

Controlled and Accelerated Hydrolysis of Polylactide (PLA) through Pentaerythritol Phosphites with Acid Scavengers

Abstract
1. Introduction
2. Materials and Methods
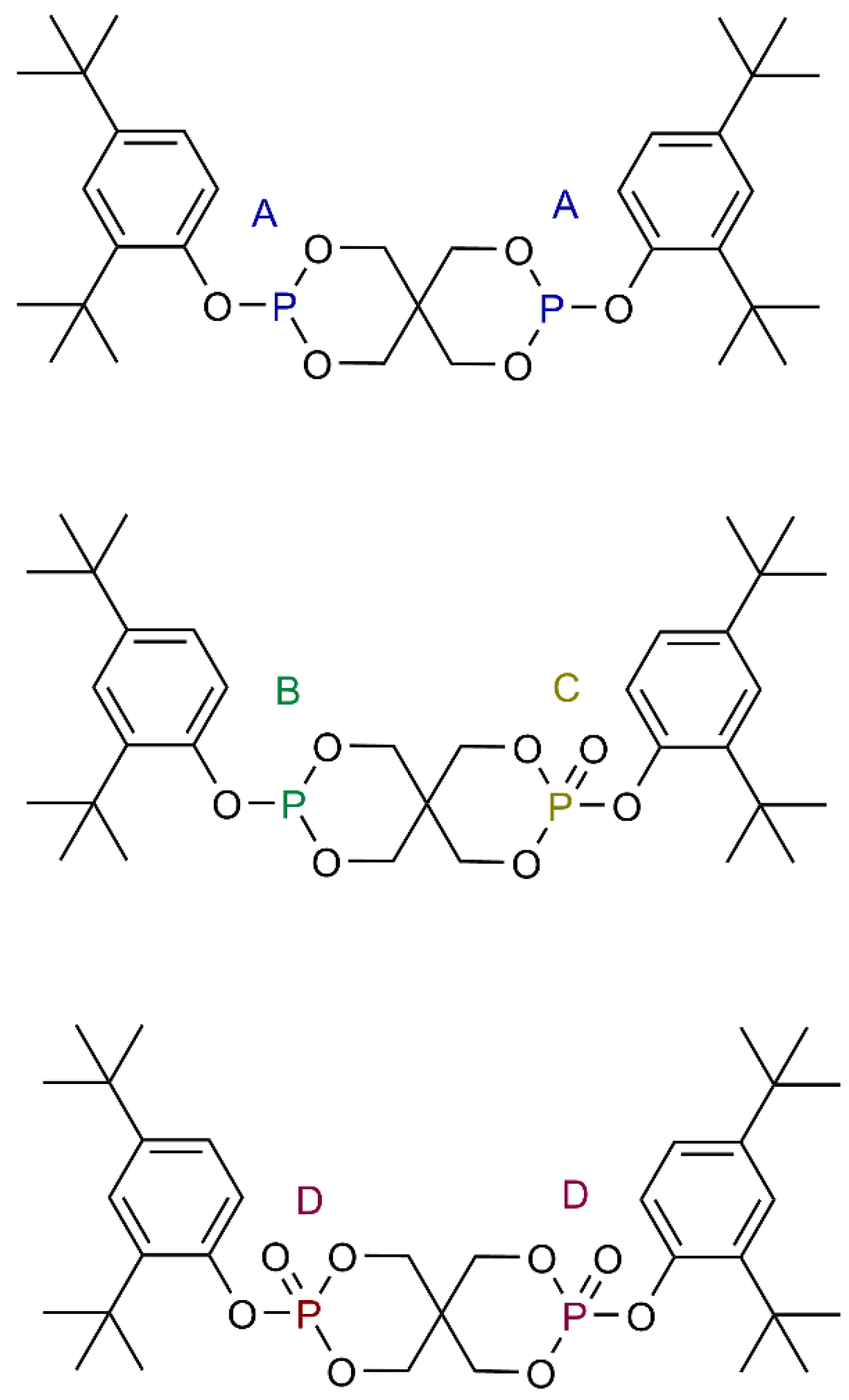
2.1. Materials
2.2. Extrusion
2.3. Hydrolysis Test
2.4. Melt Volume Rate
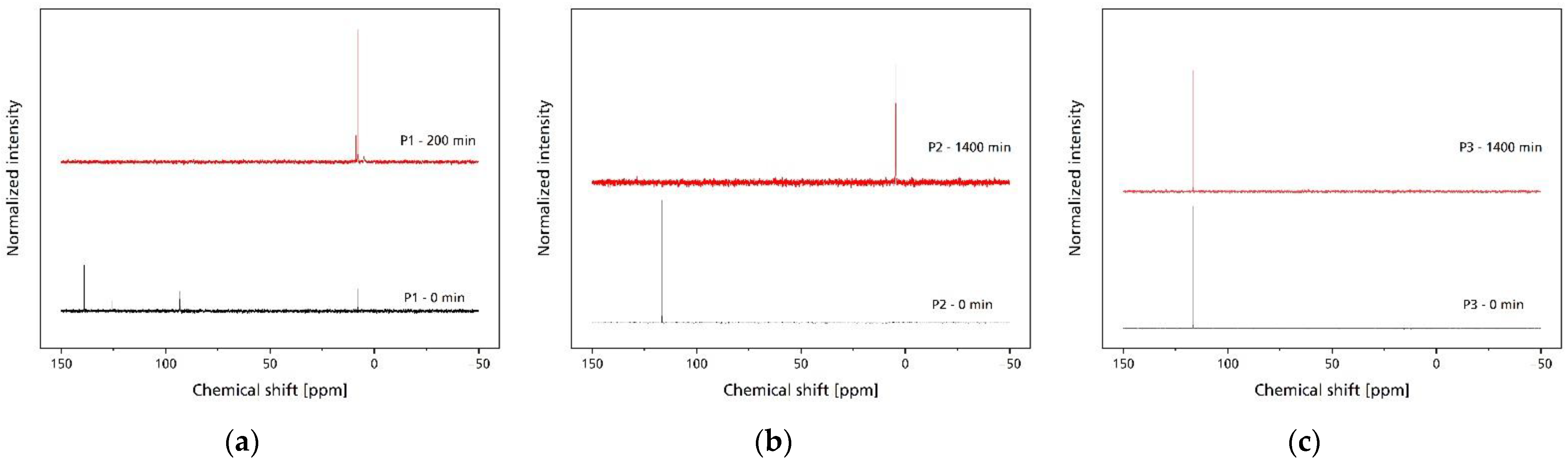
2.5. Aging of Phosphites
2.6. Size-Exclusion Chromatography (SEC)
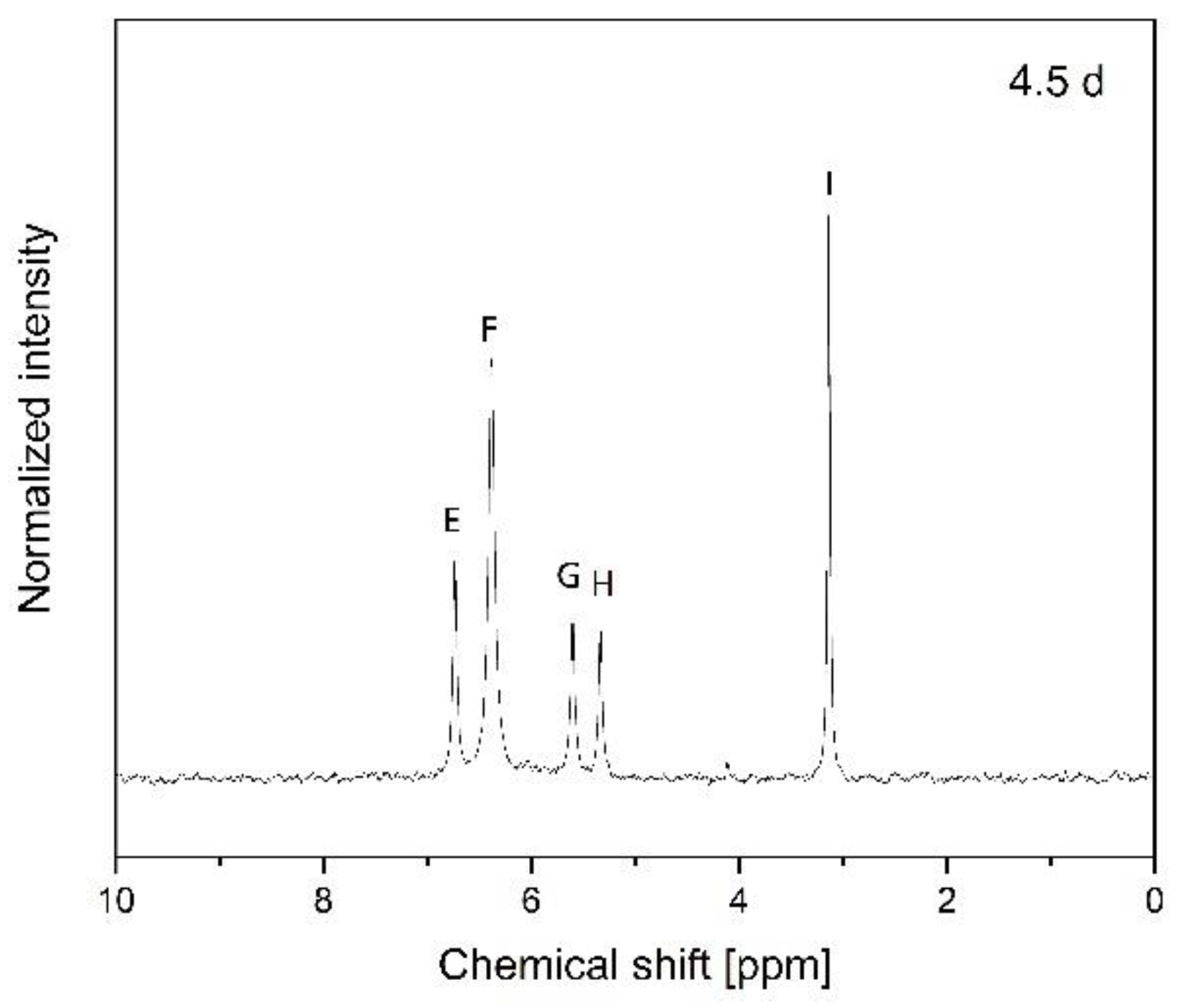
2.7. Nuclear Magnetic Resonance (NMR)
3. Results and Discussion
3.1. Processing Stability
3.2. Hydrolytic Degradation
4. Conclusions
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spierling, S.; Röttger, C.; Venkatachalam, V.; Mudersbach, M.; Herrmann, C.; Endres, H.-J. Bio-based Plastics-A Building Block for the Circular Economy? Procedia CIRP 2018, 69, 573–578. [Google Scholar] [CrossRef]
- Karan, H.; Funk, C.; Grabert, M.; Oey, M.; Hankamer, B. Green Bioplastics as Part of a Circular Bioeconomy. Trends Plant Sci. 2019, 24, 237–249. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- European Comission. A European Strategy for Plastics in a Circular Economy; European Comission: Brussels, Belgium, 2018. [Google Scholar]
- Sander, M. Biodegradation of Polymeric Mulch Films in Agricultural Soils: Concepts, Knowledge Gaps, and Future Research Directions. Environ. Sci. Technol. 2019, 53, 2304–2315. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications-A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed]
- Zaaba, N.F.; Jaafar, M. A review on degradation mechanisms of polylactic acid: Hydrolytic, photodegradative, microbial, and enzymatic degradation. Polym. Eng. Sci. 2020, 60, 2061–2075. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Preziosi, R.; Robson, G.D. Abiotic and biotic environmental degradation of the bioplastic polymer poly(lactic acid): A review. Polym. Degrad. Stab. 2017, 137, 122–130. [Google Scholar] [CrossRef]
- Kale, G.; Auras, R.; Singh, S.P. Comparison of the degradability of poly(lactide) packages in composting and ambient exposure conditions. Packag. Technol. Sci. 2007, 20, 49–70. [Google Scholar] [CrossRef]
- Rudnik, E.; Briassoulis, D. Degradation behaviour of poly(lactic acid) films and fibres in soil under Mediterranean field conditions and laboratory simulations testing. Ind. Crops Prod. 2011, 33, 648–658. [Google Scholar] [CrossRef]
- Huerta-Lwanga, E.; Mendoza-Vega, J.; Ribeiro, O.; Gertsen, H.; Peters, P.; Geissen, V. Is the Polylactic Acid Fiber in Green Compost a Risk for Lumbricus terrestris and Triticum aestivum? Polymers 2021, 13, 703. [Google Scholar] [CrossRef]
- Husárová, L.; Pekařová, S.; Stloukal, P.; Kucharzcyk, P.; Verney, V.; Commereuc, S.; Ramone, A.; Koutny, M. Identification of important abiotic and biotic factors in the biodegradation of poly(l-lactic acid). Int. J. Biol. Macromol. 2014, 71, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Li, S.M.; Garreau, H.; Vert, M. Structure-property relationships in the case of the degradation of massive poly(?-hydroxy acids) in aqueous media. J. Mater. Sci. Mater. Med. 1990, 1, 131–139. [Google Scholar] [CrossRef]
- Vert, M.; Li, S.M.; Garreau, H. More about the degradation of LA/GA-derived matrices in aqueous media. J. Control. Release 1991, 16, 15–26. [Google Scholar] [CrossRef]
- Grizzi, I.; Garreau, H.; Li, S.M.; Vert, M. Hydrolytic degradation of devices based on poly(dl-lactic acid) size-dependence. Biomaterials 1995, 16, 305–311. [Google Scholar] [CrossRef]
- Li, S.M.; Girod-Holland, S.; Vert, M. Hydrolytic degradation of poly(dl-lactic acid) in the presence of caffeine base. J. Control. Release 1996, 40, 41–53. [Google Scholar] [CrossRef]
- Saha, S.K.; Tsuji, H. Effects of molecular weight and small amounts of d-lactide units on hydrolytic degradation of poly(l-lactic acid)s. Polym. Degrad. Stab. 2006, 91, 1665–1673. [Google Scholar] [CrossRef]
- Saha, S.K.; Tsuji, H. Hydrolytic Degradation of Amorphous Films of L-Lactide Copolymers with Glycolide and D-Lactide. Macromol. Mater. Eng. 2006, 291, 357–368. [Google Scholar] [CrossRef]
- Rocca-Smith, J.R.; Whyte, O.; Brachais, C.-H.; Champion, D.; Piasente, F.; Marcuzzo, E.; Sensidoni, A.; Debeaufort, F.; Karbowiak, T. Beyond Biodegradability of Poly(lactic acid): Physical and Chemical Stability in Humid Environments. ACS Sustain. Chem. Eng. 2017, 5, 2751–2762. [Google Scholar] [CrossRef]
- Qi, X.; Ren, Y.; Wang, X. New advances in the biodegradation of Poly(lactic) acid. Int. Biodeterior. Biodegrad. 2017, 117, 215–223. [Google Scholar] [CrossRef]
- Musioł, M.; Sikorska, W.; Adamus, G.; Janeczek, H.; Richert, J.; Malinowski, R.; Jiang, G.; Kowalczuk, M. Forensic engineering of advanced polymeric materials. Part III-Biodegradation of thermoformed rigid PLA packaging under industrial composting conditions. Waste Manag. 2016, 52, 69–76. [Google Scholar] [CrossRef]
- Deroiné, M.; Le Duigou, A.; Corre, Y.-M.; Le Gac, P.-Y.; Davies, P.; César, G.; Bruzaud, S. Accelerated ageing of polylactide in aqueous environments: Comparative study between distilled water and seawater. Polym. Degrad. Stab. 2014, 108, 319–329. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Robson, G.D. The influence of biotic and abiotic factors on the rate of degradation of poly(lactic) acid (PLA) coupons buried in compost and soil. Polym. Degrad. Stab. 2013, 98, 2063–2071. [Google Scholar] [CrossRef]
- Pantani, R.; Sorrentino, A. Influence of crystallinity on the biodegradation rate of injection-moulded poly(lactic acid) samples in controlled composting conditions. Polym. Degrad. Stab. 2013, 98, 1089–1096. [Google Scholar] [CrossRef]
- Tsuji, H. Autocatalytic hydrolysis of amorphous-made polylactides: Effects of l-lactide content, tacticity, and enantiomeric polymer blending. Polymer 2002, 43, 1789–1796. [Google Scholar] [CrossRef]
- Siparsky, G.L.; Voorhees, K.J.; Miao, F. Hydrolysis of Polylactic Acid (PLA) and Polycaprolactone (PCL) in Aqueous Acetonitrile Solutions: Autocatalysis. J. Polym. Environ. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Göpferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikarashi, K. In vitro hydrolysis of poly(l-lactide) crystalline residues as extended-chain crystallites. Polym. Degrad. Stab. 2004, 85, 647–656. [Google Scholar] [CrossRef]
- Lucas, N.; Bienaime, C.; Belloy, C.; Queneudec, M.; Silvestre, F.; Nava-Saucedo, J.-E. Polymer biodegradation: Mechanisms and estimation techniques. Chemosphere 2008, 73, 429–442. [Google Scholar] [CrossRef]
- de Jong, S.J.; Arias, E.R.; Rijkers, D.; van Nostrum, C.F.; Kettenes-van den Bosch, J.J.; Hennink, W.E. New insights into the hydrolytic degradation of poly(lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. [Google Scholar] [CrossRef]
- Lyu, S.P.; Untereker, D. Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.J.; Marcos, B.; Huneault, M.A. Hydrolysis of polylactide in aqueous media. J. Appl. Polym. Sci. 2016, 133, 44152. [Google Scholar] [CrossRef]
- Lyu, S.P.; Schley, J.; Loy, B.; Lind, D.; Hobot, C.; Sparer, R.; Untereker, D. Kinetics and time-temperature equivalence of polymer degradation. Biomacromolecules 2007, 8, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Codari, F.; Lazzari, S.; Soos, M.; Storti, G.; Morbidelli, M.; Moscatelli, D. Kinetics of the hydrolytic degradation of poly(lactic acid). Polym. Degrad. Stab. 2012, 97, 2460–2466. [Google Scholar] [CrossRef]
- Gleadall, A.; Pan, J.; Kruft, M.-A.; Kellomäki, M. Degradation mechanisms of bioresorbable polyesters. Part 1. Effects of random scission, end scission and autocatalysis. Acta Biomater. 2014, 10, 2223–2232. [Google Scholar] [CrossRef]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef]
- von Burkersroda, F.; Schedl, L.; Göpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef]
- Li, S.M.; McCarthy, S. Further investigations on the hydrolytic degradation of poly (DL-lactide). Biomaterials 1999, 20, 35–44. [Google Scholar] [CrossRef]
- Agrawal, C.M.; Huang, D.; Schmitz, J.P.; Athanasiou, K.A. Elevated Temperature Degradation of a 50: 50 Copolymer of PLA-PGA. Tissue Eng. 1997, 3, 345–352. [Google Scholar] [CrossRef]
- Oyama, H.T.; Kimura, M.; Nakamura, Y.; Ogawa, R. Environmentally safe bioadditive allows degradation of refractory poly(lactic acid) in seawater: Effect of poly(aspartic acid-co-l-lactide) on the hydrolytic degradation of PLLA at different salinity and pH conditions. Polym. Degrad. Stab. 2020, 178, 109216. [Google Scholar] [CrossRef]
- Stloukal, P.; Kucharczyk, P. Acceleration of polylactide degradation under biotic and abiotic conditions through utilization of a new, experimental, highly compatible additive. Polym. Degrad. Stab. 2017, 142, 217–225. [Google Scholar] [CrossRef]
- Oyama, H.T.; Tanishima, D.; Maekawa, S. Poly(malic acid-co-L-lactide) as a superb degradation accelerator for Poly(l-lactic acid) at physiological conditions. Polym. Degrad. Stab. 2016, 134, 265–271. [Google Scholar] [CrossRef]
- Oyama, H.T.; Tanaka, Y.; Kadosaka, A. Rapid controlled hydrolytic degradation of poly(l-lactic acid) by blending with poly(aspartic acid-co-l-lactide). Polym. Degrad. Stab. 2009, 94, 1419–1426. [Google Scholar] [CrossRef]
- Valentina, I.; Haroutioun, A.; Fabrice, L.; Vincent, V.; Roberto, P. Tuning the hydrolytic degradation rate of poly-lactic acid (PLA) to more durable applications. In AIP Conference Proceedings; AIP Publishing LLC: Melville, NY, USA, 2017; Volume 1914, p. 210001. [Google Scholar] [CrossRef]
- Renouf-Glauser, A.C.; Rose, J.; Farrar, D.F.; Cameron, R.E. Comparison of the hydrolytic degradation and deformation properties of a PLLA-lauric acid based family of biomaterials. Biomacromolecules 2006, 7, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Hyon, S.-H.; Jamshidi, K.; Ikada, Y. Effects of residual monomer on the degradation of DL-lactide polymer. Polym. Int. 1998, 46, 196–202. [Google Scholar] [CrossRef]
- Mauduit, J.; Prouse, E.; Vert, M. Hydrolytic degradation of films prepared from blends of high and low molecular weight poly(DL-lactic acid)s. J. Biomed. Mater. Res. 1996, 30, 201–207. [Google Scholar] [CrossRef]
- Gorrasi, G.; Pantani, R. Effect of PLA grades and morphologies on hydrolytic degradation at composting temperature: Assessment of structural modification and kinetic parameters. Polym. Degrad. Stab. 2013, 98, 1006–1014. [Google Scholar] [CrossRef]
- Höglund, A.; Hakkarainen, M.; Edlund, U.; Albertsson, A.-C. Surface modification changes the degradation process and degradation product pattern of polylactide. Langmuir 2010, 26, 378–383. [Google Scholar] [CrossRef]
- Tsuji, H.; Muramatsu, H. Blends of aliphatic polyesters: V non-enzymatic and enzymatic hydrolysis of blends from hydrophobic poly(l-lactide) and hydrophilic poly(vinyl alcohol). Polym. Degrad. Stab. 2001, 71, 403–413. [Google Scholar] [CrossRef]
- Hakkarainen, M.; Albertsson, A.-C.; Karlsson, S. Weight losses and molecular weight changes correlated with the evolution of hydroxyacids in simulated in vivo degradation of homo- and copolymers of PLA and PGA. Polym. Degrad. Stab. 1996, 52, 283–291. [Google Scholar] [CrossRef]
- Li, Y.X.; Nothnagel, J.; Kissel, T. Biodegradable brush-like graft polymers from poly(d,l-lactide) or poly(d,l-lactide-co-glycolide) and charge-modified, hydrophilic dextrans as backbone—Synthesis, characterization and in vitro degradation properties. Polymer 1997, 38, 6197–6206. [Google Scholar] [CrossRef]
- Kim, H.J.; Hillmyer, M.A.; Ellison, C.J. Enhanced Polyester Degradation through Transesterification with Salicylates. J. Am. Chem. Soc. 2021, 143, 15784–15790. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, L.V.; Kumar, R.A.; Dav, V.; Gross, R.A.; McCarthy, S.P. Citrate esters as plasticizers for poly(lactic acid). J. Appl. Polym. Sci. 1997, 66, 1507–1513. [Google Scholar] [CrossRef]
- Höglund, A.; Hakkarainen, M.; Albertsson, A.-C. Migration and hydrolysis of hydrophobic polylactide plasticizer. Biomacromolecules 2010, 11, 277–283. [Google Scholar] [CrossRef]
- Cicero, J.A.; Dorgan, J.R.; Dec, S.F.; Knauss, D.M. Phosphite stabilization effects on two-step melt-spun fibers of polylactide. Polym. Degrad. Stab. 2002, 78, 95–105. [Google Scholar] [CrossRef]
- Meng, X.; Shi, G.; Chen, W.; Wu, C.; Xin, Z.; Han, T.; Shi, Y. Structure effect of phosphite on the chain extension in PLA. Polym. Degrad. Stab. 2015, 120, 283–289. [Google Scholar] [CrossRef]
- Meng, X.; Shi, G.; Wu, C.; Chen, W.; Xin, Z.; Shi, Y.; Sheng, Y. Chain extension and oxidation stabilization of Triphenyl Phosphite (TPP) in PLA. Polym. Degrad. Stab. 2016, 124, 112–118. [Google Scholar] [CrossRef]
- Sirisinha, K.; Samana, K. Improvement of melt stability and degradation efficiency of poly (lactic acid) by using phosphite. J. Appl. Polym. Sci. 2021, 138, 49951. [Google Scholar] [CrossRef]
- Fernandes, A.R.; Rose, M.; Charlton, C. 4-Nonylphenol (NP) in food-contact materials: Analytical methodology and occurrence. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2008, 25, 364–372. [Google Scholar] [CrossRef]
- European Chemicals Agency. Tris(Nonylphenyl) Phosphite: Substance Infocard. Available online: https://echa.europa.eu/de/substance-information/-/substanceinfo/100.043.402 (accessed on 24 April 2022).
- Ortuoste, N.; Allen, N.S.; Papanastasiou, M.; McMahon, A.; Edge, M.; Johnson, B.; Keck-Antoine, K. Hydrolytic stability and hydrolysis reaction mechanism of bis(2,4-di-tert-butyl)pentaerythritol diphosphite (Alkanox P-24). Polym. Degrad. Stab. 2006, 91, 195–211. [Google Scholar] [CrossRef]
- Costanzi, S.; Farris, R.; Girelli, D. New high performance phosphites. Polym. Degrad. Stab. 2001, 73, 425–430. [Google Scholar] [CrossRef]
- Stein, D.; Stevenson, D. High-performance phosphite stabilizer. J. Vinyl. Addit. Technol. 2000, 6, 129–137. [Google Scholar] [CrossRef]
- Palade, L.-I.; Lehermeier, H.J.; Dorgan, J.R. Melt Rheology of High l-Content Poly(lactic acid). Macromolecules 2001, 34, 1384–1390. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J.; Wood-Adams, P.M. Control of thermal degradation of polylactide (PLA)-clay nanocomposites using chain extenders. Polym. Degrad. Stab. 2012, 97, 554–565. [Google Scholar] [CrossRef]
- Jacques, B.; Devaux, J.; Legras, R.; Nield, E. Reactions induced by triphenyl phosphite addition during melt mixing of PET/PBT blends: Chromatographic evidence of a molecular weight increase due to the creation of bonds of two different natures. Polymer 1997, 38, 5367–5377. [Google Scholar] [CrossRef]
- Schwetlick, K.; Pionteck, J.; Winkler, A.; Hähner, U.; Kroschwitz, H.; Habicher, W.D. Organophosphorus antioxidants: Part X—Mechanism of antioxidant action of aryl phosphites and phosphonites at higher temperatures. Polym. Degrad. Stab. 1991, 31, 219–228. [Google Scholar] [CrossRef]
- Golovoy, A.; Cheung, M.-F.; Carduner, K.R.; Rokosz, M.J. Control of transesterification in polyester blends. Polym. Eng. Sci. 1989, 29, 1226–1231. [Google Scholar] [CrossRef]
- du Sart, G.G. Process for the Preparation of Lactide and Polylactide Mixture. U.S. Patent Application 17/252,395, 2 September 2021. [Google Scholar]
- Berger, S.; Braun, S.; Kalinowski, H.-O. 31P-NMR-Spektroskopie; Thieme: Stuttgart, Germany, 1993; ISBN 978-3-13-769201-0. [Google Scholar]
- Papanastasiou, M.; McMahon, A.W.; Allen, N.S.; Doyle, A.M.; Johnson, B.J.; Keck-Antoine, K. The hydrolysis mechanism of bis(2,4-di-tert-butyl)pentaerythritol diphosphite (Alkanox P24): An atmospheric pressure photoionisation mass spectrometric study. Polym. Degrad. Stab. 2006, 91, 2675–2682. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
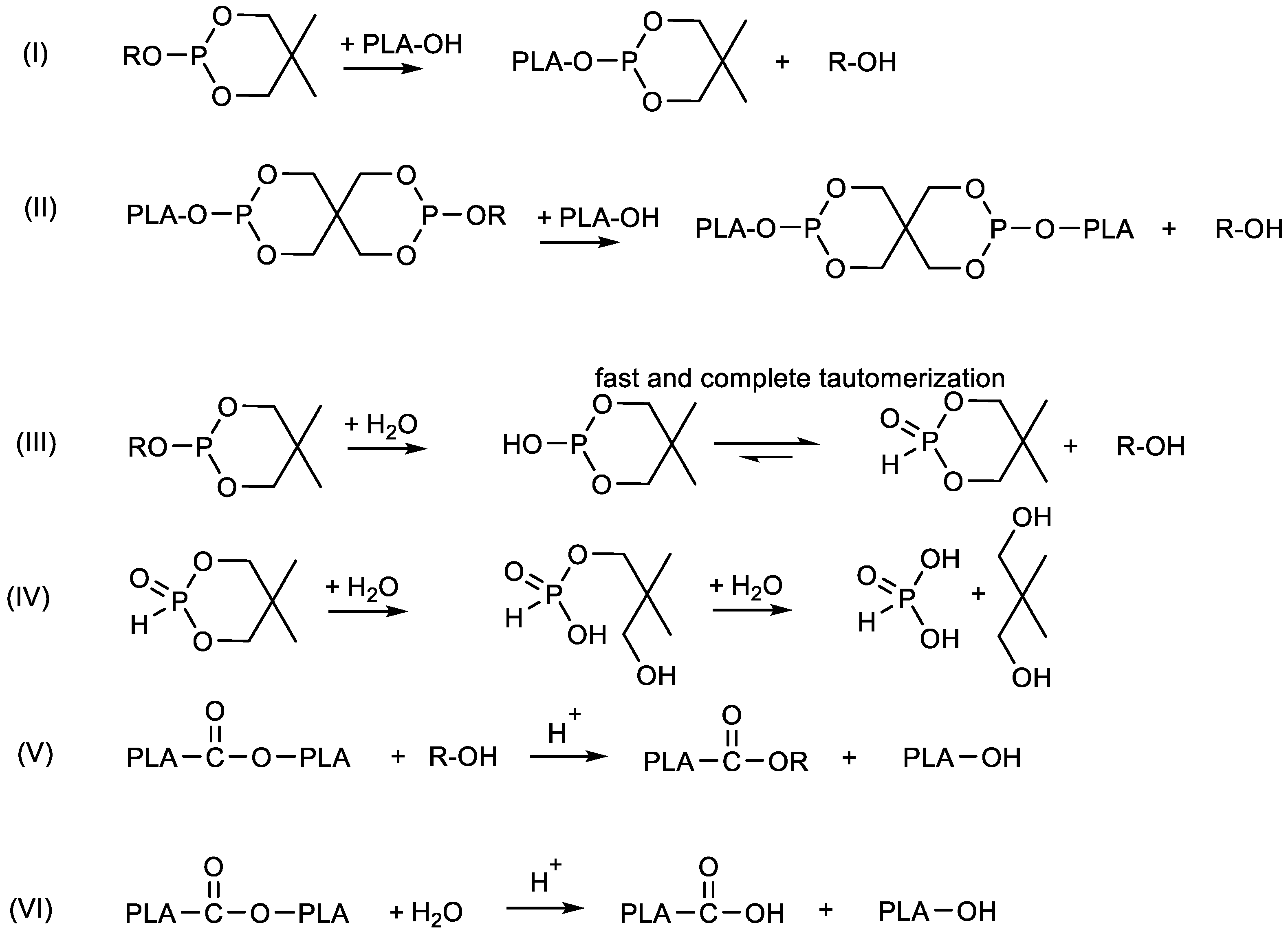{kind=link}
{kind=link}
| Compound | MVR in cm3 10 min−1 |
|---|---|
| PLA L175 | 6.62 ± 0.58 |
| 0.500% P1 | 14.28 ± 0.59 |
| 0.413% P2 | 8.40 ± 0.31 |
| 0.581% P3 | 6.89 ± 0.26 |
| 0.500% P1 + 0.25% S1 | 7.52 ± 0.86 |
| 0.500% P1 + 0.25% S2 | 6.87 ± 0.80 |
| Compound | * in kDa after Processing | * in kDa after Hydrolysis | * in kDa after Processing | * in kDa after Hydrolysis |
|---|---|---|---|---|
| PLA comparison | 61.3 | 42.1 (68.7%) | 139.0 | 91.4 (65.8%) |
| 0.200% P1 | 56.6 | 31.5 (55.7%) | 134.4 | 72.8 (54.2%) |
| 0.500% P1 | 56.6 | 27.1 (47.9%) | 134.6 | 65.0 (48.3%) |
| 0.800% P1 | 56.8 | 24.0 (42.3%) | 137.4 | 61.1 (44.5%) |
| 0.413% P2 | 54.1 | 29.6 (54.7%) | 132.2 | 73.5 (55.6%) |
| 0.581% P3 | 62.4 | 31.6 (50.6%) | 135.1 | 76.6 (56.7%) |
| 0.500% P1 + 0.250% S1 | 55.4 | 30.1 (54.3%) | 132.5 | 73.3 (55.3%) |
| 0.500% P1 + 0.250% S2 | 66.3 | 38.4 (57.9%) | 139.0 | 96.8 (69.6%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polidar, M.; Metzsch-Zilligen, E.; Pfaendner, R. Controlled and Accelerated Hydrolysis of Polylactide (PLA) through Pentaerythritol Phosphites with Acid Scavengers. Polymers 2022, 14, 4237. https://doi.org/10.3390/polym14194237
Polidar M, Metzsch-Zilligen E, Pfaendner R. Controlled and Accelerated Hydrolysis of Polylactide (PLA) through Pentaerythritol Phosphites with Acid Scavengers. Polymers. 2022; 14(19):4237. https://doi.org/10.3390/polym14194237
Chicago/Turabian StylePolidar, Matthias, Elke Metzsch-Zilligen, and Rudolf Pfaendner. 2022. "Controlled and Accelerated Hydrolysis of Polylactide (PLA) through Pentaerythritol Phosphites with Acid Scavengers" Polymers 14, no. 19: 4237. https://doi.org/10.3390/polym14194237
APA StylePolidar, M., Metzsch-Zilligen, E., & Pfaendner, R. (2022). Controlled and Accelerated Hydrolysis of Polylactide (PLA) through Pentaerythritol Phosphites with Acid Scavengers. Polymers, 14(19), 4237. https://doi.org/10.3390/polym14194237