

Carvedilol Precipitation Inhibition by the Incorporation of Polymeric Precipitation Inhibitors Using a Stable Amorphous Solid Dispersion Approach: Formulation, Characterization, and In Vitro In Vivo Evaluation

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
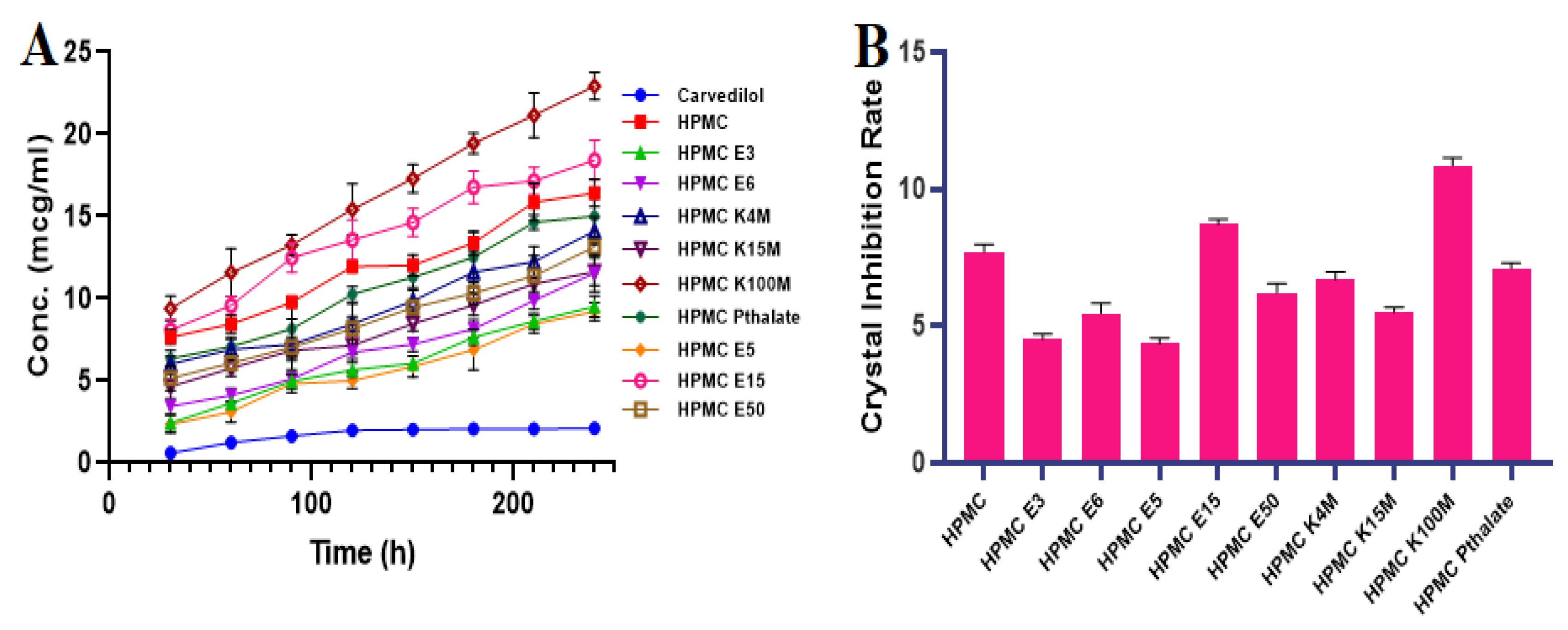
2.2.1. Inhibition Effect of Polymers on CVL Recrystallization in a Supersaturated State
2.2.2. Preparation of a CVL Solid Dispersion (CVL-SDs)
2.2.3. Practical Yield
2.2.4. Drug Content
2.3. Characterization of Solid Dispersion
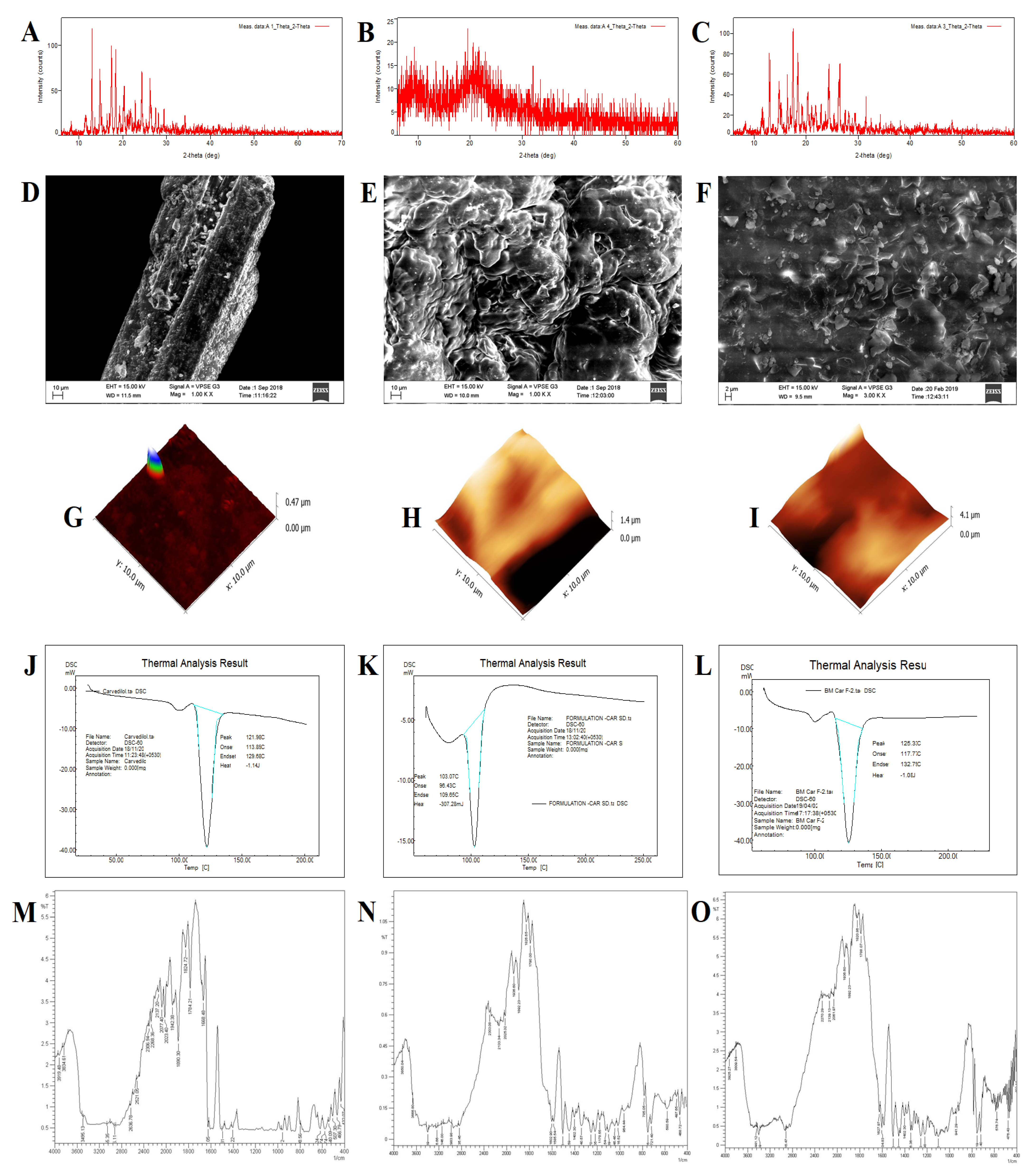
2.3.1. Powder X-ray Diffraction
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. Atomic Force Microscopy (AFM) Analysis
2.3.4. Differential Scanning Calorimetry (DSC)
2.3.5. Fourier-Transform Infrared Spectroscopy
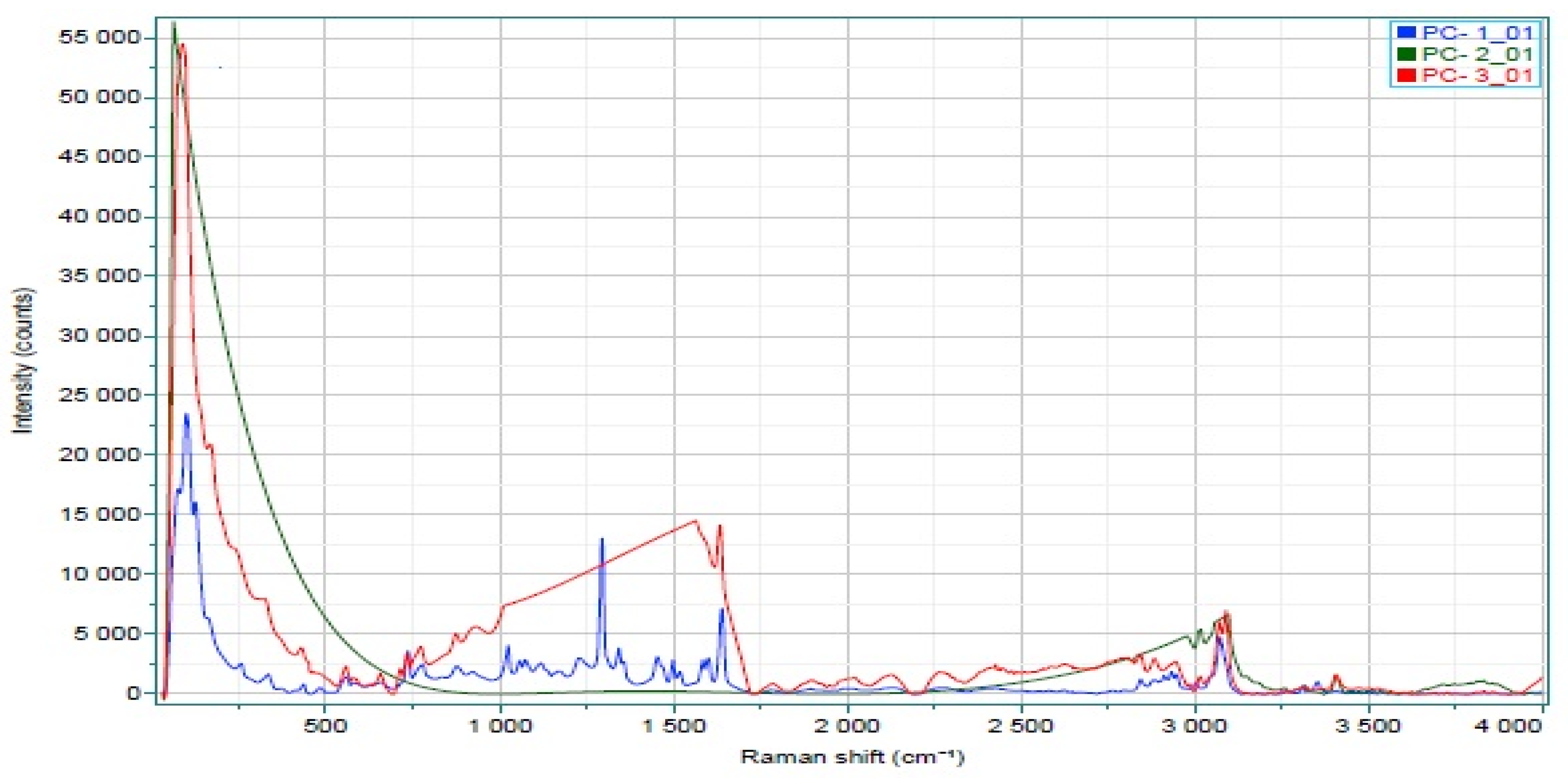
2.3.6. Raman Spectroscopy
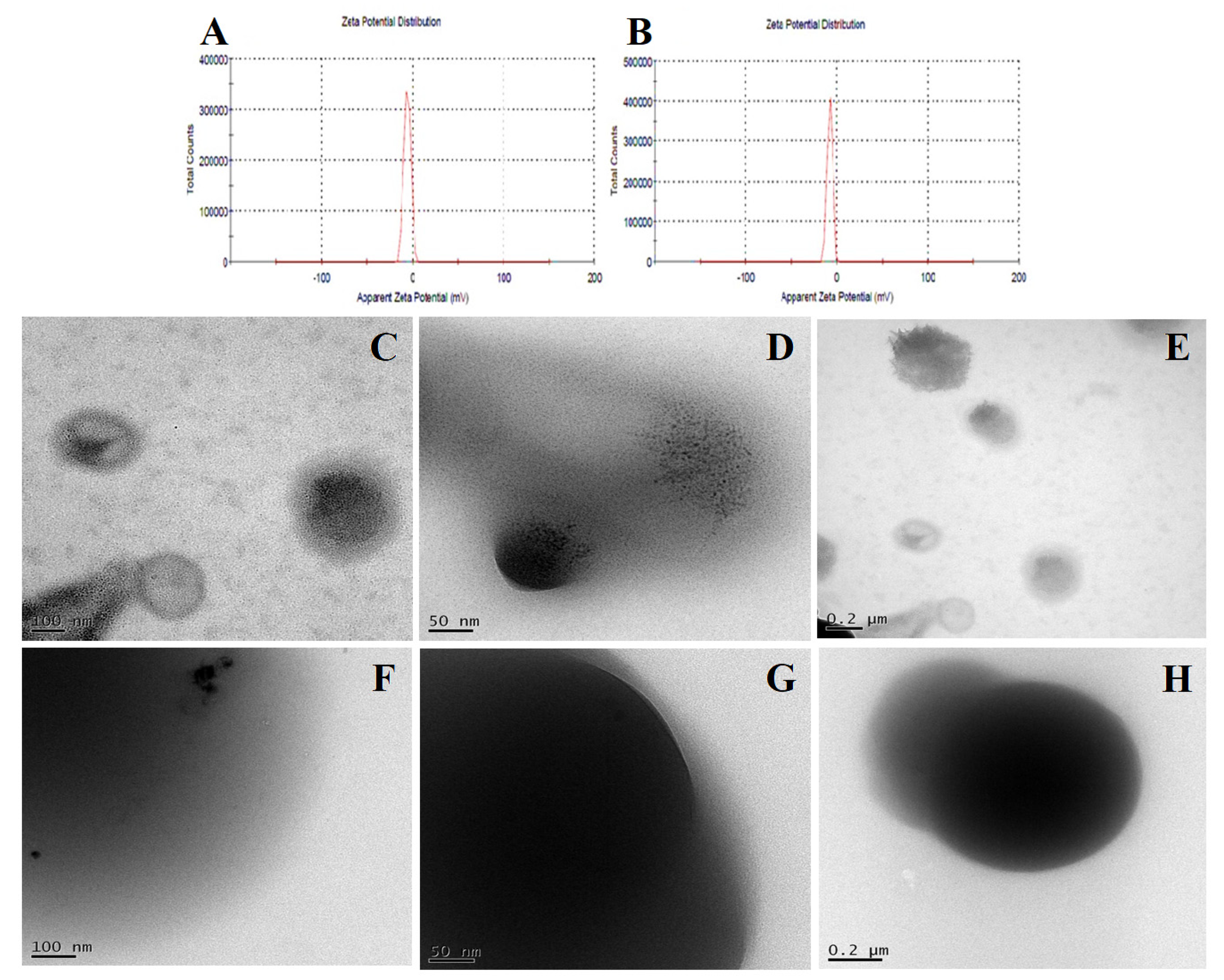
2.3.7. Dynamic Light Scattering
2.3.8. Transmission Electron Microscopy Analysis
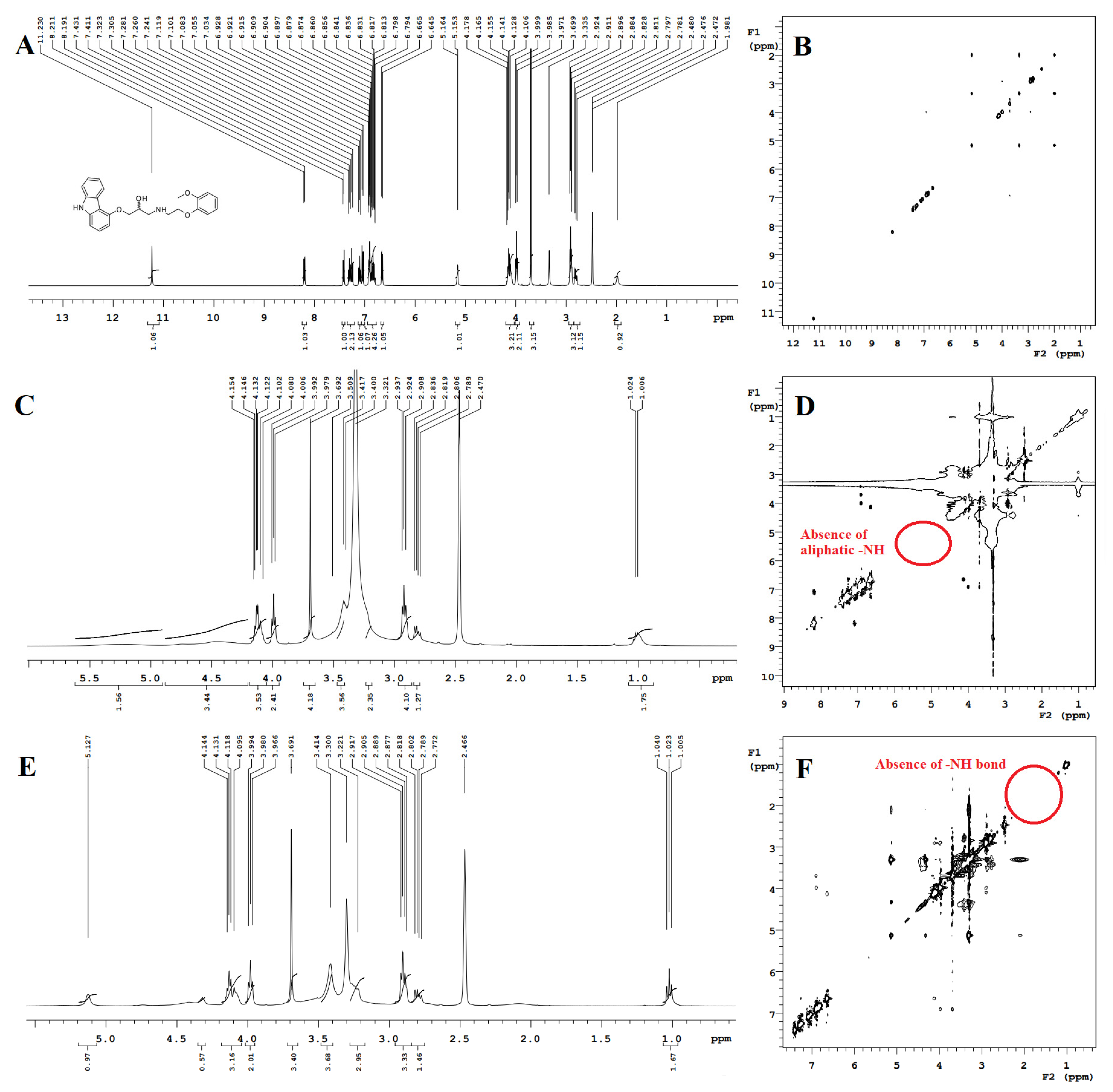
2.3.9. 1H Nuclear Magnetic Resonance
2.3.10. Nuclear Overhauser Enhancement Spectroscopy
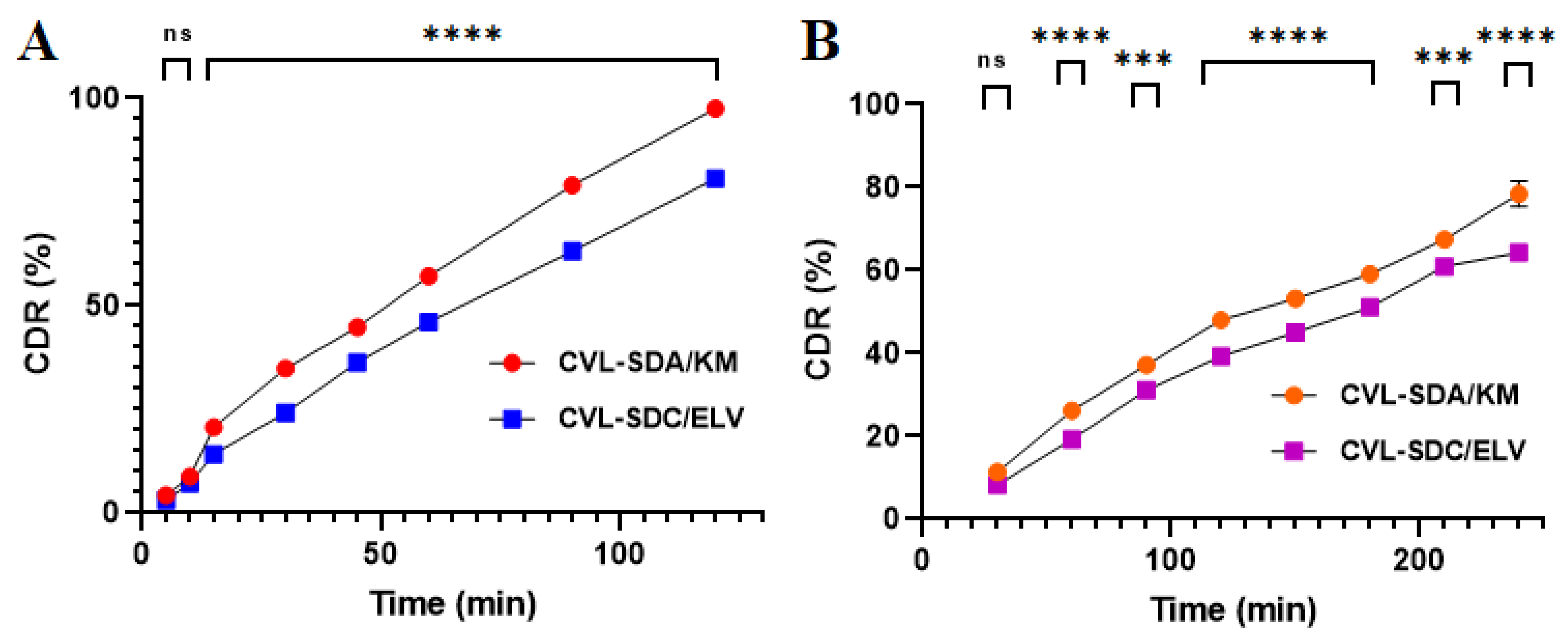
2.3.11. In Vitro Release Study
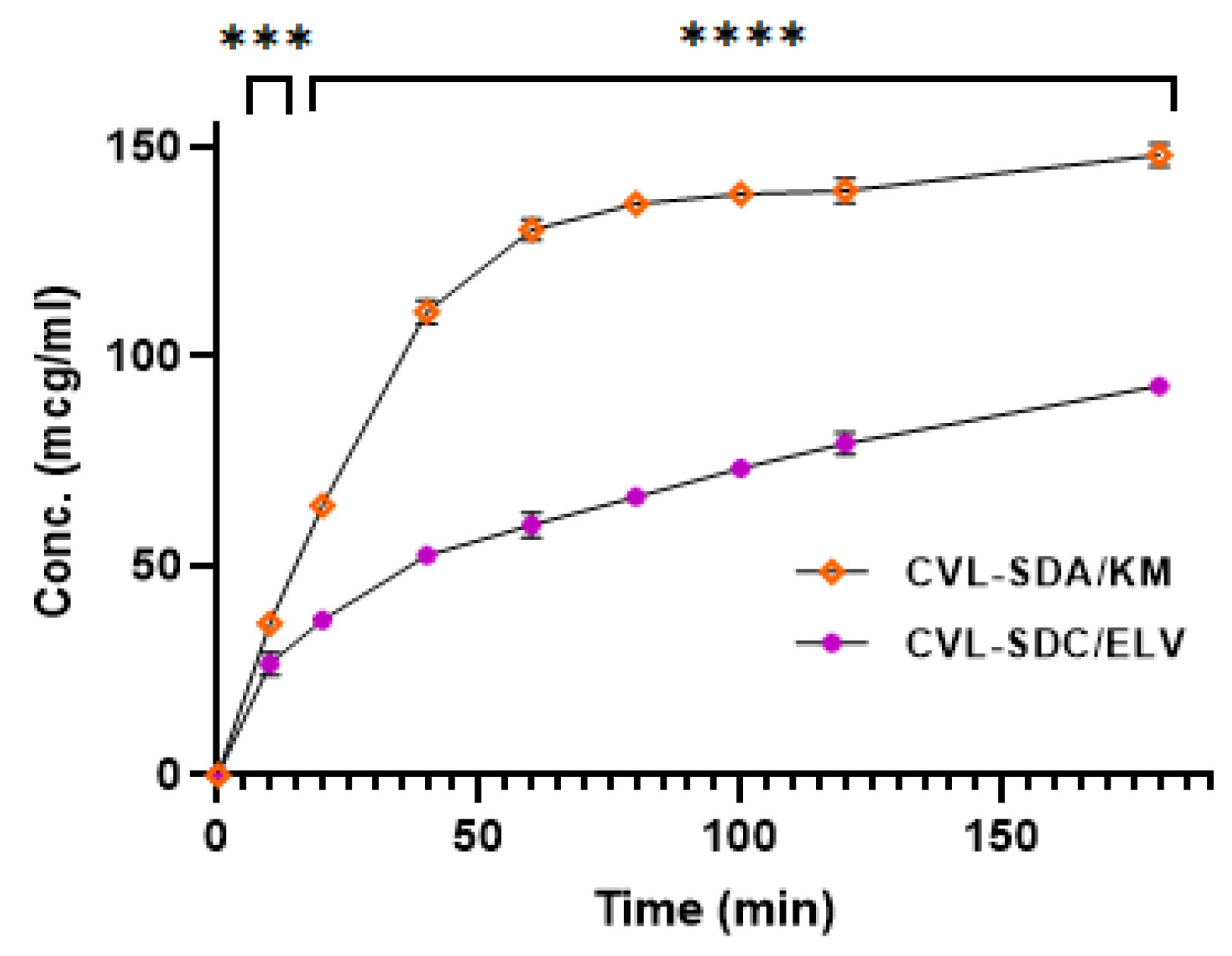
2.4. Infused Precipitation Study of Solid Dispersion
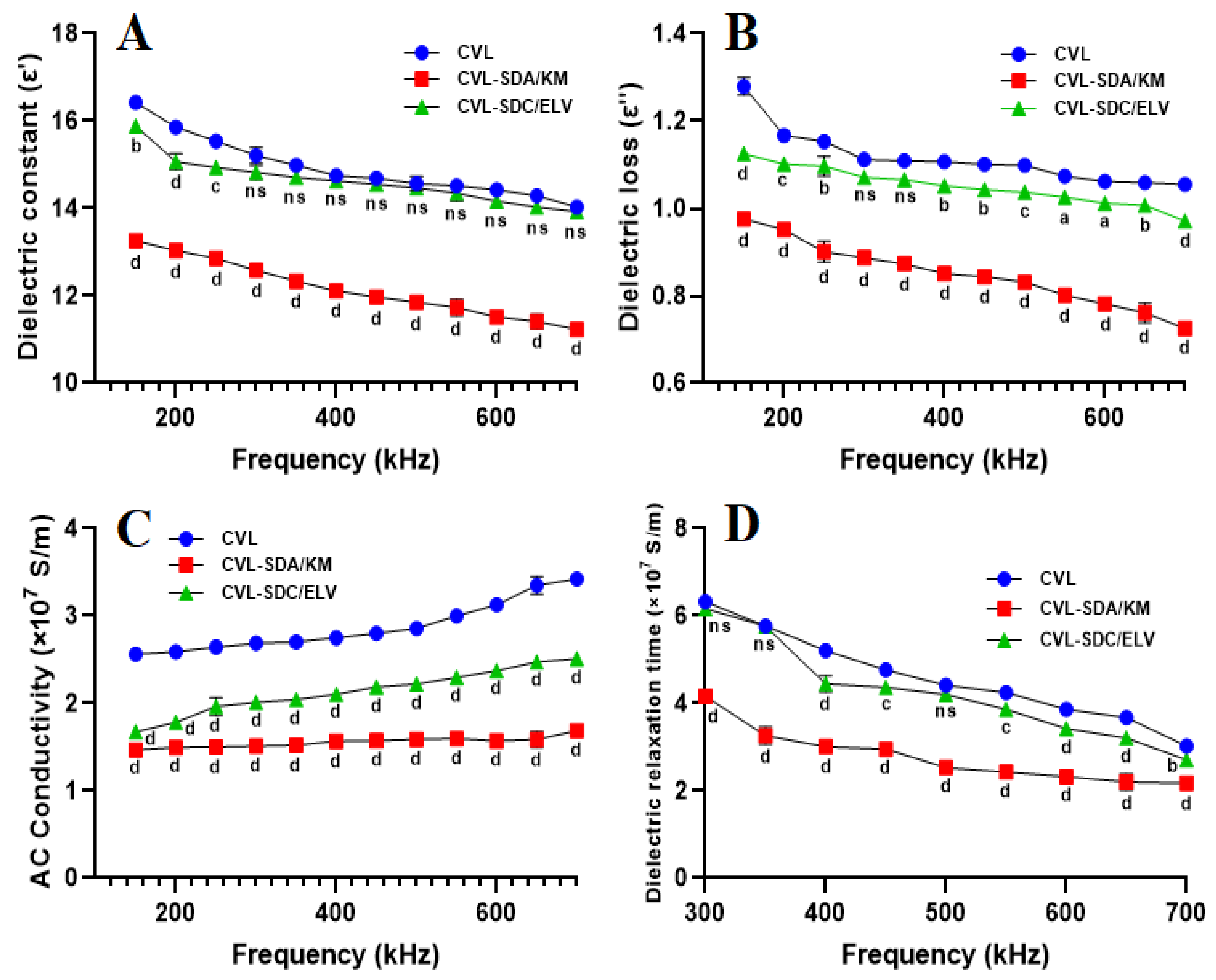
2.5. Dielectric Analysis
2.5.1. Frequency Domain Spectroscopy
2.5.2. Polarization and Depolarization Current Measurements
2.5.3. Electrical Resistivity
2.6. In Vivo Analysis
2.6.1. Oral Pharmacokinetic Studies
2.6.2. In Vivo Irritation Test
2.7. Statistical Analysis
3. Results and Discussion
3.1. Inhibition Effect of Polymers by Recrystallization
3.2. Formulation and Evaluation of CVL-SDs
Preparation of the Solid Dispersion
3.3. Characterization of CVL-SDs
3.3.1. Powder X-ray Diffraction
3.3.2. Scanning Electron Microscopy
3.3.3. Atomic Force Microscopy
3.3.4. Differential Scanning Calorimetry
3.3.5. Fourier-Transform Infrared Spectroscopy
3.3.6. Raman Spectroscopy
3.3.7. Dynamic Light Scattering
3.3.8. Transmission Electron Microscopy
3.3.9. Proton Nuclear Magnetic Resonance and NOESY
3.3.10. In Vitro Release Study
3.4. Infused Precipitation Study
3.4.1. Solution Phase Transfer (SPT)
3.4.2. Polarized Light Microscopy (PLM)
3.4.3. The pH Measurement
3.4.4. Viscosity and Particle Size Measurement
3.5. Dielectric Constant
3.5.1. Frequency of Domain Spectroscopy
3.5.2. The Relationship between Conduction Current and Crystallinity
3.5.3. Electrical Resistivity
3.6. In Vivo Analysis
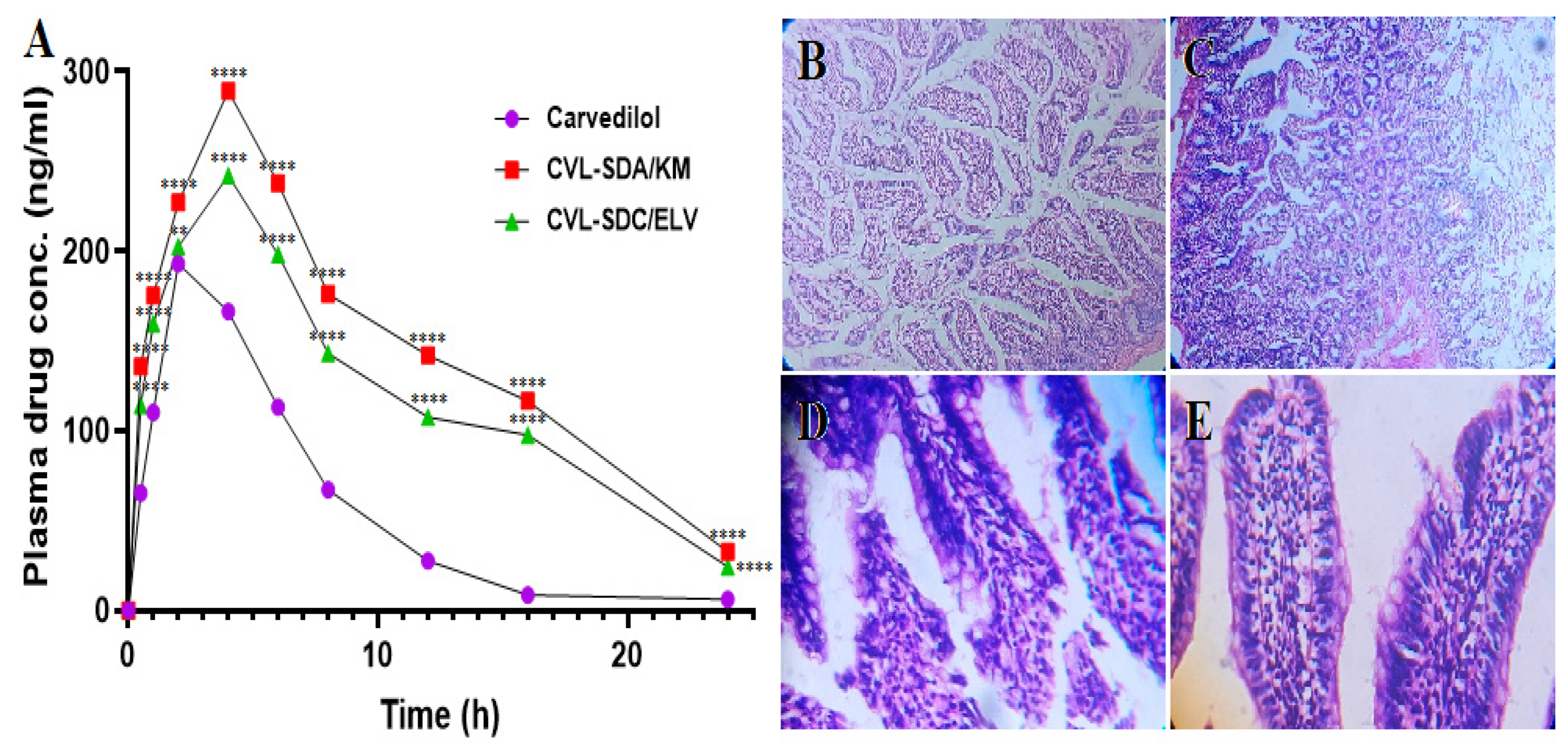
3.6.1. Oral Pharmacokinetic Studies
3.6.2. In Vivo Irritation Test
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Singh, A.; Worku, Z.A.; Mooter, G.V.D. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O., 3rd. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Han, H.-K.; Lee, B.-J.; Lee, H.-K. Enhanced dissolution and bioavailability of biochanin A via the preparation of solid dispersion: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 415, 89–94. [Google Scholar] [CrossRef]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid Dispersion as an Approach for Bioavailability Enhancement of Poorly Water-Soluble Drug Ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Raina, S.A.; Van Eerdenbrugh, B.; Alonzo, D.E.; Mo, H.; Zhang, G.G.; Gao, Y.; Taylor, L.S. Trends in the Precipitation and Crystallization Behavior of Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs Assessed Using Synchrotron Radiation. J. Pharm. Sci. 2015, 104, 1981–1992. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Imamura, K.; Maruyama, Y.; Tanaka, K.; Yokoyama, T.; Imanaka, H.; Nakanishi, K. True Density Analysis of a Freeze-Dried Amorphous Sugar Matrix. J. Pharm. Sci. 2008, 97, 2789–2797. [Google Scholar] [CrossRef]
- Caron, V.; Tajber, L.; Corrigan, O.I.; Healy, A.M. A Comparison of Spray Drying and Milling in the Production of Amorphous Dispersions of Sulfathiazole/Polyvinylpyrrolidone and Sulfadimidine/Polyvinylpyrrolidone. Mol. Pharm. 2011, 8, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Haque, K.; Roos, Y.H. Crystallization and X-ray diffraction of spray-dried and freeze-dried amorphous lactose. Carbohydr. Res. 2005, 340, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef]
- Huang, S.; Mao, C.; Williams, R.O.; Yang, C.-Y. Solubility Advantage (and Disadvantage) of Pharmaceutical Amorphous Solid Dispersions. J. Pharm. Sci. 2016, 105, 3549–3561. [Google Scholar] [CrossRef] [PubMed]
- Ahlneck, C.; Zografi, G. The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int. J. Pharm. 1990, 62, 87–95. [Google Scholar] [CrossRef]
- Feng, X.; Ye, X.; Park, J.-B.; Lu, W.; Morott, J.; Beissner, B.; Lian, Z.J.; Pinto, E.; Bi, V.; Porter, S.; et al. Evaluation of the recrystallization kinetics of hot-melt extruded polymeric solid dispersions using an improved Avrami equation. Drug Dev. Ind. Pharm. 2015, 41, 1479–1487. [Google Scholar] [CrossRef]
- Kukec, S.; Dreu, R.; Vrbanec, T.; Srčič, S.; Vrečer, F. Characterization of agglomerated carvedilol by hot-melt processes in a fluid bed and high shear granulator. Int. J. Pharm. 2012, 430, 74–85. [Google Scholar] [CrossRef]
- Prieto, C.; Evtoski, Z.; Pardo-Figuerez, M.; Lagaron, J. Bioavailability enhancement of nanostructured microparticles of carvedilol. J. Drug Deliv. Sci. Technol. 2021, 66, 102780. [Google Scholar] [CrossRef]
- Matsumoto, T.; Zografi, G. Physical Properties of Solid Molecular Dispersions of Indomethacin with Poly(vinylpyrrolidone) and Poly(vinylpyrrolidone-co-vinyl-acetate) in Relation to Indomethacin Crystallization. Pharm. Res. 1999, 16, 1722–1728. [Google Scholar] [CrossRef]
- Miyazaki, T.; Aso, Y.; Yoshioka, S.; Kawanishi, T. Differences in crystallization rate of nitrendipine enantiomers in amorphous solid dispersions with HPMC and HPMCP. Int. J. Pharm. 2011, 407, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ohárriz, M.C.; Rodríguez-Espinosa, C.; Martín, C.; Goñi, M.M.; Tros-Ilarduya, M.C.; Sánchez, M. Solid Dispersions of Diflunisal-PVP: Polymorphic and Amorphous States of the Drug. Drug Dev. Ind. Pharm. 2002, 28, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pui, Y.; Chen, H.; Wang, S.; Serno, P.; Tonnis, W.; Chen, L.; Qian, F. Polymer-Mediated Drug Supersaturation Controlled by Drug–Polymer Interactions Persisting in an Aqueous Environment. Mol. Pharm. 2018, 16, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Jasani, M.S.; Kale, D.P.; Singh, I.P.; Bansal, A.K. Influence of Drug–Polymer Interactions on Dissolution of Thermodynamically Highly Unstable Cocrystal. Mol. Pharm. 2018, 16, 151–164. [Google Scholar] [CrossRef]
- Hate, S.S.; Reutzel-Edens, S.M.; Taylor, L.S. Insight into Amorphous Solid Dispersion Performance by Coupled Dissolution and Membrane Mass Transfer Measurements. Mol. Pharm. 2018, 16, 448–461. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Kestur, U.S.; Hussain, M.A.; Taylor, L.S. Dissolution of Danazol Amorphous Solid Dispersions: Supersaturation and Phase Behavior as a Function of Drug Loading and Polymer Type. Mol. Pharm. 2016, 13, 223–231. [Google Scholar] [CrossRef]
- Wilson, V.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.; Gao, W.; Zhang, G.G.; Taylor, L.S. Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: Phase behavior during dissolution, speciation, and membrane mass transport. J. Control. Release 2018, 292, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Jamzad, S.; Fassihi, R. Role of surfactant and pH on dissolution properties of fenofibrate and glipizide—A technical note. AAPS PharmSciTech 2006, 7, E17–E22. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.; Junginger, H. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug Deliv. Rev. 2001, 50, S91–S101. [Google Scholar] [CrossRef]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A.S. Hydroxypropyl Methylcellulose Acetate Succinate-Based Spray-Dried Dispersions: An Overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef]
- Lu, Z.; Yang, Y.; Covington, R.-A.; Bi, Y.; Dürig, T.; Ilies, M.A.; Fassihi, R. Supersaturated controlled release matrix using amorphous dispersions of glipizide. Int. J. Pharm. 2016, 511, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kakumanu, V.K.; Bansal, A.K. Stability and Solubility of Celecoxib-PVP Amorphous Dispersions: A Molecular Perspective. Pharm. Res. 2004, 21, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Evaluation of the inhibitory potential of HPMC, PVP and HPC polymers on nucleation and crystal growth. RSC Adv. 2016, 6, 77569–77576. [Google Scholar] [CrossRef]
- Thenge, R.R.; Mahajan, K.G.; Sawarkar, H.S.; Adhao, V.S.; Gangane, P.S. Formulation and evaluation of transdermal drug delivery system for lercanidipine hydrochloride. Int. J. Pharm. Tech. Res. 2010, 2, 253–258. [Google Scholar]
- Prasad, D.; Chauhan, H.; Atef, E. Role of Molecular Interactions for Synergistic Precipitation Inhibition of Poorly Soluble Drug in Supersaturated Drug–Polymer–Polymer Ternary Solution. Mol. Pharm. 2016, 13, 756–765. [Google Scholar] [CrossRef]
- Shmeis, R.A.; Wang, Z.; Krill, S.L. A Mechanistic Investigation of an Amorphous Pharmaceutical and Its Solid Dispersions, Part I: A Comparative Analysis by Thermally Stimulated Depolarization Current and Differential Scanning Calorimetry. Pharm. Res. 2004, 21, 2025–2030. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.; Pouton, C. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, L.; Forssén, S.; Westergren, J.; Olsson, U. Nucleation and crystal growth in supersaturated solutions of a model drug. J. Colloid Interface Sci. 2008, 325, 404–413. [Google Scholar] [CrossRef]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating Miscibility and Molecular Mobility of Nifedipine-PVP Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy. Mol. Pharm. 2013, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheung, M.K.; Mi, Y. Miscibility and morphology in crystalline/amorphous blends of poly(caprolactone)/poly(4-vinylphenol) as studied by DSC, FTIR, and 13C solid state NMR. Polymer 2002, 43, 1357–1364. [Google Scholar] [CrossRef]
- Rumondor, A.C.F.; Ivanisevic, I.; Bates, S.; Alonzo, D.E.; Taylor, L.S. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm. Res. 2009, 26, 2523–2534. [Google Scholar] [CrossRef]
- Klein, S. The use of Biorelevant Dissolution media to forecast the in-vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef]
- Yamashita, K.; Nakate, T.; Okimoto, K.; Ohike, A.; Tokunaga, Y.; Ibuki, R.; Higaki, K.; Kimura, T. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int. J. Pharm. 2003, 267, 79–91. [Google Scholar] [CrossRef]
- Osmani, R.A.M.; Aloorkar, N.H.; Ingale, D.J.; Kulkarni, P.K.; Hani, U.; Bhosale, R.R.; Dev, D.J. Microsponges based novel drug delivery system for augmented arthritis therapy. Saudi Pharm. J. 2015, 23, 562–572. [Google Scholar] [CrossRef]
- Osmani, R.A.M.; Hani, U.; Bhosale, R.R.; Kulkarni, P.K.; Shanmuganathan, S. Nanosponge Carriers—An Archetype Swing in Cancer Therapy: A Comprehensive Review. Curr. Drug Targets 2016, 18, 108–118. [Google Scholar] [CrossRef]
- Osmani, R.A.M.; Kulkarni, P.K.; Shanmuganathan, S.; Hani, U.; Srivastava, A.; Prerana, M.; Shinde, C.G.; Bhosale, R.R. A 32 full factorial design for development and characterization of a nanosponge-based intravaginal in situ gelling system for vulvovaginal candidiasis. RSC Adv. 2016, 6, 18737–18750. [Google Scholar] [CrossRef]
- Osmani, R.A.M.; Aloorkar, N.H.; Thaware, B.U.; Kulkarni, P.K.; Moin, A.; Hani, U.; Srivastava, A.; Bhosale, R.R. Microsponge based drug delivery system for augmented gastroparesis therapy: Formulation development and evaluation. Asian J. Pharm. Sci. 2015, 10, 442–451. [Google Scholar] [CrossRef]
- Jakubiak, P.; Wagner, B.; Grimm, H.P.; Petrig-Schaffland, J.; Schuler, F.; Alvarez-Sánchez, R. Development of a Unified Dissolution and Precipitation Model and Its Use for the Prediction of Oral Drug Absorption. Mol. Pharm. 2016, 13, 586–598. [Google Scholar] [CrossRef]
- Koyama, H.; Ito, M.; Terada, K.; Sugano, K. Effect of Seed Particles on Precipitation of Weak Base Drugs in Physiological Intestinal Conditions. Mol. Pharm. 2016, 13, 2711–2717. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of Indomethacin from the Amorphous State below and above Its Glass Transition Temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef]
- Takeuchi, H.; Yasuji, T.; Yamamoto, H.; Kawashima, Y. Temperature-Induced Crystallization and Compactibility of Spray Dried Composite Particles Composed of Amorphous Lactose and Various Types of Water-Soluble Polymer. Chem. Pharm. Bull. 2000, 48, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, H.; Nambu, N.; Nagai, T. Stability and physical properties of amorphous and crystalline forms of indomethacin. Chem. Pharm. Bull. 1980, 28, 2565–2569. [Google Scholar] [CrossRef]
- Li, J.; Spivey, N.; Silchenko, S.; Gonzalez-Alvarez, I.; Bermejo, M.; Hidalgo, I.J. A differential equation based modelling approach to predict supersaturation and in vivo absorption from in vitro dissolution-absorption system (idas2) data. Eur. J. Pharm. Biopharm. 2021, 165, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Güldenpfennig, A.; Distaso, M.; Peukert, W. In situ investigations on the amorphous to crystalline phase transformation of precursors for methanol synthesis catalysts. Chem. Eng. J. 2019, 369, 996–1004. [Google Scholar] [CrossRef]
- Marsac, P.J.; Roumondor, A.C.F.; Nivens, D.E.; Kestur, U.S.; Stanciu, L.; Taylor, L.S. Effect of temparature and moisture on the miscibility of amorphous dispersions of Felodepine and Poly(vinyl pyrrolidone). J. Pharm. Sci. 2009, 99, 169–185. [Google Scholar] [CrossRef]
- Akbari, B.; Tavandashti, M.P.; Zandrahimi, M. Particle size characterization of nanoparticles—A practicalapproach. Iran. J. Mater. Sci. Eng. 2011, 8, 48–56. [Google Scholar]
- Guzmán, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, Ö.; Remenar, J.F. Combined Use of Crystalline Salt Forms and Precipitation Inhibitors to Improve Oral Absorption of Celecoxib from Solid Oral Formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Degree of Polymerization | Molecular Weight | Hydrogen Bond Acceptor | Viscosity | Hydroxy Propyl Content | Methoxyl Content |
|---|---|---|---|---|---|---|
| HPMC | 35 | 1261.4 | 6 | 2.4 | 7.3 | 27.8 |
| HPMC E3 | 37 | 10,000 | 6 | 3.2 | 8.8 | 29.4 |
| HPMC E5 | 37 | 13,000 | 6 | 5 | 9.5 | 28.4 |
| HPMC E6 | 49 | 10,000 | 6 | 6.2 | 9.1 | 28.7 |
| HPMC E15 | 49 | 13,000 | 6 | 16.6 | 9.7 | 28.2 |
| HPMC E50 | 75 | 20,000 | 6 | 48 | 9 | 28.8 |
| HPMC K4M | 155 | 41,000 | 6 | 4927 | 8.2 | 23.3 |
| HPMC K15M | 416 | 110,000 | 6 | 7382 | 8.6 | 23.1 |
| HPMC K100M | 530 | 140,000 | 6 | 113.384 | 10.5 | 22.8 |
| HPMC phthalate | 489 | 1425.15 | 9 | 136 | 8.1 | 24.4 |
| Parameters | CVL | SDA/KM | SDC/ELV |
|---|---|---|---|
| Cmax (ng/mL) | 193.07 ± 1.91 | 289.25 ± 2.11 | 242 ± 2.45 |
| Tmax (h) | 2.48 ± 0.21 | 4.31 ± 0.63 | 4.01 ± 0.41 |
| AUC0-t (ng.h/mL) | 1376.98 ± 16.32 | 2874.65 ± 49.53 | 1915.16 ± 23.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravikumar, A.A.; Kulkarni, P.K.; Osmani, R.A.M.; Hani, U.; Ghazwani, M.; Fatease, A.A.; Alamri, A.H.; Gowda, D.V. Carvedilol Precipitation Inhibition by the Incorporation of Polymeric Precipitation Inhibitors Using a Stable Amorphous Solid Dispersion Approach: Formulation, Characterization, and In Vitro In Vivo Evaluation. Polymers 2022, 14, 4977. https://doi.org/10.3390/polym14224977
Ravikumar AA, Kulkarni PK, Osmani RAM, Hani U, Ghazwani M, Fatease AA, Alamri AH, Gowda DV. Carvedilol Precipitation Inhibition by the Incorporation of Polymeric Precipitation Inhibitors Using a Stable Amorphous Solid Dispersion Approach: Formulation, Characterization, and In Vitro In Vivo Evaluation. Polymers. 2022; 14(22):4977. https://doi.org/10.3390/polym14224977
Chicago/Turabian StyleRavikumar, Akhila Akkihebbal, Parthasarathi K. Kulkarni, Riyaz Ali M. Osmani, Umme Hani, Mohammed Ghazwani, Adel Al Fatease, Ali H. Alamri, and Devegowda V. Gowda. 2022. "Carvedilol Precipitation Inhibition by the Incorporation of Polymeric Precipitation Inhibitors Using a Stable Amorphous Solid Dispersion Approach: Formulation, Characterization, and In Vitro In Vivo Evaluation" Polymers 14, no. 22: 4977. https://doi.org/10.3390/polym14224977
APA StyleRavikumar, A. A., Kulkarni, P. K., Osmani, R. A. M., Hani, U., Ghazwani, M., Fatease, A. A., Alamri, A. H., & Gowda, D. V. (2022). Carvedilol Precipitation Inhibition by the Incorporation of Polymeric Precipitation Inhibitors Using a Stable Amorphous Solid Dispersion Approach: Formulation, Characterization, and In Vitro In Vivo Evaluation. Polymers, 14(22), 4977. https://doi.org/10.3390/polym14224977