
Solvent-Free Production by Extrusion of Bio-Based Poly(glycerol-co-diacids) Sheets for the Development of Biocompatible and Electroconductive Elastomer Composites

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
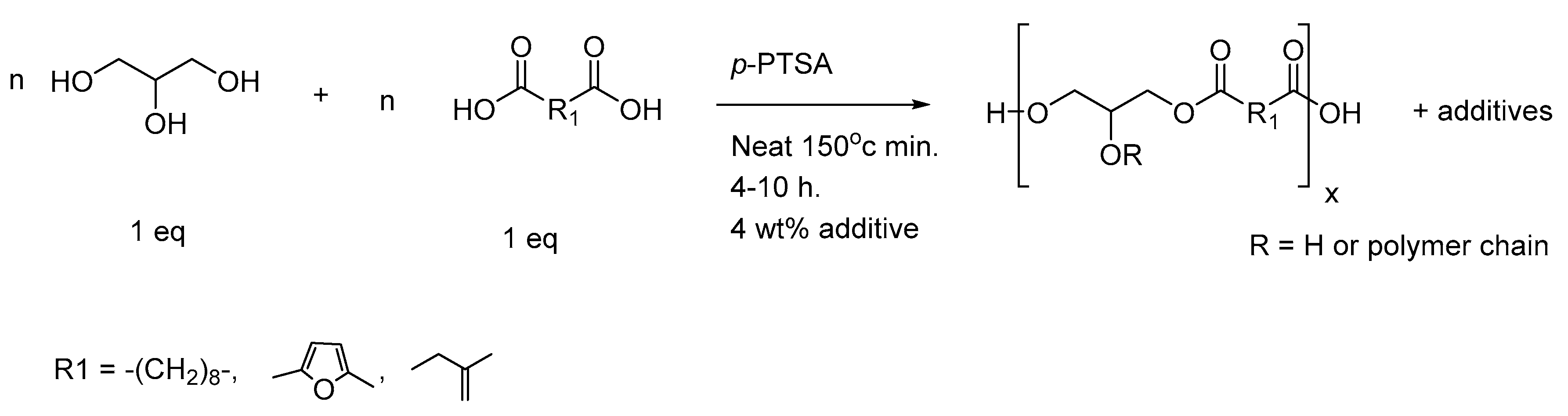
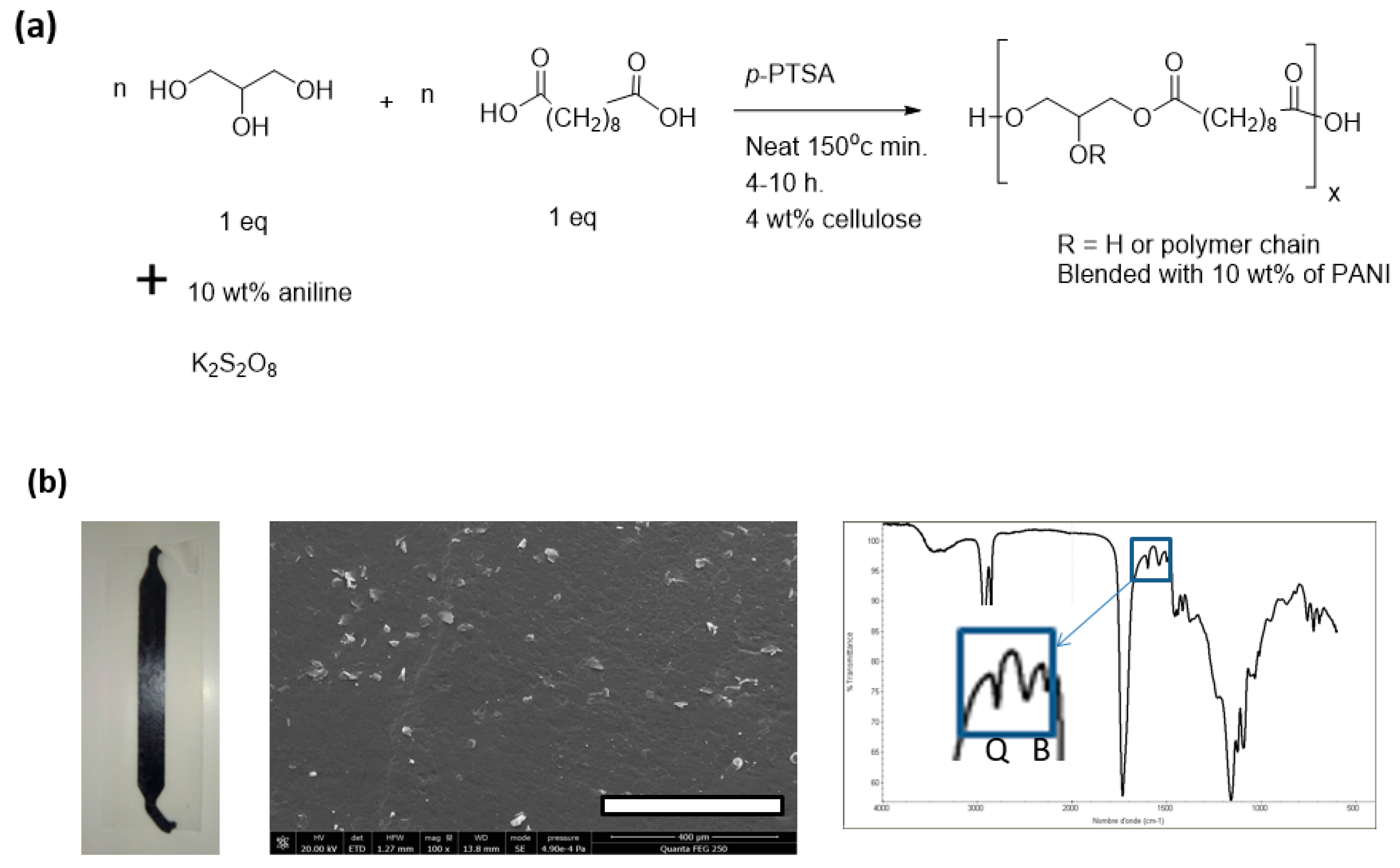
2.2. Preparation of Polyester-Based Elastomers
2.3. Extrusion of PGI, PGS, PGFS, and PGS Composites
2.4. Removal of Unreacted Monomers and Surface pH Monitoring
2.5. Fourier-Transform Infrared (FTIR)
2.6. Field-Emission Scanning Electron Microscopy (FE-SEM)
2.7. Differential Scanning Calorimetry (DSC)
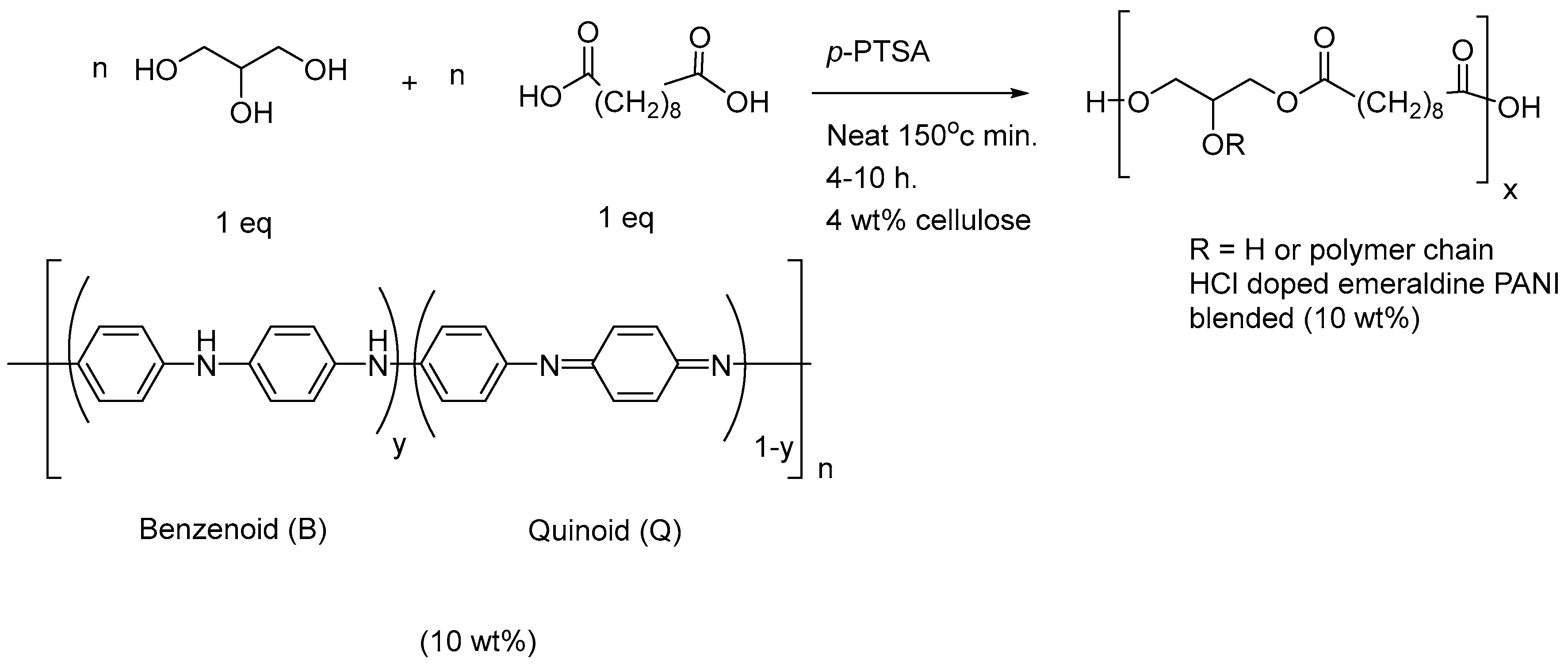
2.8. Production of Electroconductive Polymer Glycerol Solutions
2.9. Electroconductivity and pH Sensitivity Study of PANI Conductive Polymer Blends
3. Results and Discussion
3.1. Optimization of Prepolymer Synthesis and Extrusion Process
3.2. Extrusion of PGI, PGS, PGFS, and PGS Composites
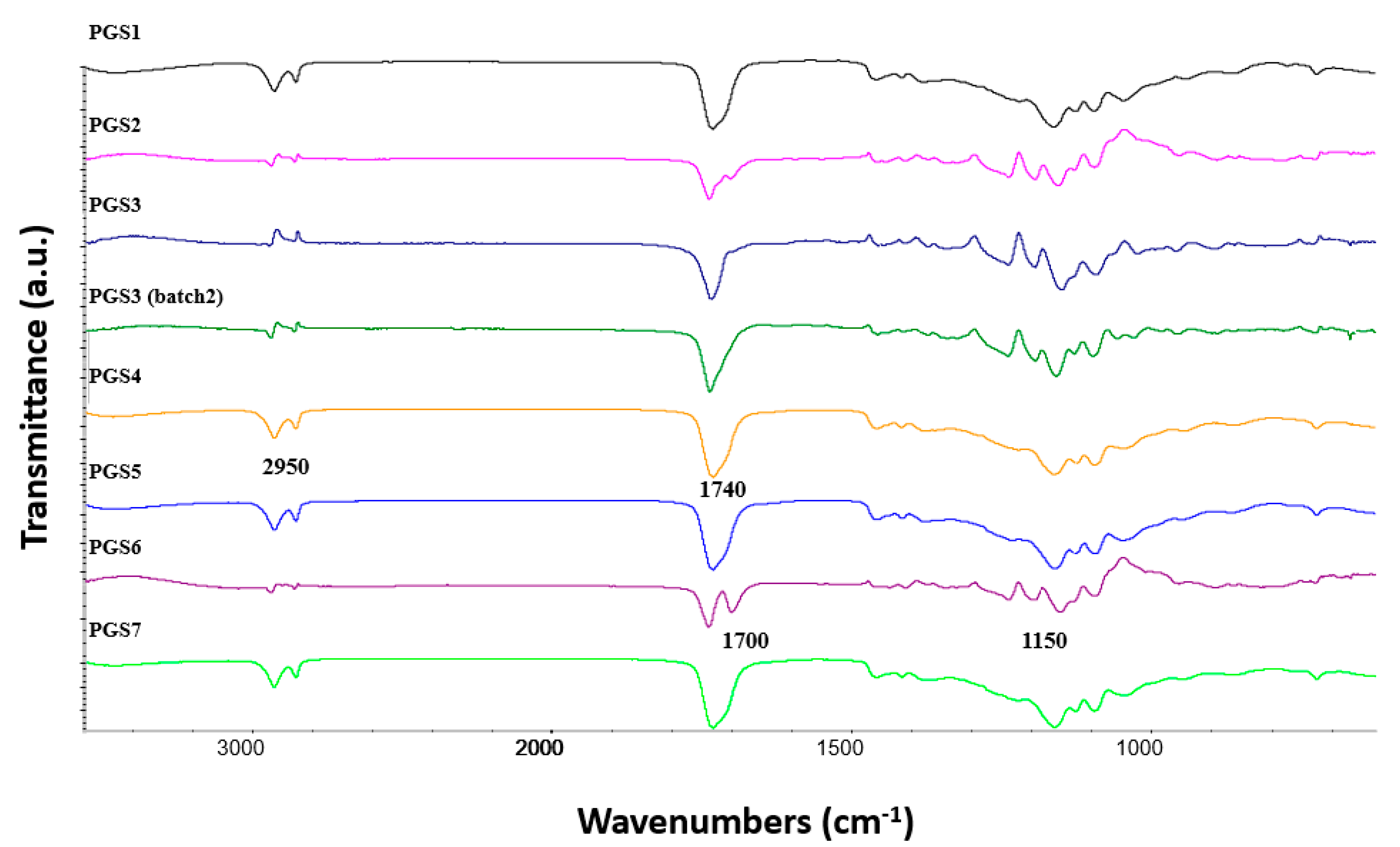
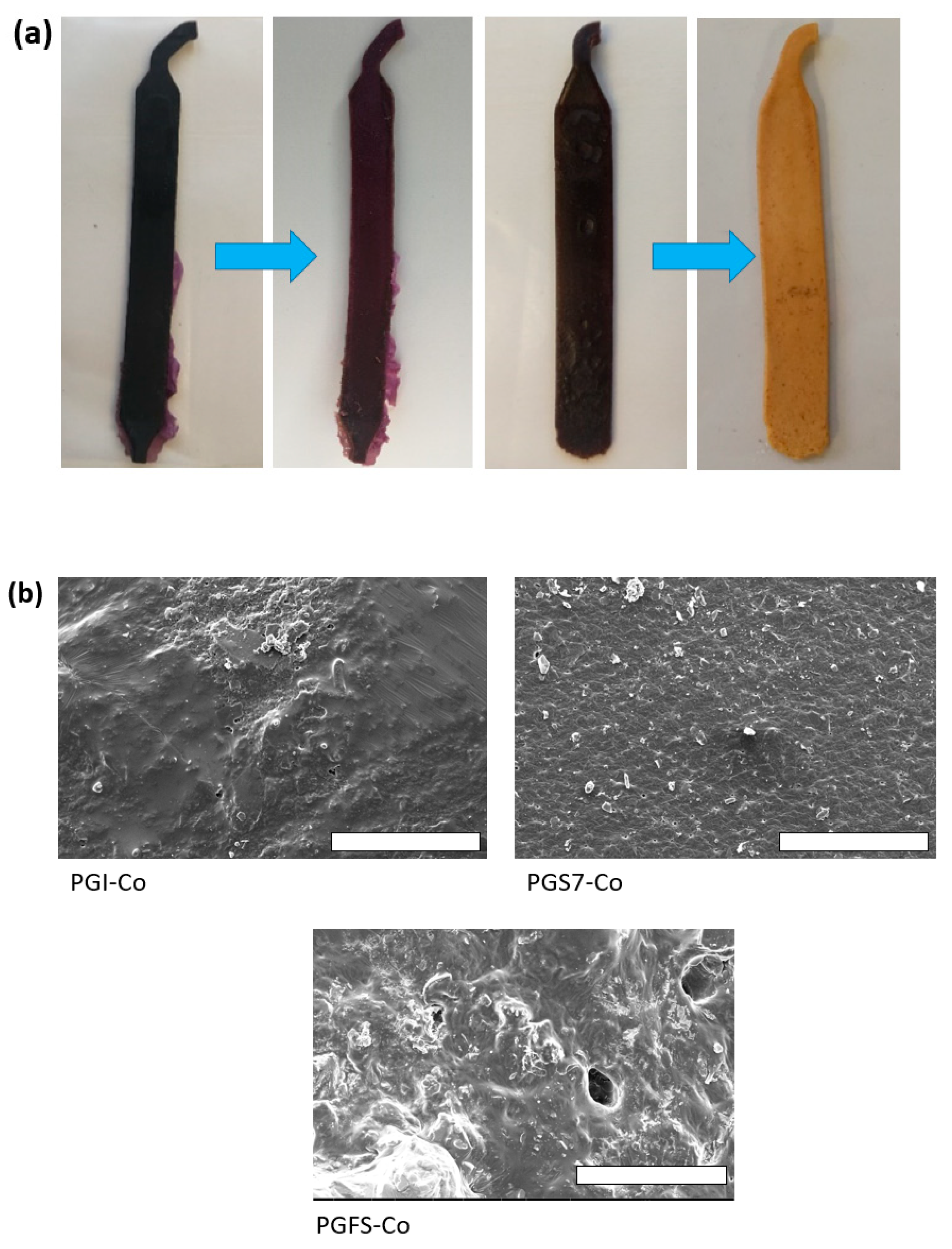
3.3. FE-SEM Observations and Spectroscopic Analysis
3.4. Calorimetry and pH Monitoring of the Materials
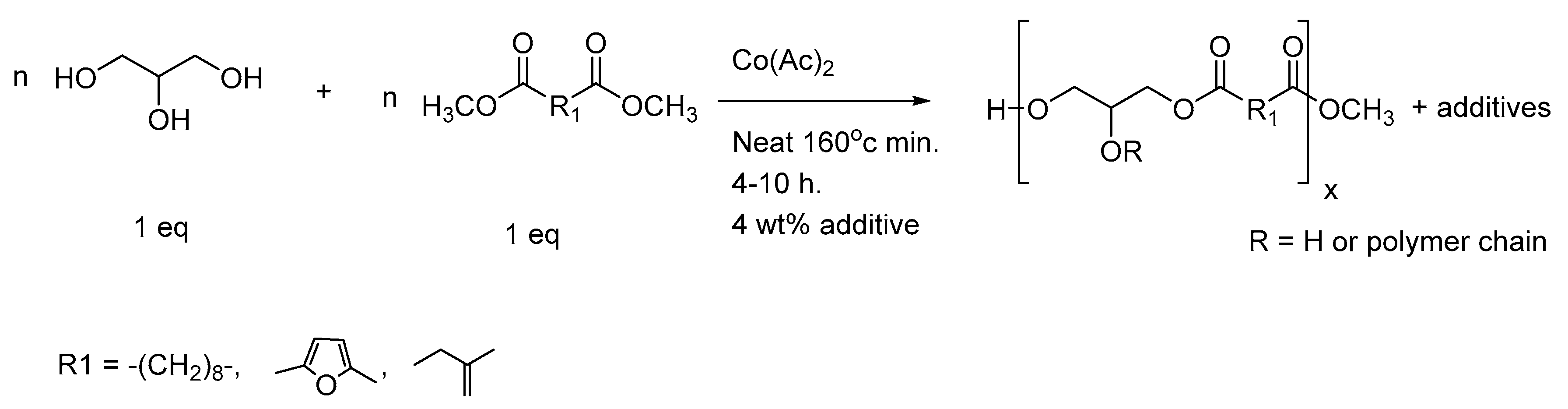
3.5. Alternative Polytransesterification for the Production of Non-Acidic Sheets
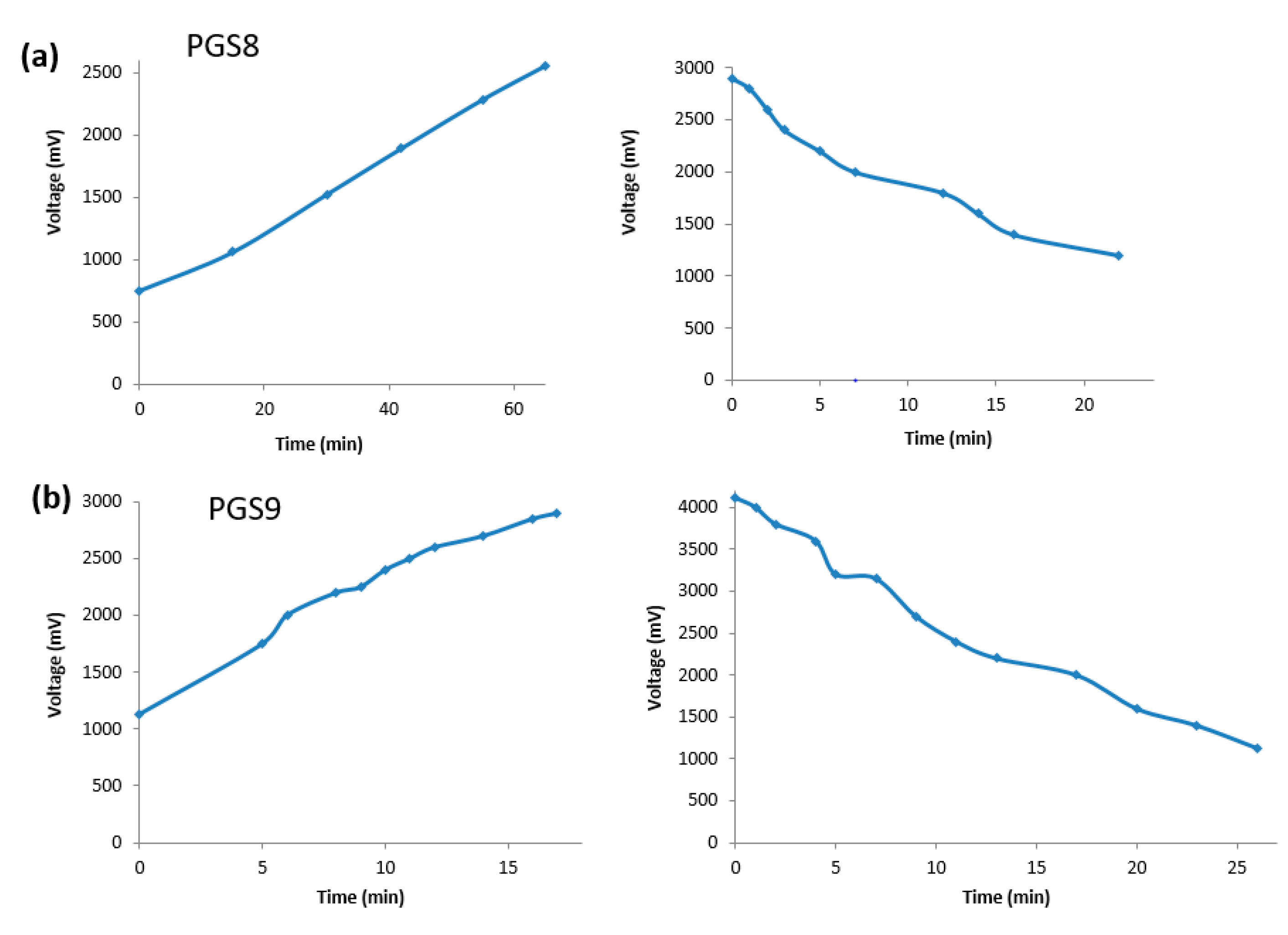
3.6. Study of a PGS Sheet Doped with 10 wt% PANI as a pH Sensor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valerio, O.; Misra, M.; Mohanty, A.K. Poly(glycerol-co-diacids) polyesters: From Glycerol Biorefinery to Sustainable Engineering Applications, A review. ACS Sustain. Chem. Eng. 2018, 6, 5681–5693. [Google Scholar] [CrossRef]
- Tham, W.H.; Wahit, M.U.; Kadir, M.R.A.; Wong, T.W.; Hassan, O. Polyol-based biodegradable polyesters: A short review. Rev. Chem. Eng. 2016, 32, 201–221. [Google Scholar] [CrossRef]
- Zhang, T.; Howell, B.A.; Dumitrascu, A.; Martin, S.J.; Smith, P.B. Synthesis and characterization of glycerol adipic acid hyperbranched polyesters. Polymer 2014, 55, 5065–5072. [Google Scholar] [CrossRef]
- Amarasekera, A.S.; Razzaq, A.; Bonham, P. Synthesis and characterization of all renewable resources based branched polyesters: Poly(2,5-furandicarboxylic acid-co-glycerol). Polym. Sci. 2013, 2013, 645169. [Google Scholar] [CrossRef]
- Neal, R.A.; Jean, A.; Park, H.; Wu, P.B.; Hsiao, J.; Engalmayr, G.C., Jr.; Langer, R.; Freed, L.E. Three-Dimensional Elastomeric Scaffolds Designed with Cardiac-Mimetic Structural and Mechanical Features. Tissue Eng. Part A 2013, 19, 793–807. [Google Scholar] [CrossRef]
- Crapo, P.M.; Wang, Y. Physiologic compliance in engineered small-diameter arterial constructs based on an elastomeric substrate. Biomaterials 2010, 31, 1626–1635. [Google Scholar] [CrossRef]
- Zaky, S.H.; Lee, K.-W.; Gao, J.; Jensen, A.; Close, J.; Wang, Y.; Almarza, A.J.; Sfeir, C. Poly(glycerol Sebacate) Elastomer: A novel Material for Mechanically Loaded Bone Regeneration. Tissue Eng. Part A 2014, 20, 45–53. [Google Scholar] [CrossRef]
- Kemppainen, J.M.; Hollister, S.J. Tailoring the Mechanical properties of 3D-designed poly(glycerol sebacate) scaffolds for cartilage applications. J. Biomed. Mater. Res. Part A 2010, 94, 9–18. [Google Scholar] [CrossRef]
- Loh, X.J.; Karim, A.A.; Owh, C. Poly(glycerol sebacate) biomaterial: Synthesis and biomedical applications. J. Mater. Chem. B 2015, 3, 7641–7652. [Google Scholar] [CrossRef]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar] [CrossRef]
- Louage, B.; Tack, L.; Wang, Y.; De Geest, B.G. Poly(glycerol Sebacate) nanoparticles for encapsulation of hydrophobic anti-cancer drugs. Polym. Chem. 2017, 8, 5033–5038. [Google Scholar] [CrossRef]
- Chen, Q.; Liang, S.; Thouas, G.A. Synthesis and characterization of poly(glycerol sebacate)-co-lactic acid as surgical sealants. Soft Matter 2011, 7, 6484–6492. [Google Scholar] [CrossRef]
- Shirazaki, P.; Varshosaz, J.; Kharazi, A.Z. Electrospun gelatin/poly(Glycerol Sebacate) Membrane with Controlled Release of antibiotics for wound dressing. Adv. Biomed. Res. 2007, 6, 205. [Google Scholar]
- Vogt, L.; Liverani, L.; Roether, J.A.; Boccaccini, A.R. Electrospun Zein Fibers Incorporating Poly(Glycerol Sebacate) for soft Tissue Engineering. Nanomaterials 2018, 8, 150. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, C.; Qiao, X.; Liu, T.; Sun, K. Silk fibroin microfibers and chitosan modified poly(glycerol sebacate) composite scaffolds for skin tissue engineering. Polym. Test. 2017, 62, 88–95. [Google Scholar] [CrossRef]
- Lee, K.-W.; Stolz, D.B.; Wang, Y. Substantial expression of mature elastin in arterial constructs. Proc. Natl. Acad. Sci. USA 2011, 108, 2705–2710. [Google Scholar] [CrossRef]
- Savarani, S.; Ebrahimian-Hoseinabadi, M.; Mohebbi-Kalhori, D. Polyglycerol sebacate/chitosan/gelatin nano-composite scaffolds for engineering neural construct. Mater. Chem. Phys. 2019, 222, 147–151. [Google Scholar] [CrossRef]
- Zhou, L.; He, H.; Jiang, C.; He, S. Preparation and Characterization of poly(glycerol sebacate)/cellulose nanocrystals elastomeric composites. J. Appl. Polym. Sci. 2015, 132, 42196. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Nikkhah, M.; Sant, S.; Khademhosseini, A. Anisotropic poly(glycerol sebacate)-poly(ε-caprolactone) electronspun fibers promote endothelial cell guidance. Biofabrication 2015, 5, 015001. [Google Scholar]
- Keirouz, A.; Fortunato, G.; Zhang, M.; Callanan, A.; Radacsi, N. Nozzle-free electrospinning of polyvinylpyrrolidone/poly(glycerol sebacate) fibrous scaffolds for skin tissue engineering applications. Med. Eng. Phys. 2019, 71, 56–67. [Google Scholar] [CrossRef]
- O’Brien, D.; Hankins, A.; Golestaneh, N.; Paranjape, M. Highly aligned and geometrically structured poly(glycerol sebacate)-polyethylene oxide composite fiber matrices towards bioscaffolding applications. Biomed. Microdevices 2019, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Roether, J.A.; Rai, R.; Wolf, R.; Tallawi, M.; Boccaccini, A.R. Biodegradable poly(glycerol sebacate)/poly(3-hydroxybutyrate)-TiO2 nanocomposites: Fabrication and characterization. Mater. Sci. Technol. 2014, 30, 574–581. [Google Scholar] [CrossRef]
- Tevlek, A.; Agacik, D.T.; Aydin, H.M. Stretchable poly(glycerol-sebacate)/β tricalcium phosphate composites with shape recovery feature by extrusion. J. Appl. Polym. Sci. 2020, 137, 48689. [Google Scholar] [CrossRef]
- Liang, S.; Cook, W.D.; Chen, Q. Physical Characterization of poly(glycerol sebacate)/Bioglass ® composites. Polym. Int. 2012, 61, 17–22. [Google Scholar] [CrossRef]
- Ferrer, C.T.; Vilarino-Feltrer, G.; Rizk, M.; Sydow, H.G.; Valles-Liunch, A. Nanocomposites based on poly(glycerol sebacate) with silica nanoparticles with potential application in dental tissue engineering. Int. J. Polym. Mater. Polym. Biomater. 2019, 69, 761–772. [Google Scholar] [CrossRef]
- Naser, R.; Khrazi, A.Z.; Dini, G. Fabrication of PGS/CaTiO3 Nano-Composite for Biomedical Application. Int. J. Nanosci. Nanotechnol. 2016, 12, 103–108. [Google Scholar]
- Rosenbalm, T.N.; Teruel, H.; Day, C.S.; Donati, G.L.; Morykwas, M.; Argenta, L.; Kuthirummal, N.; Palyachenko, N.L. Structural and Mechanical Characterization of bioresorbable, elastomeric nanocomposites from poly(glycerol sebacate)/nano hydroxyl apatite for tissue transport applications. J. Biomed. Mater. Res. B 2016, 104, 1366–1373. [Google Scholar] [CrossRef]
- Chen, Q.-Z.; Liang, S.-L.; Wang, J.; Simon, G.P. Manipulation of Mechanical compliance of elastomeric PGS by incorporation of halloysite nanotubes for tissue engineering applications. J. Mech. Behav. Biomed. Mater. 2011, 4, 1805–1818. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Patel, A.; Dolatshahi-Pirouz, A.; Zhang, H.; Rangarajan, K.; Iviglia, G.; Shin, S.-R.; Hussein, H.A.; Khademhosseini, A. Elastomeric nanocomposite scaffolds made from PGS chemically crosslinked with CNT. Biomater. Sci. 2015, 3, 45–68. [Google Scholar] [CrossRef]
- Li, C.-Y.; Hu, M.-H.; Hu, J.-J. Use of Aligned Microscale of Sacrificial fibers in Creating Biomimetic, Anisotropic Poly(glycerol sebacate) Scaffolds. Polymers 2019, 11, 1492. [Google Scholar] [CrossRef]
- Wu, H.-J.; Hu, M.-H.; Tuan-Mu, H.-Y.; Hu, J.-J. Preparation of aligned poly(glycerol sebacate) fibrous membranes for anisotropic tissue engineering. Mater. Sci. Eng. C 2019, 100, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Solazzo, M.; O’Brien, F.J.; Nicolosi, V.; Monaghan, M.G. The rationale and emergence of electroconductive biomaterials scaffolds in cardiac tissue engineering. APL Bioeng. 2019, 3, 041501. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.H.; Rai, R.; Dippold, D.; Roether, J.E.; Shubert, D.W.; Rosellini, E.; Barbani, N.; Boccaccini, A.R. Development and characterization of novel electrically conductive PANI-PGS composite for cardiac tissue engineering applications. Acta Biomater. 2014, 10, 2434–2445. [Google Scholar] [CrossRef]
- Hu, T.; Wu, Y.; Zhao, X.; Wang, L.; Bi, L.; Ma, P.X.; Guo, B. Micropatterned, electroactive, and biodegradable poly(glycerol sebacate)-aniline trimer elastomer for tissue engineering. Chem. Eng. Technol. 2019, 366, 208–222. [Google Scholar] [CrossRef]
- Cao, Y.; Smith, P.; Heeger, A.J. Counter-ion induced processability of conducting polyaniline and of conducting polyblends of polyaniline in bulk polymers. Synth. Met. 1992, 48, 91–97. [Google Scholar] [CrossRef]
- Ezazi, N.Z.; Adjary, R.; Correia, A.; Makila, E.; Salonen, J.; Kemell, M.; Hirvonen, J.; Rojas, O.J.; Ruskoaho, H.J.; Santos, H.A. Fabrication and Characterization of Drug Loaded Conductive Poly(glycerol Sebacate)/Nanoparticle-Based Composite Patch for Myocardial Infarctus Applications. ACS Appl. Mater. Interfaces 2020, 12, 6899–6909. [Google Scholar] [CrossRef]
- Gadomska-Gajadhur, A.; Wrzecionek, H.; Matyszczak, G.; Pietowski, P.; Wieclaw, M.; Ruskowski, P. Optimization of Poly(Glycerol sebacate) Synthesis for Biomedical Purposes with the Design Experiments. Org. Process Res. Dev. 2018, 22, 1793–1800. [Google Scholar] [CrossRef]
- Li, X.; Hong, A.T.-L.; Naskar, N.; Chung, H.-J. Criteria for quick and Consistent Synthesis of Poly(glycerol Sebacate) for Tailored Mechanical Properties. Biomacromolecules 2015, 16, 1525–1533. [Google Scholar] [CrossRef]
- Harris, J.J.; Lu, S.; Gabriele, P. Commercial Challenges in Developing Biomaterials for Medical Device Development. Polym. Int. 2018, 67, 969–974. [Google Scholar] [CrossRef]
- Kafouris, D.; Kossiras, F.; Constantinides, C.; Nguyen, N.Q.; Wesdemiotis, C.; Patrickios, C.S. Biosourced Amphiphilic Degradable Elastomers of Poly(glycerol Sebacate): Synthesis network and Oligomer Characterization. Macromolecules 2013, 46, 622–630. [Google Scholar] [CrossRef]
- Liu, Q.; Tian, H.; Ding, T.; Shi, R.; Feng, Y.; Zhang, L.; Chen, D.; Tian, W. Preparation and Characterization of a Thermoplastic Poly(glycerol Sebacate) Elastomer by Two-Step Method. J. Appl. Polym. Sci. 2007, 103, 1412–1419. [Google Scholar] [CrossRef]
- Martin-Cabezuelo, R.; Vilariño-Feltrer, G.; Valles-Lluch, A. Influence of prepolymerization atmosphere on the properties of pure- and poly(glycerol sebacate). Mater. Sci. Eng. C 2021, 119, 111429. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, K.-W.; Gade, P.S.; Robertson, A.M.; Wang, Y. Microwave assisted Facile Fabrication of porous poly(glycerol sebacate) scaffolds. J. Biomater. Sci. Polym. Ed. 2018, 29, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, H.H.; Salimi, K.; Rzayev, Z.M.O.; Piskin, E. Microwave-assisted rapid synthesis of poly(glycerol sebacate) elastomers. Biomater. Sci. 2013, 1, 503–509. [Google Scholar] [CrossRef]
- Lau, C.C.; Bayazit, H.K.; Knowles, J.C.; Tang, J. Tailoring degree of esterification and branching of poly(glycerol sebacate) by energy efficient microwave irradiation. Polym. Chem. 2017, 8, 3937–3947. [Google Scholar] [CrossRef]
- Zhao, D.; Delbecq, F.; Len, C. One-pot FDCA diesters synthesis from mucic acid and their solvent-free regioselective polytransesterification for production of glycerol-based furanic polyesters. Molecules 2019, 24, 1030. [Google Scholar] [CrossRef]
- Wuang, S.; Dupin, L.; Noel, H.; Carroux, C.J.; Renaud, L.; Gehin, T.; Meyer, A.; Souteyrand, E.; Vasseur, J.J.; Vergoten, G.; et al. Toward the Rational Design of Galactosylated Glycoclusters That Target Pseudomonas aeruginosa Lectin A (Lec A): Influence of a Linker Arms That lead to Low-Nanomolar Mutivalents Ligands. Chem. Eur. J. 2016, 22, 11785–11794. [Google Scholar] [CrossRef]
- Kunkuma, V.L.; Kaki, S.S.; Rao, B.V.S.K.; Prasad, R.B.N.; Prabhavathi Devi, B.L.A. A simple and facile method for the synthesis of 1-Octosanol. Eur. J. Lipid Sci. Technol. 2013, 115, 921–927. [Google Scholar] [CrossRef]
- Malferrari, D.; Armenise, N.; Decesari, S.; Galletti, P.; Tagliavini, E. Surfactants from Itaconic acids Physicochemical Properties and Assessments of the Synthetic Strategies. ACS Sustain. Chem. Eng. 2015, 3, 1579–1588. [Google Scholar] [CrossRef]
- Nijst, C.L.E.; Bruggeman, J.P.; Karp, J.M.; Ferreira, L.; Zumbuehl, A.; Bettinger, C.J.; Langer, R. Synthesis and Characterization of Photocurable Elastomers from Poly(Glycerol-co-Sebacate). Biomacromolecules 2007, 10, 3067–3073. [Google Scholar] [CrossRef]
- Konyushenko, E.N.; Reynaud, S.; Pellerin, V.; Trchova, M.; Stejskal, J.; Sapurina, I. Polyaniline prepared in ethylene glycol or glycerol. Polymer 2021, 52, 1900–1907. [Google Scholar] [CrossRef]
- Dai, J.; Ma, S.; Wu, Y.; Han, L.; Zhang, L.; Zhu, J.; Liu, X. Polyesters derived from itaconic acid for the properties and bio-based contact enhancement of soybean-oil thermosets. Green Chem. 2015, 17, 2383–2392. [Google Scholar] [CrossRef]
- Chanda, S.; Ramakrishnan, S. Poly(alkylene itaconate)s-an interesting class of polyesters with periodically located exo-chains double bonds susceptible to Michael addition. Polym. Chem. 2015, 6, 2108–2114. [Google Scholar] [CrossRef]
- Moore, O.B.; Hanson, P.-A.; Comerford, J.W.; Pellis, A.; Farmer, T.J. Improving the Post-Polymerization Modification of Biobased Itaconate Unsaturated Polyesters; Catalyzing Aza-Michael Additions with Reusable iodine on acidic Alumina. Front. Chem. 2019, 7, 501. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Chen, F.; Liu, Z.-Y.; Yang, W.; Yang, M. Preparation of cellulose-graft-polylactic acid via melt polycondensation for use in polylactic acid based composites: Synthesis, characterization and properties. RSC Adv. 2016, 6, 1973–1983. [Google Scholar] [CrossRef]
- Humpolicek, P.; Kasparkova, V.; Saha, P.; Stejskal, J. Biocompatibility of polyaniline. Synth. Met. 2012, 162, 722–727. [Google Scholar] [CrossRef]
- Neelakandan, R.; Giridev, V.R.; Murugesan, M.; Madhusoothanan, M. Surface Resistivity and Shear Characteristics of Polyaniline Coated Polyester Fabric. J. Ind. Text. 2009, 39, 175–185. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, J.; Xiao, H.; Lu, Y.; Lu, M. Creation of polyaniline-coated polyester fabrics with conductive, electrothermal and energy-storage properties via micro-dissolution method. Mater. Today Commun. 2020, 24, 101042. [Google Scholar] [CrossRef]
- Li, Y.; Mao, Y.; Xiao, C.; Xu, X.; Li, X. Flexible pH sensor based on a conductive PANI membrane for pH monitoring. RSC Adv. 2021, 10, 21–28. [Google Scholar] [CrossRef]
- Dejeu, J.; Cot, A.; Rougeot, P.; Lakard, B.; Lakard, S.; Gauthier, M. Development of new sticky and conducting polymer surfaces for MEMS applications. Synth. Met. 2021, 276, 116757. [Google Scholar] [CrossRef]
- Ba, Y.; Zhou, S.; Jiao, S.; Pan, W. Fabrication of polyaniline/copper sulfide/polyethylene (terephthalate) thread electrode for flexible fiber-shaped supercapacitors. J. Appl. Polym. Sci. 2018, 135, 46769. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Additive (4 wt%) | Temp (°C) | Original Characteristics |
|---|---|---|---|
| Pre-PGI1 | - | 160 | Not sticky, elastic |
| Pre-PGI2 | - | 150 | Not sticky, elastic, translucent |
| Pre-PGS1 | - | 160 | Sticky, soft, translucent |
| Pre-PGS2 | NaCl 1 | 160 | Sticky, brown, soft, translucent |
| Pre-PGS3 | Microcrystalline cellulose | 150 | Not sticky, soft, translucent |
| Pre-PGS4 | Nanocellulose | 150 | Sticky, soft, translucent |
| Pre-PGS5 | Pluronic® F127 | 150 | Sticky, soft, translucent |
| Pre-PGS6 | PSSNa-PEDOT 2 | 150 | Sticky, rigid, dark blue |
| Pre-PGS7 | PANI 2 | 150 | Sticky, dark brown |
| Pre-PGFS1 | - | 150 | Sticky, elastic, white |
| Pre-PGFS1 | NaCl 1 | 150 | Sticky, elastic, cream |
| Event (°C) | PGI1 | PGI2 | PGFS1 | PGFS2 | PGS1 | PGS2 | PGS3 | PGS4 | PGS5 | PGS6 | PGS7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tm1 | 55 | 37 | 16 | 35 | 9 | 6 | 11 | ||||
| Tm2 | 83 | 74 | 38 | 36 | |||||||
| Ttc | 177 | 136 | 190 | 56 | 141 | 124 | 128 | 130 | 183 | 241 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, S.; Stricher, M.; Nadaud, F.; Guenin, E.; Egles, C.; Delbecq, F. Solvent-Free Production by Extrusion of Bio-Based Poly(glycerol-co-diacids) Sheets for the Development of Biocompatible and Electroconductive Elastomer Composites. Polymers 2022, 14, 3829. https://doi.org/10.3390/polym14183829
Ji S, Stricher M, Nadaud F, Guenin E, Egles C, Delbecq F. Solvent-Free Production by Extrusion of Bio-Based Poly(glycerol-co-diacids) Sheets for the Development of Biocompatible and Electroconductive Elastomer Composites. Polymers. 2022; 14(18):3829. https://doi.org/10.3390/polym14183829
Chicago/Turabian StyleJi, Shengzhi, Mathilde Stricher, Frédéric Nadaud, Erwann Guenin, Christophe Egles, and Frédéric Delbecq. 2022. "Solvent-Free Production by Extrusion of Bio-Based Poly(glycerol-co-diacids) Sheets for the Development of Biocompatible and Electroconductive Elastomer Composites" Polymers 14, no. 18: 3829. https://doi.org/10.3390/polym14183829
APA StyleJi, S., Stricher, M., Nadaud, F., Guenin, E., Egles, C., & Delbecq, F. (2022). Solvent-Free Production by Extrusion of Bio-Based Poly(glycerol-co-diacids) Sheets for the Development of Biocompatible and Electroconductive Elastomer Composites. Polymers, 14(18), 3829. https://doi.org/10.3390/polym14183829