Nanofiber Electrodes Prepared by Electrospinning for Electrochemical Supercapacitors. Electrochem. Solid-State Lett. 2004, 7, 397–399. [Google Scholar] [CrossRef]
- Josef, E.; Yan, R.; Guterman, R.; Oschatz, M. Electrospun Carbon Fibers Replace Metals as a Current Collector in Supercapacitors. ACS Appl. Energy Mater. 2019, 2, 5724–5733. [Google Scholar] [CrossRef]
- Jo, E.; Yeo, J.G.; Kim, D.K.; Oh, J.S.; Hong, C.K. Preparation of Well-Controlled Porous Carbon Nanofiber Materials by Varying the Compatibility of Polymer Blends. Polym. Int. 2014, 63, 1471–1477. [Google Scholar] [CrossRef]
- He, T.; Fu, Y.; Meng, X.; Yu, X.; Wang, X. A Novel Strategy for the High Performance Supercapacitor Based on Polyacrylonitrile-Derived Porous Nanofibers as Electrode and Separator in Ionic Liquid Electrolyte. Electrochim. Acta 2018, 282, 97–104. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Wang, H.; Jin, X.; Niu, H.; Wang, H.; Zhou, H.; Lin, T. High Performance Supercapacitor Electrode Materials from Electrospun Carbon Nanofibers in Situ Activated by High Decomposition Temperature Polymer. ACS Appl. Energy Mater. 2018, 1, 431–439. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Khan, A.U.; Liu, G. Block Copolymer–Based Porous Carbon Fibers. Sci. Adv. 2019, 5, eaau6852. [Google Scholar] [CrossRef]
- Ponnusamy, K.; Babu, R.P.; Dhamodharan, R. Synthesis of Block and Graft Copolymers of Styrene by Raft Polymerization, Using Dodecyl-Based Trithiocarbonates as Initiators and Chain Transfer Agents. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1066–1078. [Google Scholar] [CrossRef]
- Zhong, M.; Tang, C.; Kim, E.K.; Kruk, M.; Celer, E.B.; Jaroniec, M.; Matyjaszewski, K.; Kowalewski, T. Preparation of Porous Nanocarbons with Tunable Morphology and Pore Size from Copolymer Templated Precursors. Mater. Horizons 2014, 1, 121–124. [Google Scholar] [CrossRef]
- Kopeć, M.; Lamson, M.; Yuan, R.; Tang, C.; Kruk, M.; Zhong, M.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile-Derived Nanostructured Carbon Materials. Prog. Polym. Sci. 2019, 92, 89–134. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Melting Behavior of Acrylonitrile Polymers. J. Appl. Polym. Sci. 1998, 70, 2703–2709. [Google Scholar] [CrossRef]
- Dang, W.; Liu, J.; Wang, X.; Yan, K.; Zhang, A.; Yang, J.; Chen, L.; Liang, J. Structural Transformation of Polyacrylonitrile (PAN) Fibers during Rapid Thermal Pretreatment in Nitrogen Atmosphere. Polymers 2020, 12, 63. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, G. Controlling the Pore Size of Mesoporous Carbon Thin Films through Thermal and Solvent Annealing. Small 2017, 13, 19–21. [Google Scholar] [CrossRef]
- Cho, J. Analysis of Phase Separation in Compressible Polymer Blends and Block Copolymers. Macromolecules 2000, 33, 2228–2241. [Google Scholar] [CrossRef]
- Majewski, P.W.; Yager, K.G. Rapid Ordering of Block Copolymer Thin Films. J. Phys. Condens. Matter 2016, 28, 403002. [Google Scholar] [CrossRef] [PubMed]
- Matsen, M.W.; Bates, F.S. Unifying Weak- and Strong-Segregation Block Copolymer Theories. Macromolecules 1996, 29, 1091–1098. [Google Scholar] [CrossRef]
- Cowie, J.M.; Lath, D. Miscibility mapping in some blends involving poly (styrene-co-acrylonitrile). In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1988; Volume 16, pp. 103–112. [Google Scholar]
- Liu, T.; Liu, G. Block Copolymers for Supercapacitors, Dielectric Capacitors and Batteries. J. Phys. Condens. Matter 2019, 31, 233001. [Google Scholar] [CrossRef]
- Seo, M.; Hillmyer, M.A. Reticulated Nanoporous Polymers by Controlled Polymerization-Induced Microphase Separation. Science 2012, 336, 1422–1425. [Google Scholar] [CrossRef]
- Tong, Y.X.; Li, X.M.; Xie, L.J.; Su, F.Y.; Li, J.P.; Sun, G.H.; Gao, Y.D.; Zhang, N.; Wei, Q.; Chen, C.M. Nitrogen-Doped Hierarchical Porous Carbon Derived from Block Copolymer for Supercapacitor. Energy Storage Mater. 2016, 3, 140–148. [Google Scholar] [CrossRef]
- Zhang, H.; Ling, Y.; Peng, Y.; Zhang, J.; Guan, S. Nitrogen-Doped Porous Carbon Materials Derived from Ionic Liquids as Electrode for Supercapacitor. Inorg. Chem. Commun. 2020, 115, 107856. [Google Scholar] [CrossRef]
- Shimodaira, N.; Masui, A. Raman Spectroscopic Investigations of Activated Carbon Materials. J. Appl. Phys. 2002, 92, 902–909. [Google Scholar] [CrossRef]
- Hulicova-Jurcakova, D.; Seredych, M.; Lu, G.Q.; Bandosz, T.J. Combined Effect of Nitrogen- and Oxygen-Containing Functional Groups of Microporous Activated Carbon on Its Electrochemical Performance in Supercapacitors. Adv. Funct. Mater. 2009, 19, 438–447. [Google Scholar] [CrossRef]
- Lee, S.W.; Gallant, B.M.; Lee, Y.; Yoshida, N.; Kim, D.Y.; Yamada, Y.; Noda, S.; Yamada, A.; Yang, S.H. Self-Standing Positive Electrodes of Oxidized Few-Walled Carbon Nanotubes for Light-Weight and High-Power Lithium Batteries. Energy Environ. Sci. 2012, 5, 5437–5444. [Google Scholar] [CrossRef]
- Rosenthal, D.; Ruta, M.; Schlögl, R.; Kiwi-Minsker, L. Combined XPS and TPD Study of Oxygen-Functionalized Carbon Nanofibers Grown on Sintered Metal Fibers. Carbon N. Y. 2010, 48, 1835–1843. [Google Scholar] [CrossRef]
- He, H.; Hu, Y.; Chen, S.; Zhuang, L.; Ma, B.; Wu, Q. Preparation and Properties of A Hyperbranch-Structured Polyamine Adsorbent for Carbon Dioxide Capture. Sci. Rep. 2017, 7, 3913. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Yamashita, A.; Minoura, S.; Okamoto, M.; Morimoto, T. Modification of the Oxygen-Containing Functional Group on Activated Carbon Fiber in Electrodes of an Electric Double-Layer Capacitor. J. Power Sources 2006, 158, 1510–1516. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Frackowiak, E. Carbon Materials for Supercapacitor Application. Phys. Chem. Chem. Phys. 2007, 9, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Mei, B.A.; Munteshari, O.; Lau, J.; Dunn, B.; Pilon, L. Physical Interpretations of Nyquist Plots for EDLC Electrodes and Devices. J. Phys. Chem. C 2018, 122, 194–206. [Google Scholar] [CrossRef]
- Yuan, R.; Wang, H.; Sun, M.; Damodaran, K.; Gottlieb, E.; Kopeć, M.; Eckhart, K.; Li, S.; Whitacre, J.; Matyjaszewski, K.; et al. Well-Defined N/S Co-Doped Nanocarbons from Sulfurized PAN- b-PBA Block Copolymers: Structure and Supercapacitor Performance. ACS Appl. Nano Mater. 2019, 2, 2467–2474. [Google Scholar] [CrossRef]
- Cao, S.; Qu, T.; Zhang, A.; Zhao, Y.; Chen, A. N-Doped Hierarchical Porous Carbon with Open-Ended Structure for High-Performance Supercapacitors. ChemElectroChem 2019, 6, 1696–1703. [Google Scholar] [CrossRef]
- Bhoyate, S.; Kahol, P.K.; Sapkota, B.; Mishra, S.R.; Perez, F.; Gupta, R.K. Polystyrene Activated Linear Tube Carbon Nanofiber for Durable and High-Performance Supercapacitors. Surf. Coatings Technol. 2018, 345, 113–122. [Google Scholar] [CrossRef]
- Zhang, L.; Han, L.; Liu, S.; Zhang, C.; Liu, S. High-Performance Supercapacitors Based on Electrospun Multichannel Carbon Nanofibers. RSC Adv. 2015, 5, 107313–107317. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Shanmugam, S. Nitrogen-Doped Porous Multi-Nano-Channel Nanocarbons for Use in High-Performance Supercapacitor Applications. ACS Sustain. Chem. Eng. 2016, 4, 2439–2448. [Google Scholar] [CrossRef]
- He, G.; Song, Y.; Chen, S.; Wang, L. Porous Carbon Nanofiber Mats from Electrospun Polyacrylonitrile/Polymethylmethacrylate Composite Nanofibers for Supercapacitor Electrode Materials. J. Mater. Sci. 2018, 53, 9721–9730. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Gómez, A.; Yuan, J.; Fernández-Blázquez, J.P.; San-Miguel, V.; Serrano, M.B. Polyacrylonitrile-b-Polystyrene Block Copolymer-Derived Hierarchical Porous Carbon Materials for Supercapacitor. Polymers 2022, 14, 5109. https://doi.org/10.3390/polym14235109
Álvarez-Gómez A, Yuan J, Fernández-Blázquez JP, San-Miguel V, Serrano MB. Polyacrylonitrile-b-Polystyrene Block Copolymer-Derived Hierarchical Porous Carbon Materials for Supercapacitor. Polymers. 2022; 14(23):5109. https://doi.org/10.3390/polym14235109
Chicago/Turabian StyleÁlvarez-Gómez, Ainhoa, Jiayin Yuan, Juan P. Fernández-Blázquez, Verónica San-Miguel, and María B. Serrano. 2022. "Polyacrylonitrile-b-Polystyrene Block Copolymer-Derived Hierarchical Porous Carbon Materials for Supercapacitor" Polymers 14, no. 23: 5109. https://doi.org/10.3390/polym14235109
APA StyleÁlvarez-Gómez, A., Yuan, J., Fernández-Blázquez, J. P., San-Miguel, V., & Serrano, M. B. (2022). Polyacrylonitrile-b-Polystyrene Block Copolymer-Derived Hierarchical Porous Carbon Materials for Supercapacitor. Polymers, 14(23), 5109. https://doi.org/10.3390/polym14235109