
Molecular Insights into the Wall Slip Behavior of Pseudoplastic Polymer Melt in Nanochannels during Micro Injection Molding



Abstract
:1. Introduction
2. Simulation Methodology
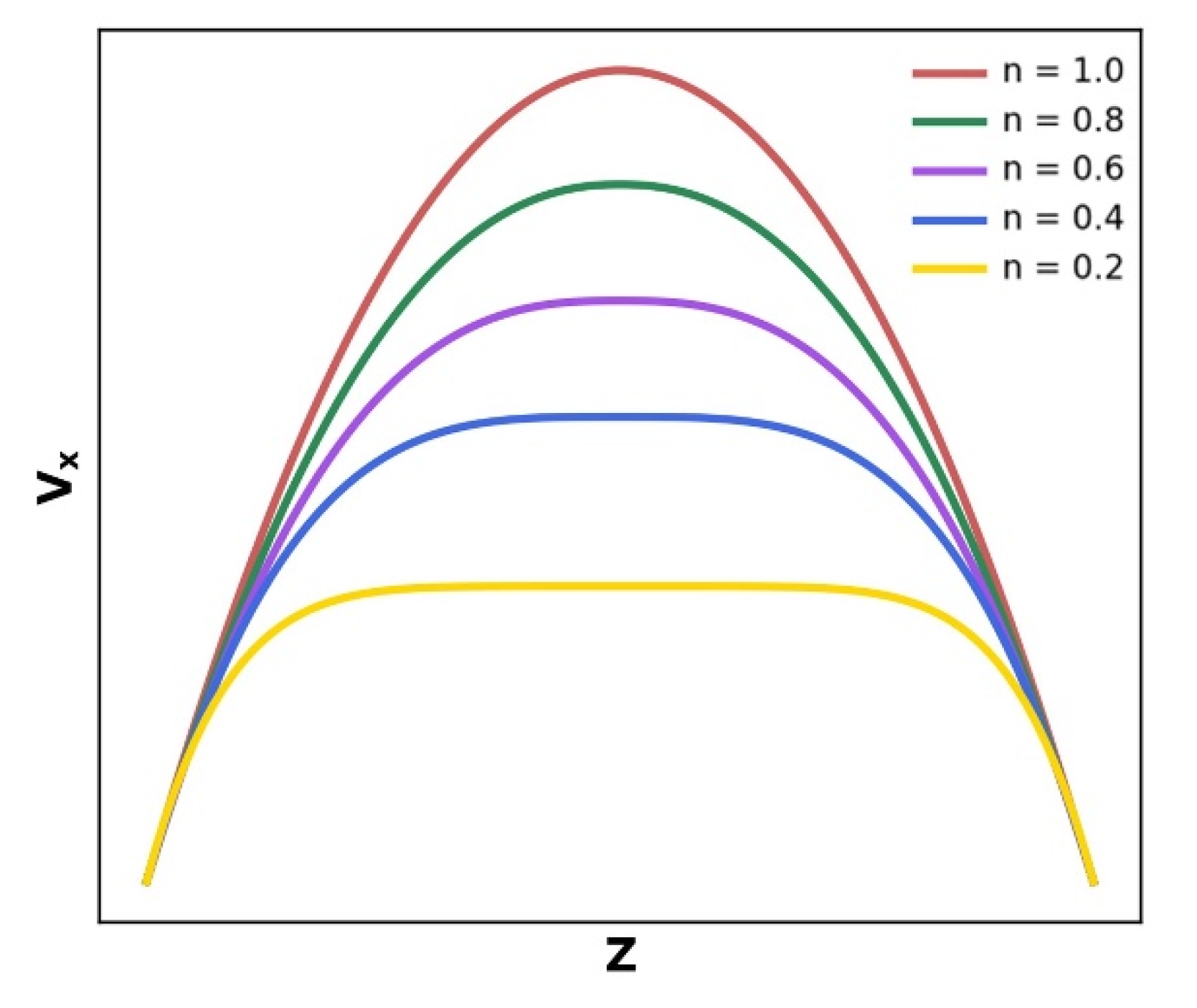
2.1. Pseudoplastic Fluid Flow Characteristics
2.2. Interatomic Potential and Normalized Units
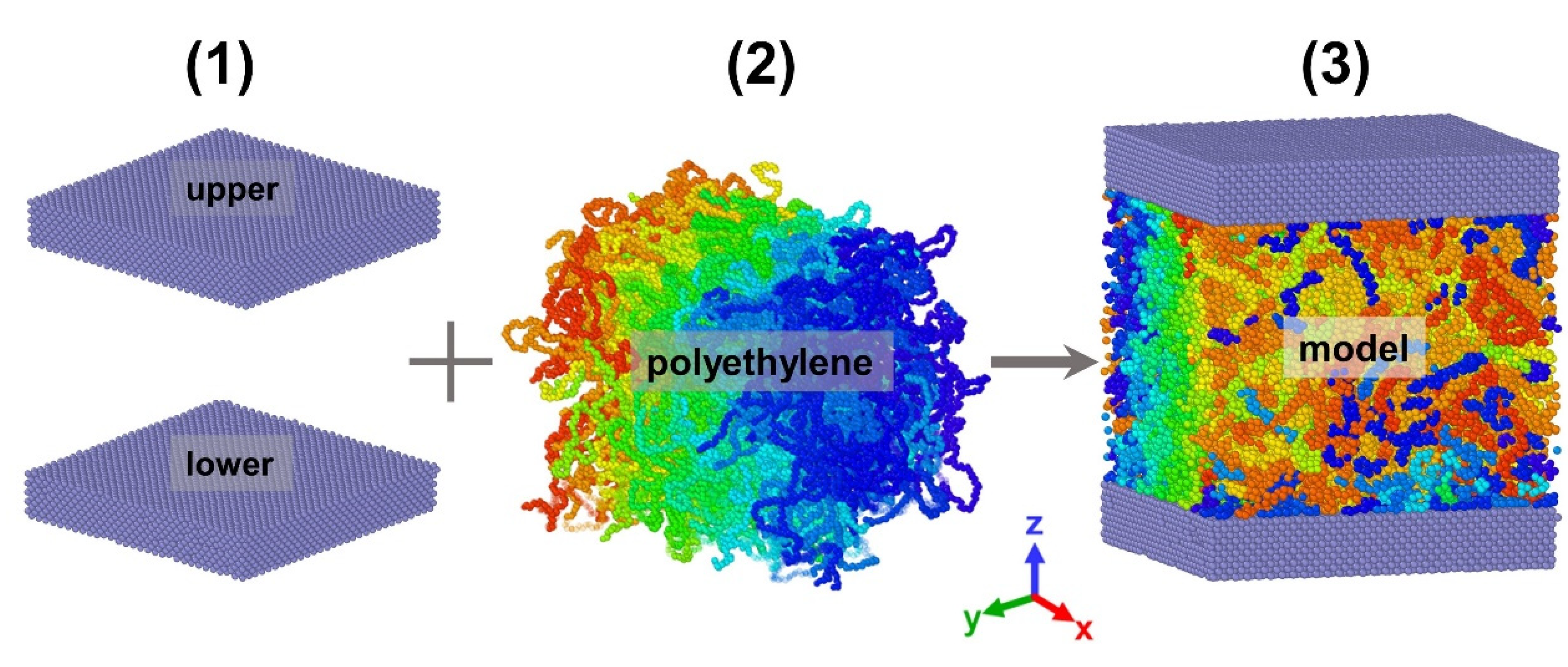
2.3. Model Construction
- (1)
- The EAM potential was utilized to construct the Ni wall surface. Meanwhile, the wall surface as a whole was rigid, and its atoms were fixed in place at the lattice point location and employed to neutralize wall atom interactions. The rigid wall surface not only increases simulation efficiency but also enhances the accuracy of the fluid flow in the channel at low shear rates;
- (2)
- The modeling of polyethylene molecular chains was constructed in moltemplate [25]. It contains 200 chains with a degree of polymerization of 130. Afterwards, the generated file was subjected to systemic relaxation in open-source code Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) to obtain a stable polyethylene in the initial state with a time step of ;
- (3)
- The entire model was composed using the potential parameters stated above, and the systemic relaxation procedure was re-performed to make the system more balanced and homogenous, using the final created file in steps (1) and (2). It is vital that the simulation temperature computation can only compute atomic thermal motion. However, the generation of kinetic energy during a non-equilibrium molecular dynamics simulation devastated the temperature calculation due to the macroscopic motion generated during the simulation; the temperature calculation must be modified to remove the influence of macroscopic kinetic energy [26,27]. As a result, an impartial temperature control strategy based on velocity profiles was employed. The system’s final measurements were in the X, Y, and Z directions. In order to more clearly show the morphology of different molecular chains and the stratification between molecular layers, we used the Open Visualization Tool to set different colors for the model.
2.4. Research Scheme
3. Results and Discussion
3.1. Influence of Atomic Driving Force
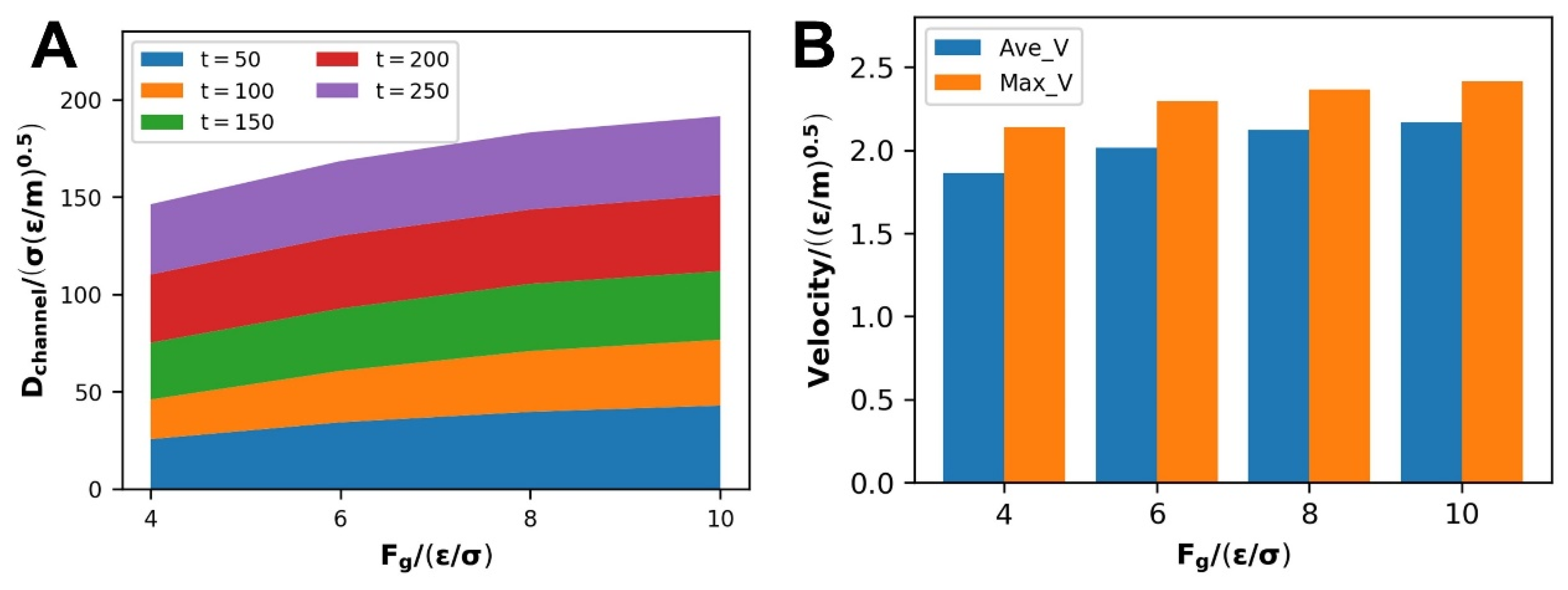
3.1.1. Velocity and Density Distribution
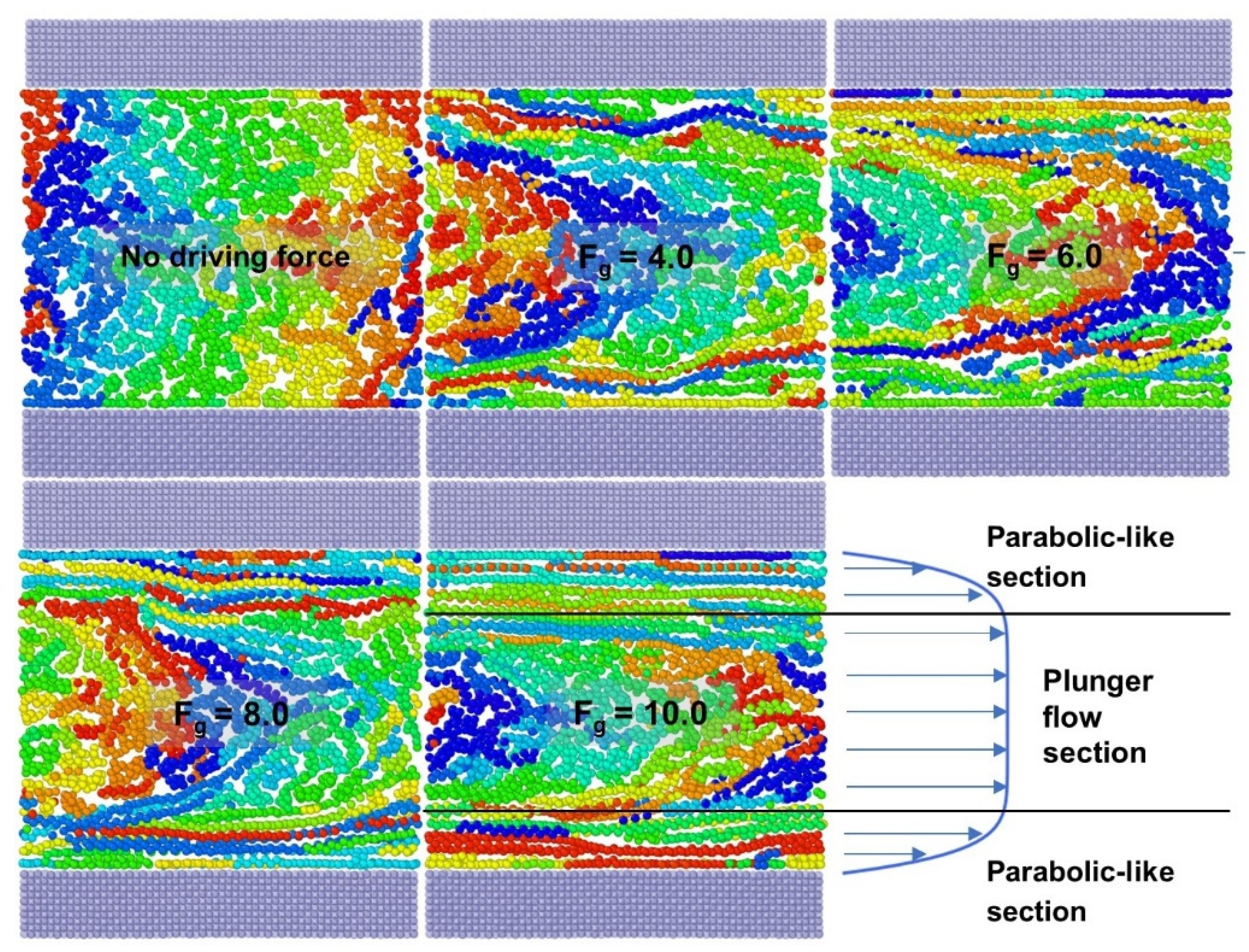
3.1.2. Dynamic Information of Flow Process
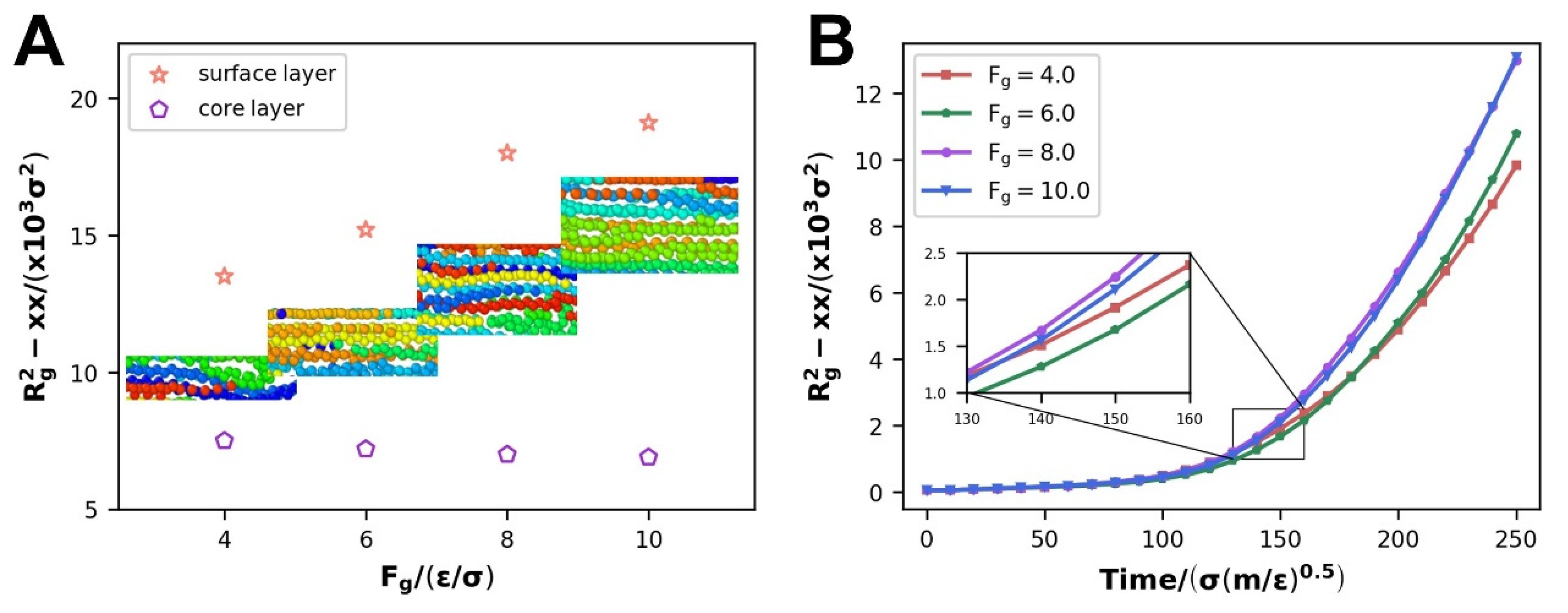
3.1.3. Evolution of Internal Molecular Chain Structure
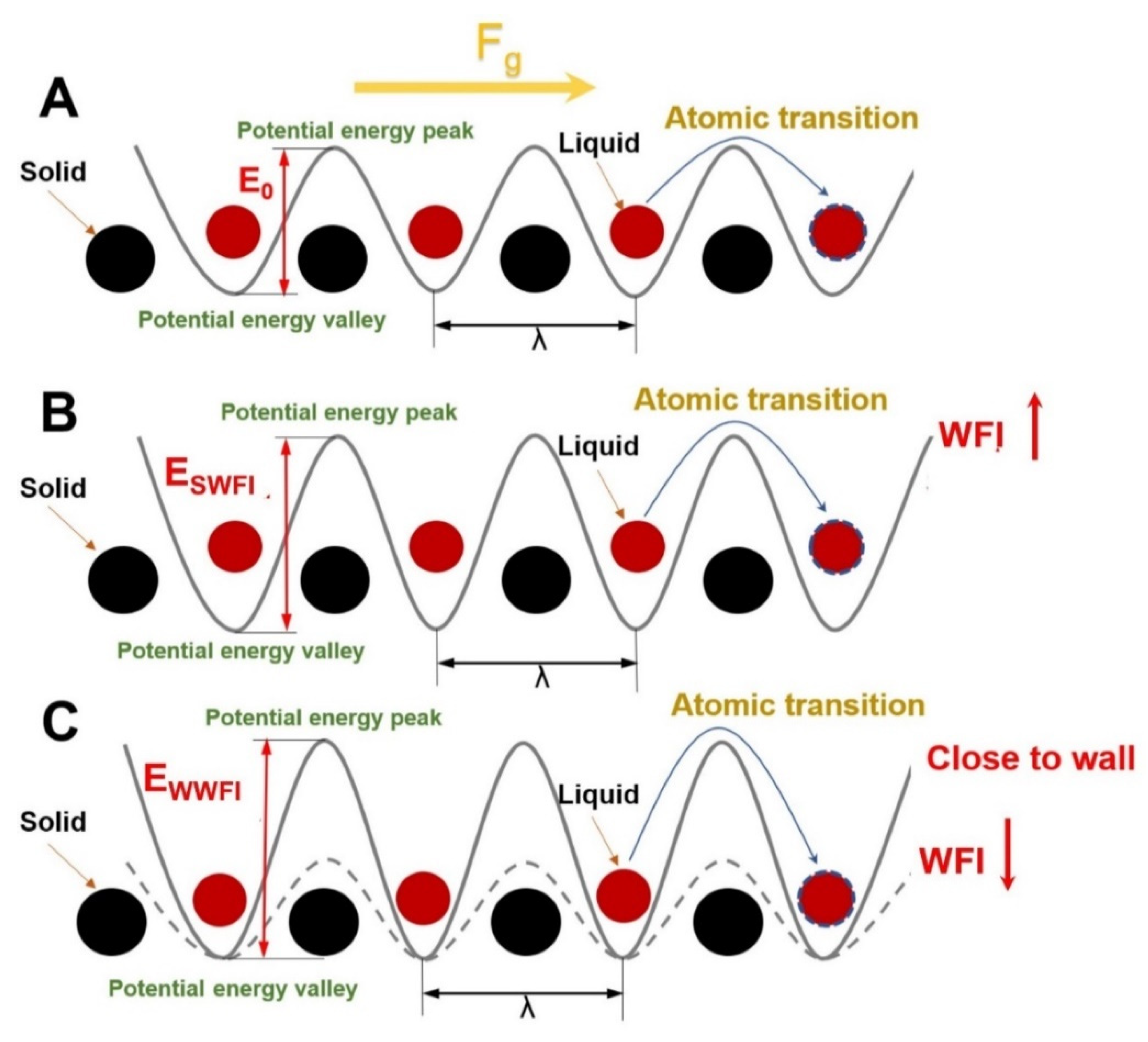
3.2. Influence of Wall–Fluid Interaction
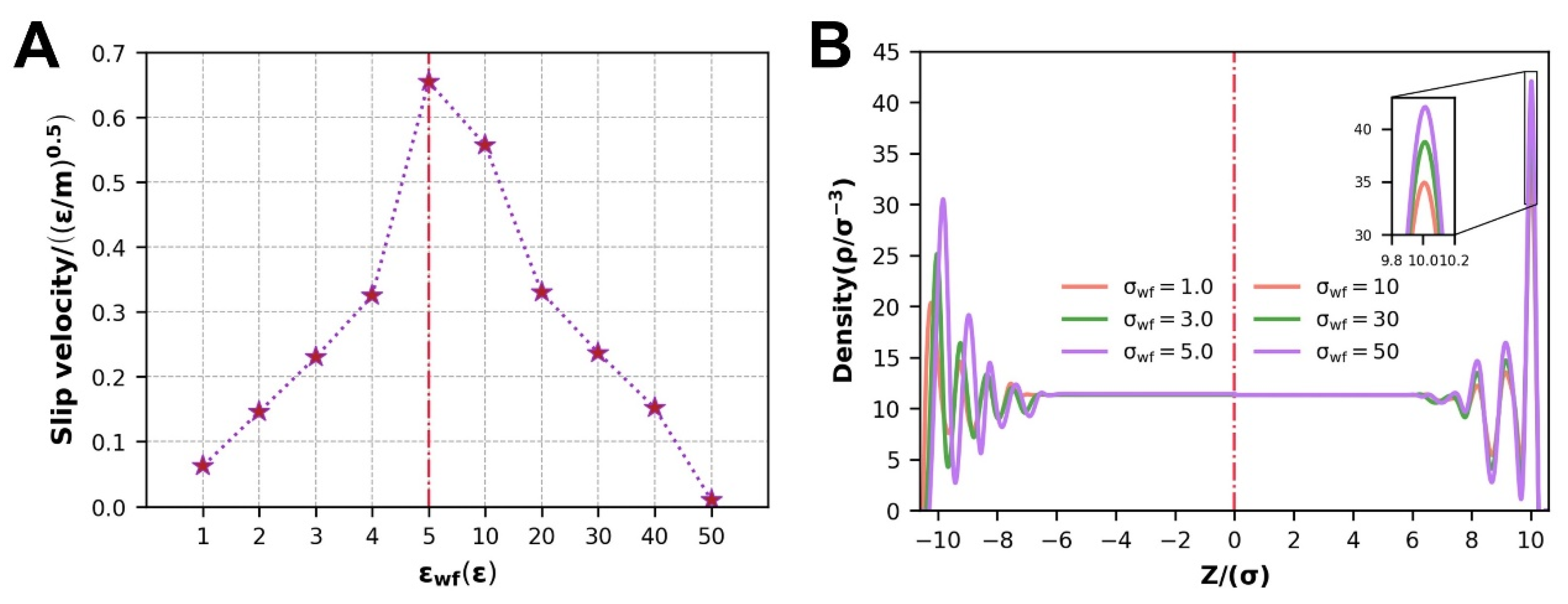
3.2.1. Slip Velocity and Density Distribution
3.2.2. Dynamic information of Flow Process
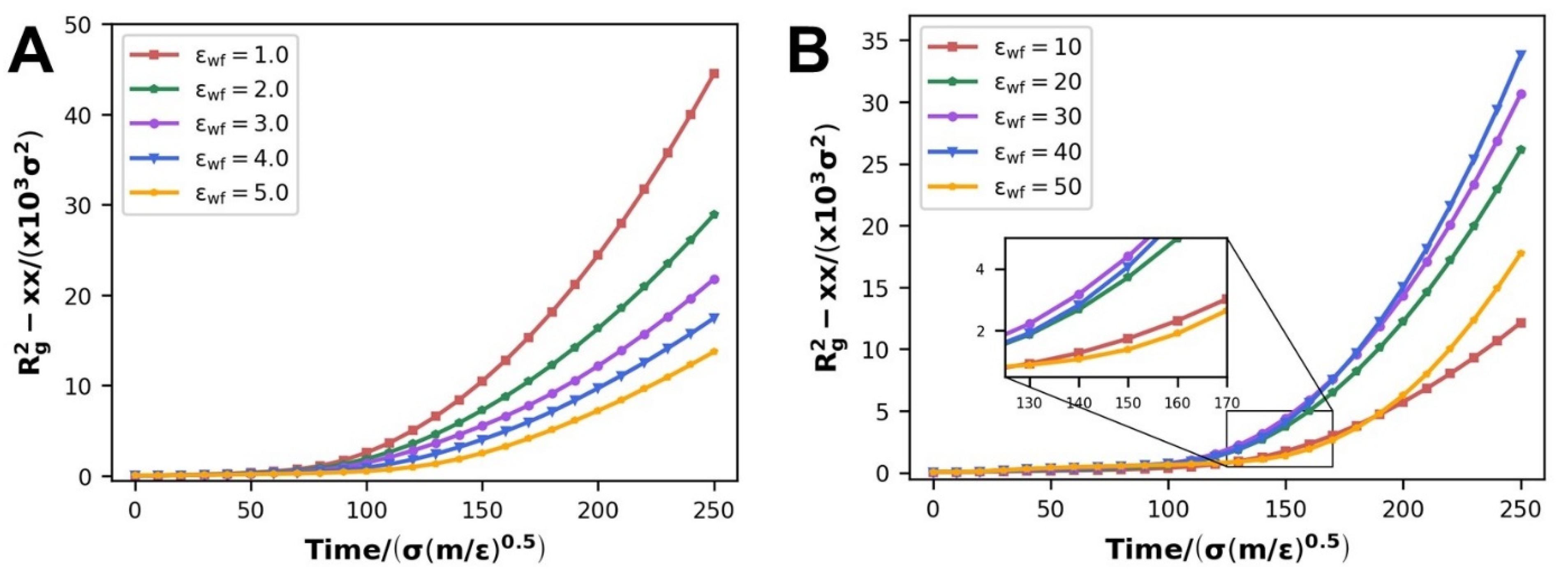
3.2.3. Evolution of Internal Molecular Chain Structure
4. Conclusions
- (1)
- In the flow process, the velocity distribution of the polymer melt represents its pseudoplasticity—that is, a plunger flow domain in the middle and two parabolic-like domains in the upper and lower side of the nanochannels rather than a parabolic flow—and the simulation findings match the high-density polyethylene flow experiments. The difference in the relative proportion of parabolic-like and plunger sections about velocity distribution is the result of the different thicknesses of the oriented thin layer;
- (2)
- Increasing the driving force improves the flow of the polymer melt while simultaneously reinforcing the amount of slip in the polymer melt, which makes it easier for the molecular chains to diffuse. The disentanglement of molecular chains and the development of an orientated state along the direction of driving pressures create this process, which lessens the barrier to flow caused by molecular chain mutual restriction;
- (3)
- At different scales of WFI, the slip velocity displays two entirely opposing states. At the scale of WWFI, the slip velocity decreases as the WFI strengthens. At the scale of WWFI, the contact density layer moves toward the wall when the WFI weakens, enhancing the atomic transition energy barrier and decaying the net atomic transition rate, resulting in a decreased slip velocity;
- (4)
- The specificity of slip velocity variation at different scales of WFI impacts not only molecular chain diffusion throughout the entire flow process but also causes certain changes in the polymer molecular chain conformation. With the strengthening of WFI, the disentanglement, orientation, and radius of the gyration Rg2-xx parameters have been elevated at the scale of WWFI; on the contrary, they all decay at the scale of SWFI.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stormonth-Darling, J.M.; Pedersen, R.H.; How, C.; Gadegaard, N. Injection Moulding of Ultra High Aspect Ratio Nanostructures Using Coated Polymer Tooling. J. Micromech. Microeng. 2014, 24, 075019. [Google Scholar] [CrossRef]
- Piotter, V.; Bauer, W.; Benzler, T.; Emde, A. Injection Molding of Components for Microsystems. Microsyst. Technol. 2001, 7, 99–102. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Huang, C.-C.; Lin, S.-K.; Tao, J. Characteristics Analysis and Mold Design for Ultrasonic-Assisted Injection Molding. J. Polym. Eng. 2014, 34, 673–681. [Google Scholar] [CrossRef]
- Kim, D.S.; Lee, S.H.; Ahn, C.H.; Lee, J.Y.; Kwon, T.H. Disposable Integrated Microfluidic Biochip for Blood Typing by Plastic Microinjection Moulding. Lab Chip 2006, 6, 794. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Qiu, Z.; Ma, Z.; Yang, Y. Flow Properties of Polymer Melt in Longitudinal Ultrasonic-Assisted Microinjection Molding: Flow Properties of Polymer Melt in Longitudinal Ultrasonic-Assisted Microinjection Molding. Polym. Eng. Sci. 2017, 57, 797–805. [Google Scholar] [CrossRef]
- Shit, G.C.; Mondal, A.; Sinha, A.; Kundu, P.K. Effects of Slip Velocity on Rotating Electro-Osmotic Flow in a Slowly Varying Micro-Channel. Colloids Surf. Physicochem. Eng. Asp. 2016, 489, 249–255. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q.; Zhu, W.; Shen, C. Scale Effect on Filling Stage in Micro-Injection Molding for Thin Slit Cavities. Microsyst. Technol. 2012, 18, 2085–2091. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y. Slip Behavior of Liquid Flow in Rough Nanochannels. Chem. Eng. Process. Process Intensif. 2014, 85, 203–208. [Google Scholar] [CrossRef]
- Priezjev, N.V. Relationship between Induced Fluid Structure and Boundary Slip in Nanoscale Polymer Films. Phys. Rev. E. 2010, 82, 051603. [Google Scholar] [CrossRef] [Green Version]
- Niavarani, A.; Priezjev, N.V. Modeling the Combined Effect of Surface Roughness and Shear Rate on Slip Flow of Simple Fluids. Phys. Rev. E. 2010, 81, 011606. [Google Scholar] [CrossRef] [Green Version]
- Baig, C.; Mavrantzas, V.G.; Kröger, M. Flow Effects on Melt Structure and Entanglement Network of Linear Polymers: Results from a Nonequilibrium Molecular Dynamics Simulation Study of a Polyethylene Melt in Steady Shear. Macromolecules 2010, 43, 6886–6902. [Google Scholar] [CrossRef]
- Duan, C.; Zhou, F.; Jiang, K.; Yu, T. Molecular Dynamics Simulation of Planar Poiseuille Flow for Polymer Melts in Atomically Flat Nanoscale Channel. Int. J. Heat Mass Transf. 2015, 91, 1088–1100. [Google Scholar] [CrossRef]
- Nagayama, G.; Cheng, P. Effects of Interface Wettability on Microscale Flow by Molecular Dynamics Simulation. Int. J. Heat Mass Transf. 2004, 47, 501–513. [Google Scholar] [CrossRef]
- Gratton, Y.; Slater, G.W. Molecular Dynamics Study of Tethered Polymers in Shear Flow. Eur. Phys. J. E. 2005, 17, 455–465. [Google Scholar] [CrossRef]
- Zhang, J.; Hansen, J.S.; Todd, B.D. Structural and Dynamical Properties for Confined Polymers Undergoing Planar Poiseuille Flow. J Chem Phys. 2007, 126, 144907. [Google Scholar] [CrossRef]
- Münstedt, H.; Schmidt, M.; Wassner, E. Stick and Slip Phenomena during Extrusion of Polyethylene Melts as Investigated by Laser-Doppler Velocimetry. J. Rheol. 2000, 44, 413–427. [Google Scholar] [CrossRef]
- Robert, L.; Demay, Y.; Vergnes, B. Stick-Slip Flow of High Density Polyethylene in a Transparent Slit Die Investigated by Laser Doppler Velocimetry. Rheol. Acta 2004, 43, 89–98. [Google Scholar] [CrossRef]
- Kloziński, A.; Jakubowska, P. The Application of an Extrusion Slit Die in the Rheological Measurements of Polyethylene Composites With Calcium Carbonate Using an In-Line Rheometer. Polym. Eng. Sci. 2019, 59, E16–E24. [Google Scholar] [CrossRef]
- Webster, M.A.; Yeomans, J.M. Modeling a Tethered Polymer in Poiseuille Flow. J. Chem. Phys. 2005, 122, 164903. [Google Scholar] [CrossRef]
- Peter, C.; Kremer, K. Multiscale Simulation of Soft Matter Systems—from the Atomistic to the Coarse-Grained Level and Back. Soft Matter 2009, 5, 4357. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A Generic Force Field for Molecular Simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Hossain, D.; Tschopp, M.A.; Ward, D.K.; Bouvard, J.L.; Wang, P.; Horstemeyer, M.F. Molecular Dynamics Simulations of Deformation Mechanisms of Amorphous Polyethylene. Polymers 2010, 51, 6071–6083. [Google Scholar] [CrossRef]
- Bao, Q.; Yang, Z.; Lu, Z. Molecular Dynamics Simulation of Amorphous Polyethylene (PE) under Cyclic Tensile-Compressive Loading below the Glass Transition Temperature. Polymers 2020, 186, 121968. [Google Scholar] [CrossRef]
- Wu, J.; Nagao, S.; Zhang, Z.; He, J. Deformation and Fracture of Nano-Sized Metal-Coated Polymer Particles: A Molecular Dynamics Study. Eng. Fract. Mech. 2015, 150, 209–221. [Google Scholar] [CrossRef]
- Jewett, A.I.; Zhuang, Z.; Shea, J.-E. Moltemplate a Coarse-Grained Model Assembly Tool. Biophys. J. 2013, 104, 169a. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Bao, L.; Priezjev, N.V.; Luo, K. Identifying Two Regimes of Slip of Simple Fluids over Smooth Surfaces with Weak and Strong Wall-Fluid Interaction Energies. J. Chem. Phys. 2017, 146, 034701. [Google Scholar] [CrossRef] [PubMed]
- Grønbech-Jensen, N.; Farago, O. A Simple and Effective Verlet-Type Algorithm for Simulating Langevin Dynamics. Mol. Phys. 2013, 111, 983–991. [Google Scholar] [CrossRef]
- Lou, Y.; Wu, G.; Feng, Y. Wall Slip Behaviour of Polymers Based on Molecular Dynamics at the Micro/Nanoscale and Its Effect on Interface Thermal Resistance. Polymers 2020, 12, 2182. [Google Scholar] [CrossRef]
- Voronov, R.S.; Papavassiliou, D.V.; Lee, L.L. Boundary Slip and Wetting Properties of Interfaces: Correlation of the Contact Angle with the Slip Length. J. Chem. Phys. 2006, 124, 204701. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, A.P.; Theodorou, D.N. Atomistic Simulations of Long-Chain Polyethylene Melts Flowing Past Gold Surfaces: Structure and Wall-Slip. Mol. Phys. 2020, 118, e1706775. [Google Scholar] [CrossRef]
- Kasiteropoulou, D.; Karakasidis, T.E.; Liakopoulos, A. Dissipative Particle Dynamics Investigation of Parameters Affecting Planar Nanochannel Flows. Mater. Sci. Eng. B. 2011, 176, 1574–1579. [Google Scholar] [CrossRef]
- Sendner, C.; Horinek, D.; Bocquet, L.; Netz, R.R. Interfacial Water at Hydrophobic and Hydrophilic Surfaces: Slip, Viscosity, and Diffusion. Langmuir 2009, 25, 10768–10781. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Cho, S.; Kim, J.M.; Baig, C. Interfacial Molecular Structure and Dynamics of Confined Ring Polymer Melts under Shear Flow. Macromolecules 2018, 51, 4670–4677. [Google Scholar] [CrossRef]
- Yashiro, K.; Ito, T.; Tomita, Y. Molecular Dynamics Simulation of Deformation Behavior in Amorphous Polymer: Nucleation of Chain Entanglements and Network Structure under Uniaxial Tension. Int. J. Mech. Sci. 2003, 45, 1863–1876. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Cho, S.; Kim, J.M.; Baig, C. Molecular Mechanisms of Interfacial Slip for Polymer Melts under Shear Flow. J. Rheol. 2017, 61, 253–264. [Google Scholar] [CrossRef]
- Lee, S.; Rutledge, G.C. Plastic Deformation of Semicrystalline Polyethylene by Molecular Simulation. Macromolecules 2011, 44, 3096–3108. [Google Scholar] [CrossRef]
- Yeh, I.-C.; Andzelm, J.W.; Rutledge, G.C. Mechanical and Structural Characterization of Semicrystalline Polyethylene under Tensile Deformation by Molecular Dynamics Simulations. Macromolecules 2015, 48, 4228–4239. [Google Scholar] [CrossRef] [Green Version]
- Zhan, S.; Xu, H.; Duan, H.; Pan, L.; Jia, D.; Tu, J.; Liu, L.; Li, J. Molecular Dynamics Simulation of Microscopic Friction Mechanisms of Amorphous Polyethylene. Soft Matter 2019, 15, 8827–8839. [Google Scholar] [CrossRef]
- Huang, D.M.; Sendner, C.; Horinek, D.; Netz, R.R.; Bocquet, L. Water Slippage versus Contact Angle: A Quasiuniversal Relationship. Phys. Rev. Lett. 2008, 101, 226101. [Google Scholar] [CrossRef] [Green Version]
- Yong, X.; Zhang, L.T. Investigating Liquid-Solid Interfacial Phenomena in a Couette Flow at Nanoscale. Phys. Rev. E. 2010, 82, 056313. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh Pahlavan, A.; Freund, J.B. Effect of Solid Properties on Slip at a Fluid-Solid Interface. Phys. Rev. E. 2011, 83, 021602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.-C.; Zhao, Y.-P. Slip Boundary Conditions Based on Molecular Kinetic Theory: The Critical Shear Stress and the Energy Dissipation at the Liquid–Solid Interface. Soft Matter 2011, 7, 8628. [Google Scholar] [CrossRef] [Green Version]
- Lichter, S.; Martini, A.; Snurr, R.Q.; Wang, Q. Liquid Slip in Nanoscale Channels as a Rate Process. Phys. Rev. Lett. 2007, 98, 226001. [Google Scholar] [CrossRef] [PubMed]
- Yang, F. Slip boundary condition for viscous flow over solid surfaces. Chem. Eng. Commun. 2009, 197, 544–550. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kröger, M.; Clarke, N. Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review. Polymers 2019, 11, 876. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | Type | Parameters |
|---|---|---|
| Bond length | X-CH2 | |
| Bond angle | X-CH2-CH2 | |
| Dihedral angle | X-CH2-CH2-CH2 | C0) |
| Non-bonded | CH2 | |
| Ni |
| Physical Quantity | Normalized Unit | International Unit |
|---|---|---|
| Mass | m | 14 g/mol |
| Distance | ||
| Energy | ||
| Time | ||
| Temperature | 56.52 K | |
| Velocity | ||
| Driving force |
| Fg | n | |||
|---|---|---|---|---|
| 4 | 1.676 | 0.46 | 2.136 | 0.15 |
| 6 | 1.605 | 0.69 | 2.295 | 0.17 |
| 8 | 1.526 | 0.84 | 2.366 | 0.18 |
| 10 | 1.533 | 0.88 | 2.413 | 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Duan, F.; Zhao, B.; Qiang, Y.; Zhou, M.; Jiang, B. Molecular Insights into the Wall Slip Behavior of Pseudoplastic Polymer Melt in Nanochannels during Micro Injection Molding. Polymers 2022, 14, 3218. https://doi.org/10.3390/polym14153218
Wu W, Duan F, Zhao B, Qiang Y, Zhou M, Jiang B. Molecular Insights into the Wall Slip Behavior of Pseudoplastic Polymer Melt in Nanochannels during Micro Injection Molding. Polymers. 2022; 14(15):3218. https://doi.org/10.3390/polym14153218
Chicago/Turabian StyleWu, Wangqing, Fengnan Duan, Baishun Zhao, Yuanbao Qiang, Mingyong Zhou, and Bingyan Jiang. 2022. "Molecular Insights into the Wall Slip Behavior of Pseudoplastic Polymer Melt in Nanochannels during Micro Injection Molding" Polymers 14, no. 15: 3218. https://doi.org/10.3390/polym14153218
APA StyleWu, W., Duan, F., Zhao, B., Qiang, Y., Zhou, M., & Jiang, B. (2022). Molecular Insights into the Wall Slip Behavior of Pseudoplastic Polymer Melt in Nanochannels during Micro Injection Molding. Polymers, 14(15), 3218. https://doi.org/10.3390/polym14153218