
Solvent Evaporation Rate as a Tool for Tuning the Performance of a Solid Polymer Electrolyte Gas Sensor

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
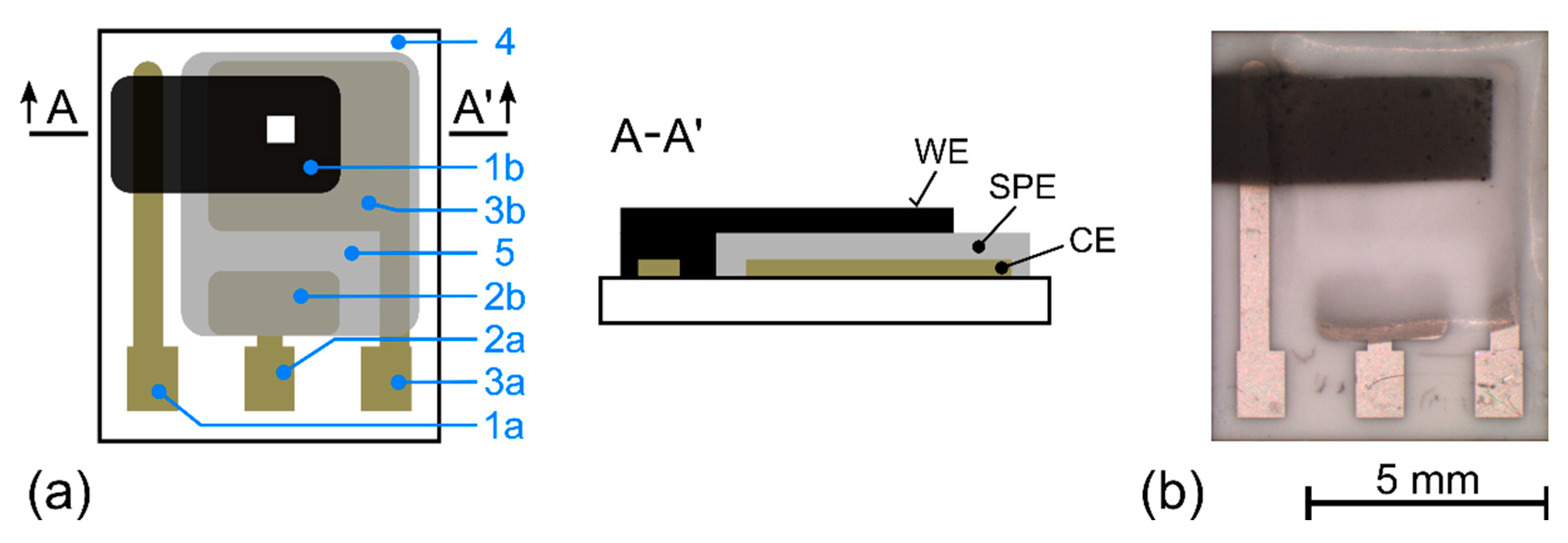
2.1. Sensor Preparation
2.2. Characterization
2.3. Experimental Setup
2.3.1. Measurement of Steady-State and Fluctuations
2.3.2. Standard Measurement of Sensor Response
3. Results and Discussion
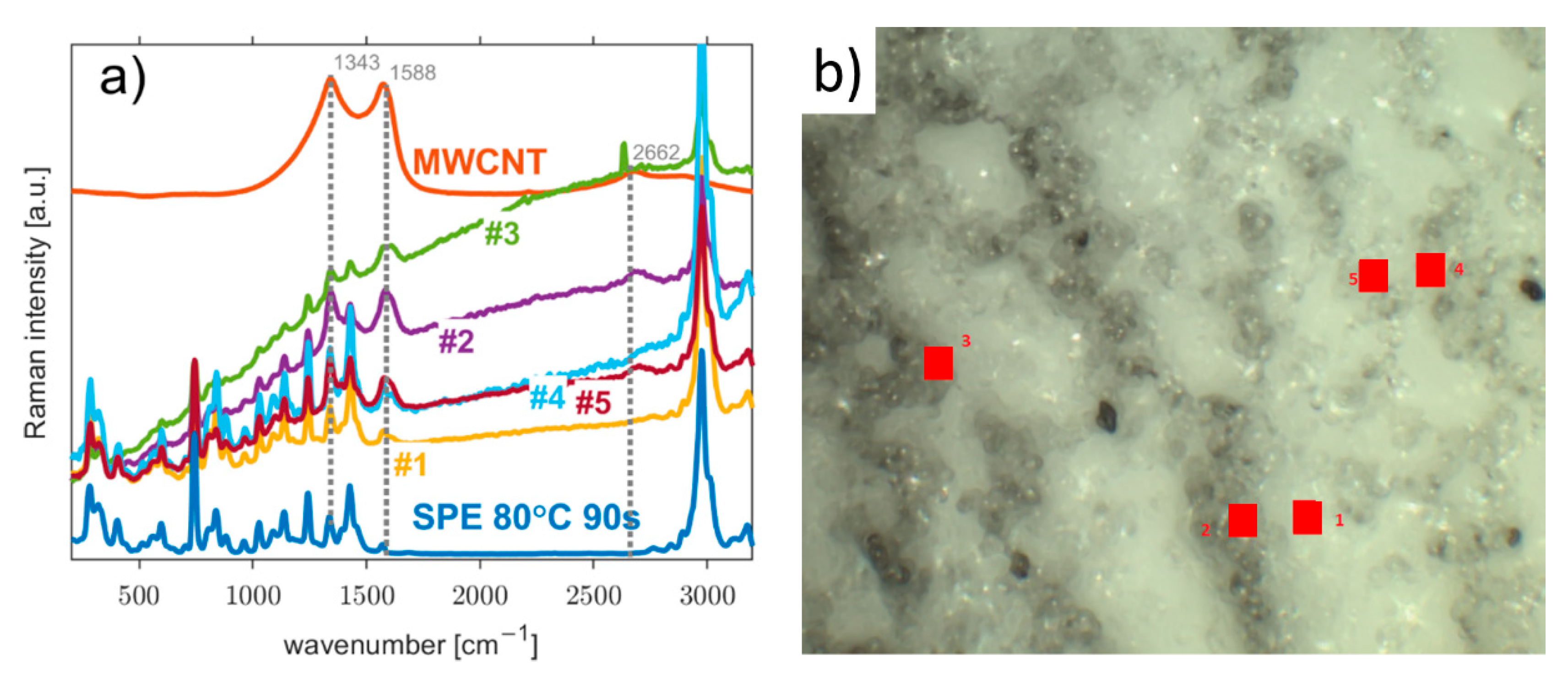
3.1. Morphology of the Working Electrode/Solid Polymer Electrolyte (WE–SPE) Interface
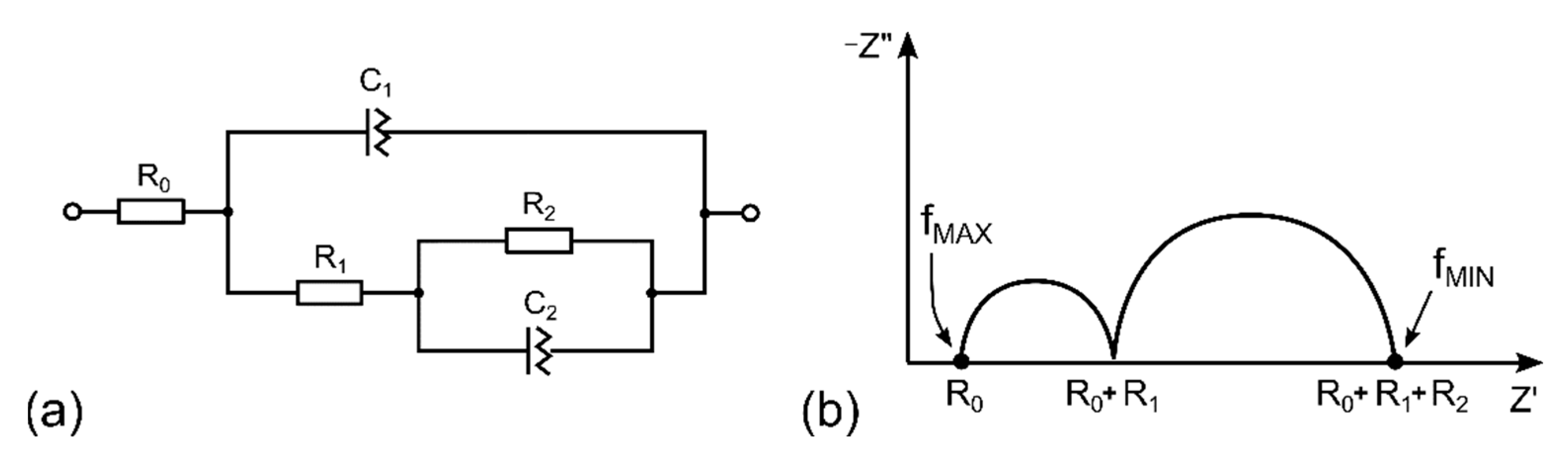
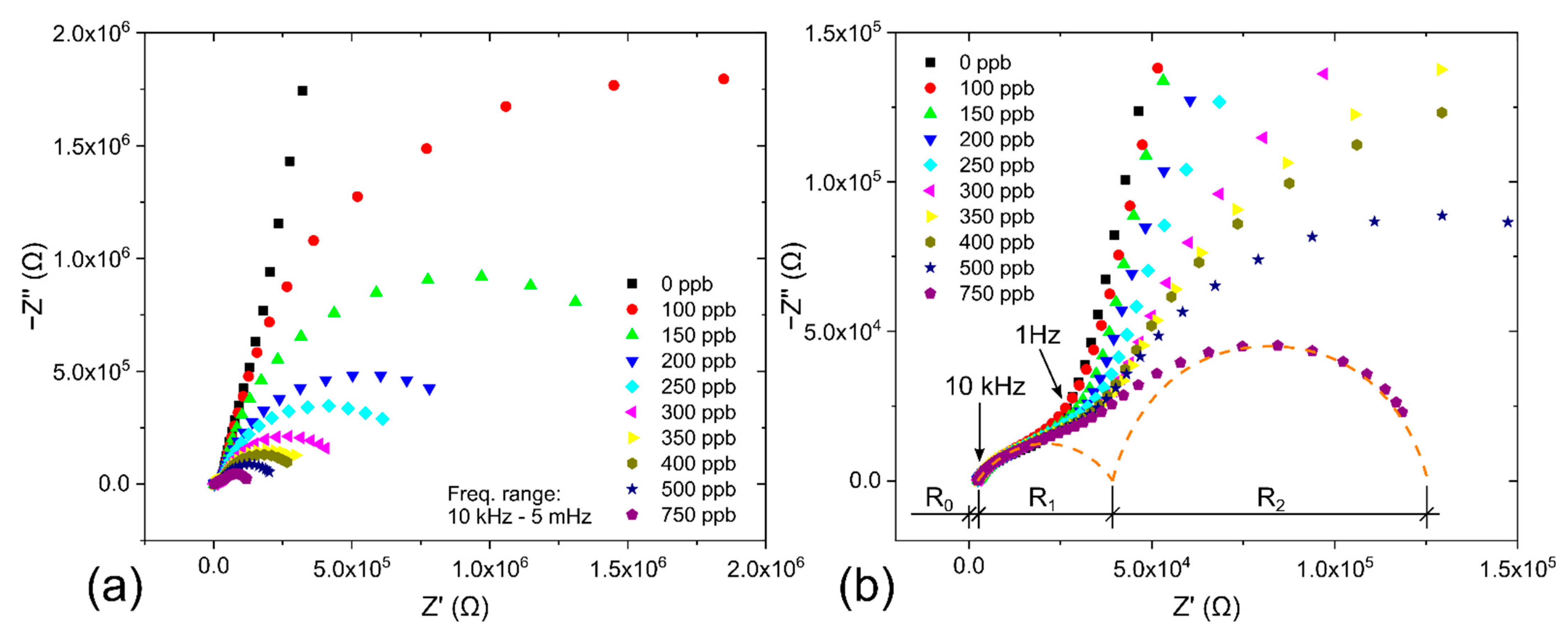
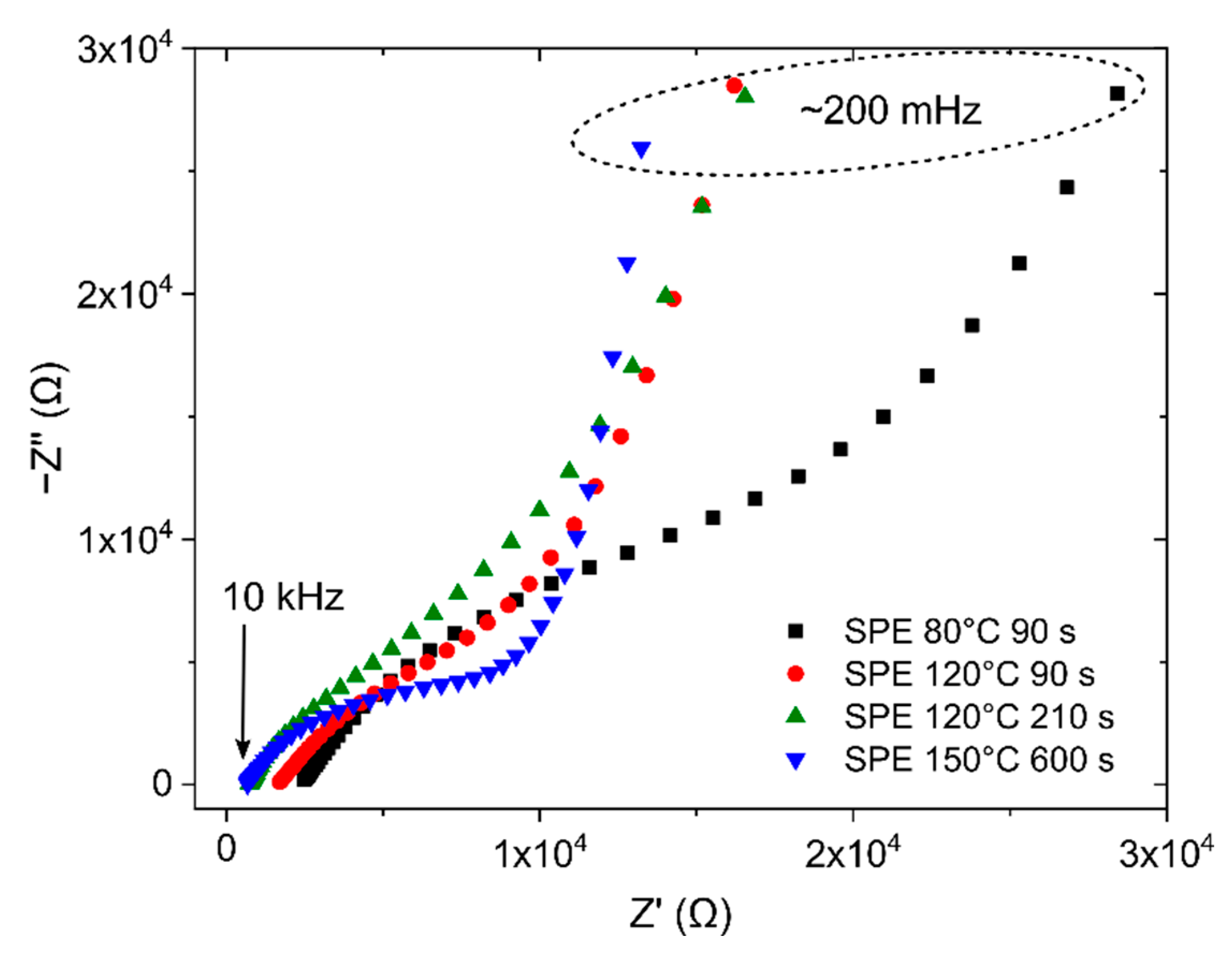
3.2. Description of the Sensor by Equivalent Electric Circuit
3.3. Effect of Solvent Evaporation Rate during SPE Preparation on the Performance of an Amperometric Gas Sensor
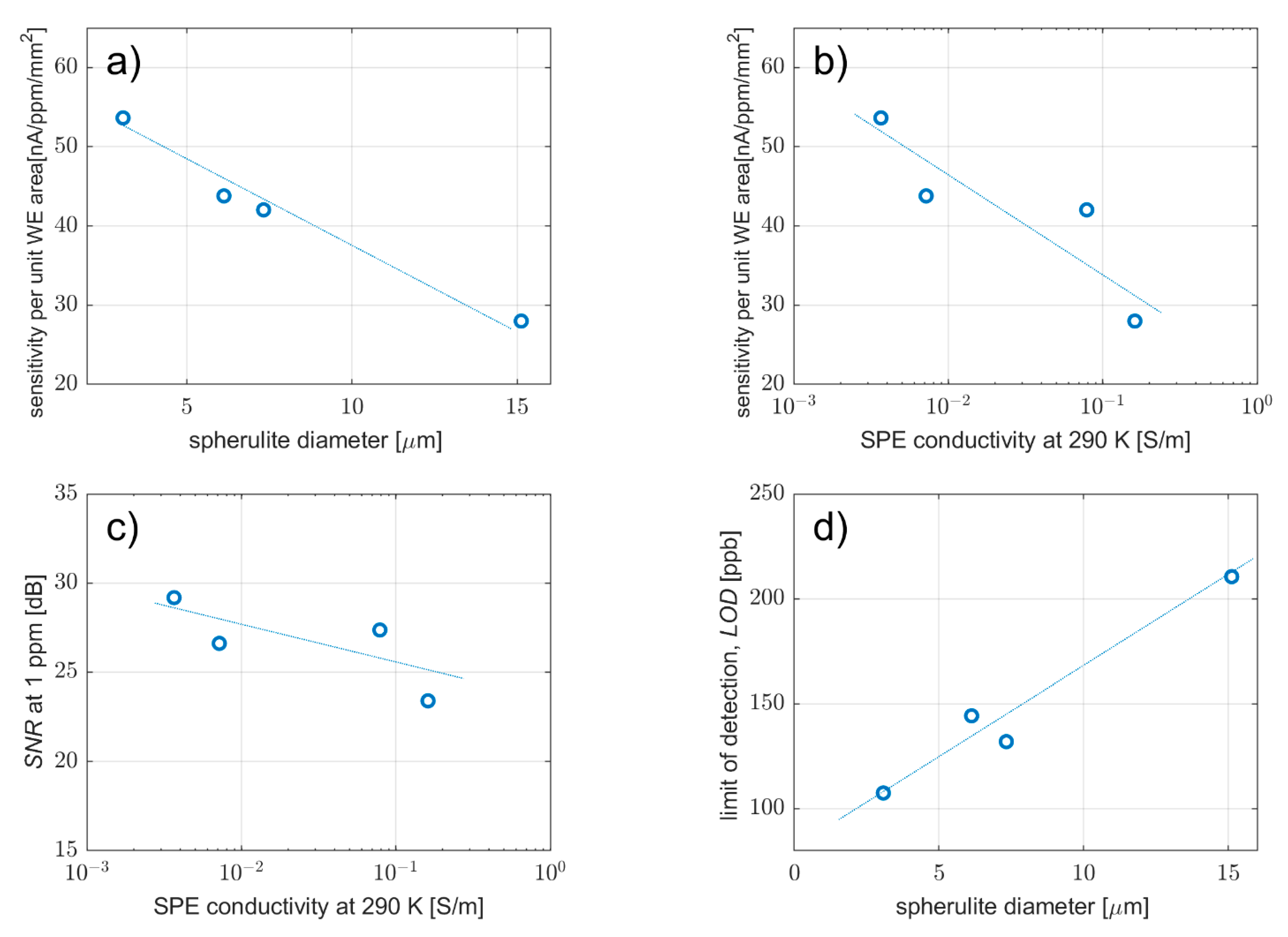
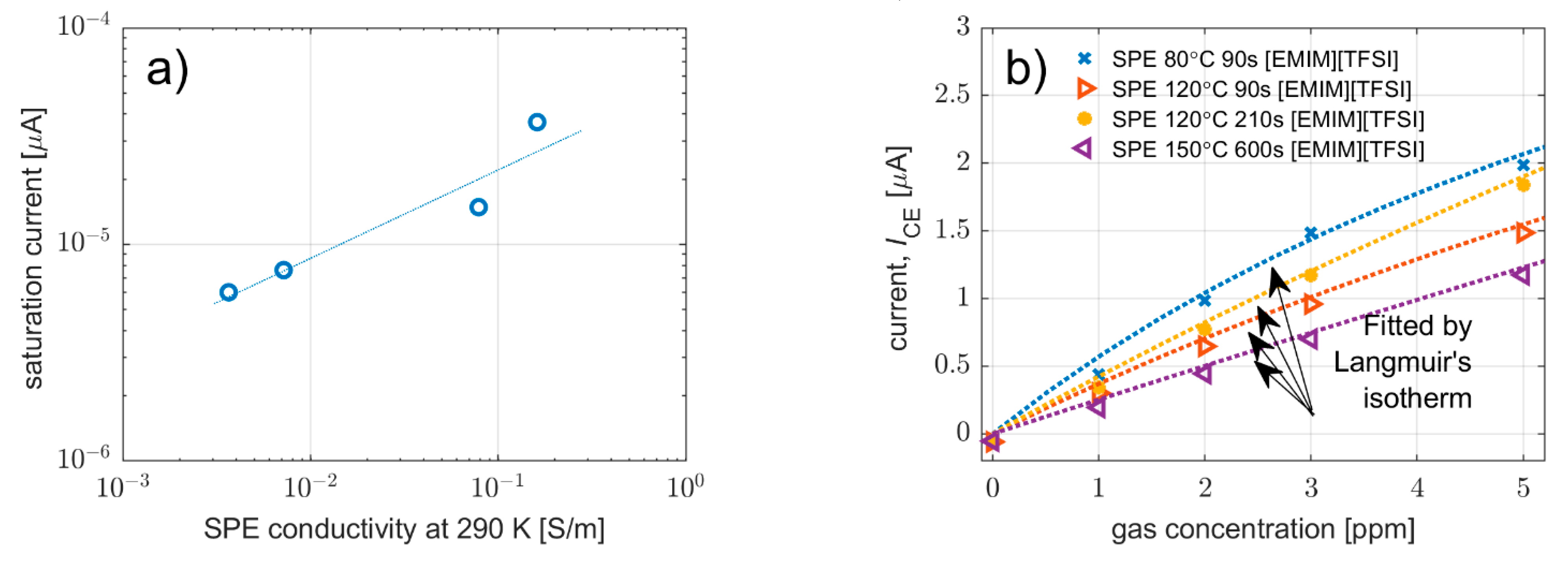
3.3.1. Effect on Sensitivity, Signal-to-Noise Ratio, Limit-of-Detection and Saturation Current
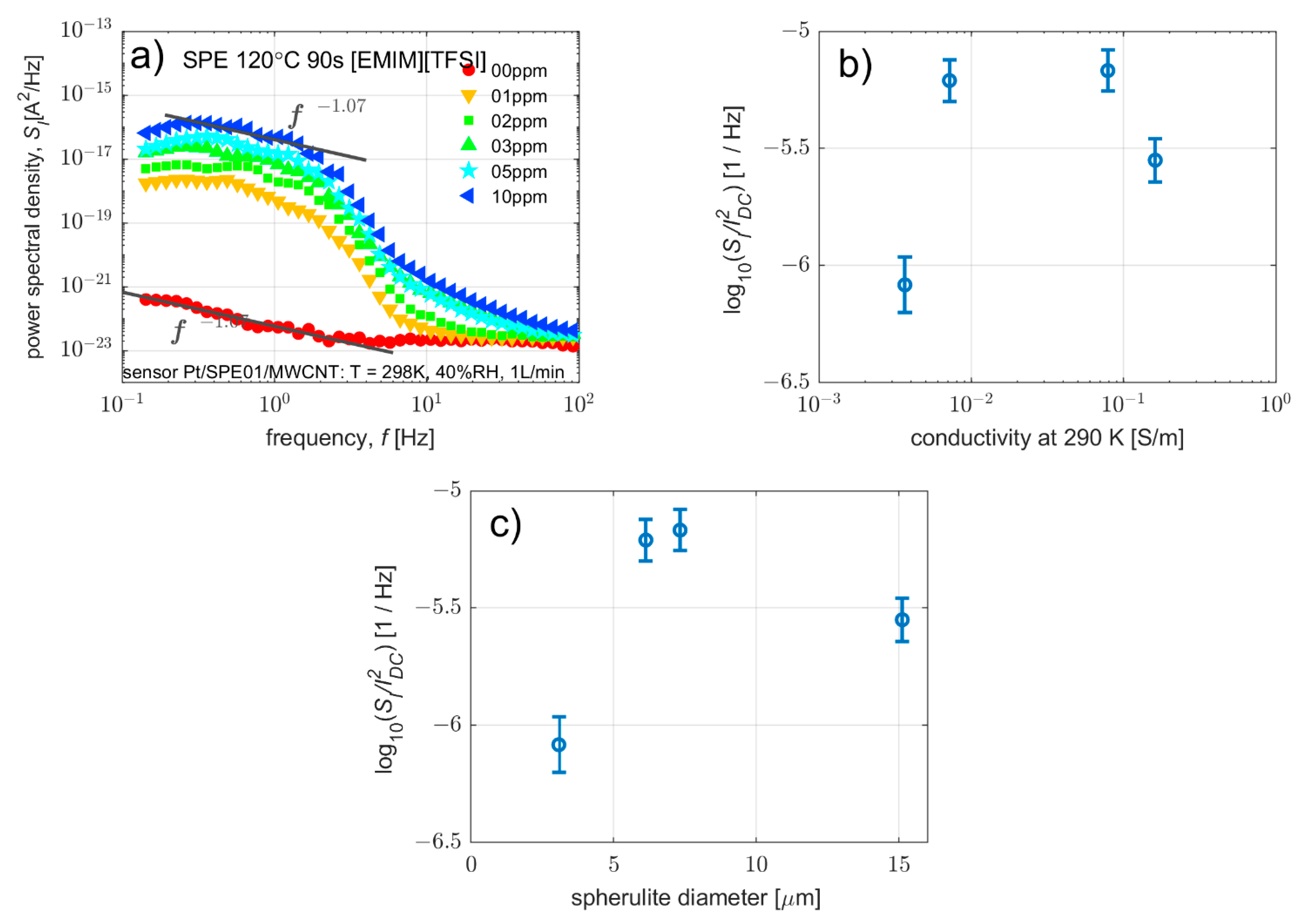
3.3.2. Effect on Current Fluctuations
3.3.3. Complete Summary of Sensor Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, Y.-S.; Rick, J.; Hwang, B.-J. Ionic liquid polymer electrolytes. J. Mater. Chem. A 2013, 1, 2719–2743. [Google Scholar] [CrossRef]
- Correia, D.M.; Fernandes, L.C.; Martins, P.M.; García-Astrain, C.; Costa, C.M.; Reguera, J.; Lanceros-Méndez, S. Ionic Liquid–Polymer Composites: A New Platform for Multifunctional Applications. Adv. Funct. Mater. 2020, 30, 1909736. [Google Scholar] [CrossRef]
- Josef, E.; Yan, Y.; Stan, M.C.; Wellmann, J.; Vizintin, A.; Winter, M.; Johansson, P.; Dominko, R.; Guterman, R. Ionic Liquids and their Polymers in Lithium-Sulfur Batteries. Isr. J. Chem. 2019, 59, 832–842. [Google Scholar] [CrossRef]
- Austin Suthanthiraraj, S.; Johnsi, M. Nanocomposite polymer electrolytes. Ionics (Kiel) 2017, 23, 2531–2542. [Google Scholar] [CrossRef]
- Xia, W.; Zhang, Z. PVDF-based dielectric polymers and their applications in electronic materials. IET Nanodielectrics 2018, 1, 17–31. [Google Scholar] [CrossRef]
- Kammoun, M.; Berg, S.; Ardebili, H. Flexible thin-film battery based on graphene-oxide embedded in solid polymer electrolyte. Nanoscale 2015, 7, 17516–17522. [Google Scholar] [CrossRef]
- Korotcenkov, G.; Cho, B.K. Instability of metal oxide-based conductometric gas sensors and approaches to stability improvement (short survey). Sens. Actuators B Chem. 2011, 156, 527–538. [Google Scholar] [CrossRef]
- Varshney, P.K.; Gupta, S. Natural polymer-based electrolytes for electrochemical devices: A review. Ionics (Kiel) 2011, 17, 479–483. [Google Scholar] [CrossRef]
- Sedlak, P.; Gajdos, A.; Macku, R.; Majzner, J.; Sedlakova, V.; Holcman, V.; Kuberský, P. The effect of thermal treatment on ac/dc conductivity and current fluctuations of PVDF/NMP/[EMIM][TFSI] solid polymer electrolyte. Sci. Rep. 2020, 10, 21140. [Google Scholar] [CrossRef]
- Wang, F.; Li, L.; Yang, X.; You, J.; Xu, Y.; Wang, H.; Ma, Y.; Gao, G. Influence of additives in a PVDF-based solid polymer electrolyte on conductivity and Li-ion battery performance. Sustain. Energy Fuels 2018, 2, 492–498. [Google Scholar] [CrossRef]
- Subba Reddy, C.V.; Chen, M.; Jin, W.; Zhu, Q.Y.; Chen, W.; Mho, S. Il Characterization of (PVDF + LiFePO4) solid polymer electrolyte. J. Appl. Electrochem. 2007, 37, 637–642. [Google Scholar] [CrossRef]
- Tjong, S.C.; Li, Y.C.; Li, R.K.Y. Frequency and temperature dependences of dielectric dispersion and electrical properties of polyvinylidene fluoride/expanded graphite composites. J. Nanomater. 2010, 2010. [Google Scholar]
- Puértolas, J.A.; García-García, J.F.; Pascual, F.J.; González-Domínguez, J.M.; Martínez, M.T.; Ansón-Casaos, A. Dielectric behavior and electrical conductivity of PVDF filled with functionalized single-walled carbon nanotubes. Compos. Sci. Technol. 2017, 152, 263–274. [Google Scholar] [CrossRef]
- Li, X.; Xuan, T.; Yin, G.; Gao, Z.; Zhao, K.; Yan, P.; He, D. Highly sensitive amperometric CO sensor using nanocomposite C-loaded PdCl2–CuCl2 as sensing electrode materials. J. Alloys Compd. 2015, 645, 553–558. [Google Scholar] [CrossRef]
- Tofel, P.; Částková, K.; Říha, D.; Sobola, D.; Papež, N.; Kaštyl, J.; Ţălu, Ş.; Hadaš, Z. Triboelectric Response of Electrospun Stratified PVDF and PA Structures. Nanomaterials 2022, 12, 349. [Google Scholar] [CrossRef] [PubMed]
- Černohorský, P.; Pisarenko, T.; Papež, N.; Sobola, D.; Ţălu, Ş.; Částková, K.; Kaštyl, J.; Macků, R.; Škarvada, P.; Sedlák, P. Structure Tuning and Electrical Properties of Mixed PVDF and Nylon Nanofibers. Materials 2021, 14, 6096. [Google Scholar] [CrossRef]
- Nespurek, S.; Mracek, L.; Kubersky, P.; Syrovy, T.; Hamacek, A. Ionic liquids in electrochemical gas sensors and transistors. Mol. Cryst. Liq. Cryst. 2019, 694, 1–20. [Google Scholar] [CrossRef]
- Ohno, H. Electrochemical Aspects of Ionic Liquids, 2nd ed.; Wiley: Hoboken, NJ, USA, 2011; ISBN 978-0-470-64781-3. [Google Scholar]
- Torriero, A.A.J.; Shiddiky, M.J.A. Electrochemical Properties and Applications of Ionic Liquids; Nova Science: Hauppauge, NY, USA, 2011; ISBN 9781617289422. [Google Scholar]
- Kirchner, B.; Perlt, E. Ionic Liquids II; Topics in Current Chemistry Collections; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-89793-6. [Google Scholar]
- Paluch, M. Dielectric Properties of Ionic Liquids; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-32487-6. [Google Scholar]
- Brandt, A.; Pohlmann, S.; Varzi, A.; Balducci, A.; Passerini, S. Ionic liquids in supercapacitors. MRS Bull. 2013, 38, 554–559. [Google Scholar] [CrossRef]
- Kuberský, P.; Navrátil, J.; Syrový, T.; Sedlák, P.; Nešpůrek, S.; Hamáček, A. An Electrochemical Amperometric Ethylene Sensor with Solid Polymer Electrolyte Based on Ionic Liquid. Sensors 2021, 21, 711. [Google Scholar] [CrossRef]
- Kuberský, P.; Altšmíd, J.; Hamáček, A.; Nešpůrek, S.; Zmeškal, O. An electrochemical NO2 sensor based on ionic liquid: Influence of the morphology of the polymer electrolyte on sensor sensitivity. Sensors 2015, 15, 28421–28434. [Google Scholar] [CrossRef]
- Luo, R.; Li, H.; Du, B.; Zhou, S.; Chen, Y. A printed and flexible NO2 sensor based on a solid polymer electrolyte. Front. Chem. 2019, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Li, Q.; Du, B.; Zhou, S.; Chen, Y. Preparation and Characterization of Solid Electrolyte Doped With Carbon Nanotubes and its Preliminary Application in NO2 Gas Sensors. Front. Mater. 2019, 6, 113. [Google Scholar] [CrossRef]
- Luo, R.; Jiang, H.; Du, B.; Zhou, S.; Zhu, Y. Preparation and application of solid polymer electrolyte based on deep eutectic solvent. AIP Adv. 2019, 9, 035341. [Google Scholar] [CrossRef]
- Nádherná, M.; Opekar, F.; Reiter, J. Ionic liquid–polymer electrolyte for amperometric solid-state NO2 sensor. Electrochim. Acta 2011, 56, 5650–5655. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Jasinski, G.; Chachulski, B. Investigation of solid polymer electrolyte gas sensor with different electrochemical techniques. IOP Conf. Ser. Mater. Sci. Eng. 2016, 104, 012029. [Google Scholar] [CrossRef]
- Dosi, M.; Lau, I.; Zhuang, Y.; Simakov, D.S.A.; Fowler, M.W.; Pope, M.A. Ultrasensitive Electrochemical Methane Sensors Based on Solid Polymer Electrolyte-Infused Laser-Induced Graphene. ACS Appl. Mater. Interfaces 2019, 11, 6166–6173. [Google Scholar] [CrossRef]
- Satapathy, S.; Pawar, S.; Gupta, P.K.; RVarma, K.B. Effect of annealing on phase transition in poly(vinylidene fluoride) films prepared using polar solvent. Bull. Mater. Sci. 2011, 34, 727–733. [Google Scholar] [CrossRef]
- Ting, Y.; Suprapto; Bunekar, N.; Sivasankar, K.; Aldori, Y.R. Using Annealing Treatment on Fabrication Ionic Liquid-Based PVDF Films. Coatings 2020, 10, 44. [Google Scholar] [CrossRef]
- Xu, P.; Fu, W.; Hu, Y.; Ding, Y. Effect of annealing treatment on crystalline and dielectric properties of PVDF/PEG-containing ionic liquid composites. Compos. Sci. Technol. 2018, 158, 1–8. [Google Scholar] [CrossRef]
- Gregorio, R.; Borges, D.S. Effect of crystallization rate on the formation of the polymorphs of solution cast poly(vinylidene fluoride). Polymer 2008, 49, 4009–4016. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, Q.; Yu, C.; Peng, J.; Ma, J.; Ju, X.; Zhai, M. Effect of ionic liquid on the properties of poly(vinylidene fluoride)-based gel polymer electrolytes. Ionics (Kiel) 2013, 19, 1587–1593. [Google Scholar] [CrossRef]
- Correia, D.M.; Costa, C.M.; Rodríguez-Hernández, J.C.; Tort Ausina, I.; Biosca, L.T.; Torregrosa Cabanilles, C.; Meseguer-Duenãs, J.M.; Lanceros-Méndez, S.; Gomez Ribelles, J.L. Effect of Ionic Liquid Content on the Crystallization Kinetics and Morphology of Semicrystalline Poly(vinylidene Fluoride)/Ionic Liquid Blends. Cryst. Growth Des. 2020, 20, 4967–4979. [Google Scholar] [CrossRef]
- Sedlak, P.; Sobola, D.; Gajdos, A.; Dallaev, R.; Nebojsa, A.; Kubersky, P. Surface analyses of PVDF/NMP/[EMIM][TFSI] solid polymer electrolyte. Polymers 2021, 13, 2678. [Google Scholar] [CrossRef] [PubMed]
- Altšmíd, J.; Syrový, T.; Syrová, L.; Kuberský, P.; Hamáček, A.; Zmeškal, O.; Nešpůrek, S. Ionic Liquid based polymer electrolytes for electrochemical sensors. Mater. Sci. 2015, 21, 415–418. [Google Scholar] [CrossRef]
- Luo, R.; Wu, Y.; Li, Q.; Du, B.; Zhou, S.; Li, H. Rational synthesis and characterization of IL-CNTs-PANI microporous polymer electrolyte film. Synth. Met. 2021, 274, 116720. [Google Scholar] [CrossRef]
- Kuberský, P.; Sedlák, P.; Hamáček, A.; Nešpůrek, S.; Kuparowitz, T.; Šikula, J.; Majzner, J.; Sedlaková, V.; Grmela, L.; Syrový, T. Quantitative fluctuation-enhanced sensing in amperometric NO2 sensors. Chem. Phys. 2015, 456, 111–117. [Google Scholar] [CrossRef]
- Sedlák, P.; Kuberský, P.; Mívalt, F. Effect of various flow rate on current fluctuations of amperometric gas sensors. Sens. Actuators B Chem. 2019, 283, 321–328. [Google Scholar] [CrossRef]
- Sedlák, P.; Kuberský, P. The Effect of the Orientation Towards Analyte Flow on Electrochemical Sensor Performance and Current Fluctuations. Sensors 2020, 20, 1038. [Google Scholar] [CrossRef]
- Kuberský, P.; Hamáček, A.; Nešpůrek, S.; Soukup, R.; Vik, R. Effect of the geometry of a working electrode on the behavior of a planar amperometric NO2 sensor based on solid polymer electrolyte. Sens. Actuators B Chem. 2013, 187, 546–552. [Google Scholar] [CrossRef]
- Sedlak, P.; Kubersky, P.; Skarvada, P.; Hamacek, A.; Sedlakova, V.; Majzner, J.; Nespurek, S.; Sikula, J. Current Fluctuation Measurements of Amperometric Gas Sensors Constructed with Three Different Technology Procedures. Metrol. Meas. Syst. 2016, 23, 531–543. [Google Scholar] [CrossRef]
- Kuberský, P.; Syrový, T.; Hamáček, A.; Nešpůrek, S.; Syrová, L. Towards a fully printed electrochemical NO2 sensor on a flexible substrate using ionic liquid based polymer electrolyte. Sens. Actuators B Chem. 2015, 209, 1084–1090. [Google Scholar] [CrossRef]
- Ahmadi, M.M.; Jullien, G.A. Current-Mirror-Based Potentiostats for Three-Electrode Amperometric Electrochemical Sensors. IEEE Trans. Circuits Syst. I Regul. Pap. 2009, 56, 1339–1348. [Google Scholar] [CrossRef]
- Cui, Z.; Hassankiadeh, N.T.; Zhuang, Y.; Drioli, E.; Lee, Y.M. Crystalline polymorphism in poly(vinylidenefluoride) membranes. Prog. Polym. Sci. 2015, 51, 94–126. [Google Scholar] [CrossRef]
- California, A.; Cardoso, V.F.; Costa, C.M.; Sencadas, V.; Botelho, G.; Gómez-Ribelles, J.L.; Lanceros-Mendez, S. Tailoring porous structure of ferroelectric poly(vinylidene fluoride-trifluoroethylene) by controlling solvent/polymer ratio and solvent evaporation rate. Eur. Polym. J. 2011, 47, 2442–2450. [Google Scholar] [CrossRef]
- Magalhães, R.; Durães, N.; Silva, M.; Silva, J.; Sencadas, V.; Botelho, G.; Gómez Ribelles, J.L.; Lanceros-Méndez, S. The Role of Solvent Evaporation in the Microstructure of Electroactive β-Poly(Vinylidene Fluoride) Membranes Obtained by Isothermal Crystallization. Soft Mater. 2010, 9, 1–14. [Google Scholar] [CrossRef]
- Li, C.L.; Wang, D.M.; Deratani, A.; Quémener, D.; Bouyer, D.; Lai, J.Y. Insight into the preparation of poly(vinylidene fluoride) membranes by vapor-induced phase separation. J. Memb. Sci. 2010, 361, 154–166. [Google Scholar] [CrossRef]
- Crist, B.; Schultz, J.M. Polymer spherulites: A critical review. Prog. Polym. Sci. 2016, 56, 1–63. [Google Scholar] [CrossRef]
- Sheiko, S.S.; Magonov, S.N. Scanning Probe Microscopy of Polymers. In Polymer Science: A Comprehensive Reference; Martin Moeller, K.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 10, pp. 559–605. ISBN 9780080878621. [Google Scholar]
- Noor, N.A.M.; Kamarudin, S.K.; Darus, M.; Yunos, N.F.D.M.; Idris, M.A. Photocatalytic Properties and Graphene Oxide Additional Effects in TiO2. Solid State Phenom. 2018, 280, 65–70. [Google Scholar] [CrossRef]
- Randles, J.E.B. Kinetics of rapid electrode reactions. Faraday Discuss. 1947, 1, 11–19. [Google Scholar] [CrossRef]
- Lvovich, V.F. Impedance spectroscopy: Applications to electrochemical and dielectric phenomena; Wiley: Hoboken, NJ, USA, 2015; ISBN 9781118164099. [Google Scholar]
- Chang, S.-C.; Stetter, J.R. Electrochemical NO2 gas sensors: Model and mechanism for the electroreduction of NO2. Electroanalysis 1990, 2, 359–365. [Google Scholar] [CrossRef]
- Hassibi, A.; Navid, R.; Dutton, R.W.; Lee, T.H. Comprehensive study of noise processes in electrode electrolyte interfaces. J. Appl. Phys. 2004, 96, 1074–1082. [Google Scholar] [CrossRef]
- Musha, T.; Higuchi, H. Traffic Current Fluctuation and the Burgers Equation. Jpn. J. Appl. Phys. 1978, 17, 811–816. [Google Scholar] [CrossRef]
- Roach, P.E. The generation of nearly isotropic turbulence by means of grids. Int. J. Heat Fluid Flow 1987, 8, 82–92. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Condition | DC Conductivity [S/m] | Mean Spherulite Diameter [μm] |
|---|---|---|
| 80 °C 90 s | 0.003647 | 3.08 |
| 120 °C 90 s | 0.007162 | 6.13 |
| 120 °C 210 s | 0.078600 | 7.33 |
| 160 °C 600 s | 0.161200 | 15.12 |
| Parameter | SPE at 80 °C for 90 s | SPE at 120 °C for 90 s | SPE at 120 °C for 210 s | SPE at 150 °C for 600 s |
|---|---|---|---|---|
| Sensitivity (pA/ppb/mm2) | 55.0 | 42.0 | 31.9 | 30.6 |
| LOD (ppb) | 1.07 | 1.36 | 1.72 | 2.04 |
| LOQ (ppb) | 3.57 | 4.53 | 5.73 | 6.78 |
| Hysteresis (%) * | 3.9 | 4.2 | 4.9 | 3.9 |
| Repeatability (%) | 1.2 | 2.4 | 1.3 | 1.3 |
| Response time (s) | 47 | 58 | 59 | 55 |
| Recovery time (s) | 22 | 22 | 23 | 21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedlak, P.; Kaspar, P.; Sobola, D.; Gajdos, A.; Majzner, J.; Sedlakova, V.; Kubersky, P. Solvent Evaporation Rate as a Tool for Tuning the Performance of a Solid Polymer Electrolyte Gas Sensor. Polymers 2022, 14, 4758. https://doi.org/10.3390/polym14214758
Sedlak P, Kaspar P, Sobola D, Gajdos A, Majzner J, Sedlakova V, Kubersky P. Solvent Evaporation Rate as a Tool for Tuning the Performance of a Solid Polymer Electrolyte Gas Sensor. Polymers. 2022; 14(21):4758. https://doi.org/10.3390/polym14214758
Chicago/Turabian StyleSedlak, Petr, Pavel Kaspar, Dinara Sobola, Adam Gajdos, Jiri Majzner, Vlasta Sedlakova, and Petr Kubersky. 2022. "Solvent Evaporation Rate as a Tool for Tuning the Performance of a Solid Polymer Electrolyte Gas Sensor" Polymers 14, no. 21: 4758. https://doi.org/10.3390/polym14214758
APA StyleSedlak, P., Kaspar, P., Sobola, D., Gajdos, A., Majzner, J., Sedlakova, V., & Kubersky, P. (2022). Solvent Evaporation Rate as a Tool for Tuning the Performance of a Solid Polymer Electrolyte Gas Sensor. Polymers, 14(21), 4758. https://doi.org/10.3390/polym14214758