1. Introduction
In situ fibrillated composites can be used to overcome many the challenges of long-aspect ratio reinforcements [
1]. With many long-aspect ratio fibers, the high shear needed for dispersing and distributing the fibers can have detrimental effects upon the reinforcing fibers themselves. Fiber breakage will often result in short fibers and lower mechanical performance when compared to properly compounded long-aspect fibers. Using fibrillated reinforcements, several of these drawbacks can be circumvented. By distributing and dispersing an immiscible domain before fibrillation, long-aspect ratio fibrils are not broken as they are not yet formed [
2]. The production of in situ fibrillated materials have been studied using several methods, however, their applications in subsequent processes have rarely been studied.
In situ fibrillated reinforced composites have garnered a great deal of attention ever since their advent [
3,
4,
5]. In situ fibrillated composites refer to a class of composites where the secondary, reinforcing phase of the fibril shape is created within the primary matrix [
6]. By combining two immiscible polymers together and applying high extensional stresses, high strength composites can be achieved. Primarily uniaxial extensional stresses have been used, but shear and planar extensional stresses have also been used [
7]. New and innovative polymeric composites that are stronger, lighter, and more functional are always in demand in both engineered and consumer goods. The ability to produce parts with lower costs will increase the profits for the manufacturer. Keeping the life cycle and final disposal of the polymer composites as design considerations is important to combat the waste and environmental impacts [
8,
9].
In this work, the term fiber refers to the shape of the primary PP matrix after spunbonding, is typically a few microns in diameter, and is a long continuous strand [
10]. The term fibril refers to the secondary micro- or nano-sized fibrillated PET found within the fiber (primary matrix).
As previously mentioned, there are several distinct advantages over traditional methods of fiber reinforcement in the areas of dispersion, distribution, and fiber breakage. While compounding traditional fillers (e.g., nanoclay, glass fibers, CNTs, etc.), high shear is often used to ensure that the filler is distributed thoroughly throughout the matrix by intensive mixing [
11,
12]. Moreover, the high shear compounding is also used to breakdown large filler agglomerations to increase dispersion [
13]. However, due to the high shear used, it will induce the severe breakage of fiber fillers due to mechanical degradation [
14]. Unfortunately, fiber breakage is detrimental to the mechanical properties as the reinforcement performance is lowered. By fibrillating the fibers in situ within the matrix itself, these drawbacks can be overcome. Because the fibrillation stage is decoupled from the compounding stage, the micro/nano-fibrils are not yet formed, and thus, there is no risk of breakage occurring [
5].
Typically, creating an in situ fibrillated composite consists of three stages: (1) compounding, (2) drawing, and (3) isotropization or end-use application [
15]. The matrix and reinforcement are blended in a twin-screw compounder (TSE) and a sea-island morphology is created when their respective proportions are suitable [
16]. Adding excessive amounts of the reinforcement can often result in a co-continuous blend. To create the smallest fibrillated fibrils, the sea-island morphology should have domains (islands) that are small and uniformly dispersed [
16,
17]. Having a proper compounding stage lays the foundation for better results downstream in the fibrillation process. With in situ fibrillation, the materials are selected so that they have differing melting temperatures (T
m), with the reinforcement melting temperature (T
m,Rei) 40 °C above the matrix melting temperature (T
m,Mat). This temperature difference allows for the reinforcement polymer to be melted while compounding, but rigid during the drawing and isotropization stages.
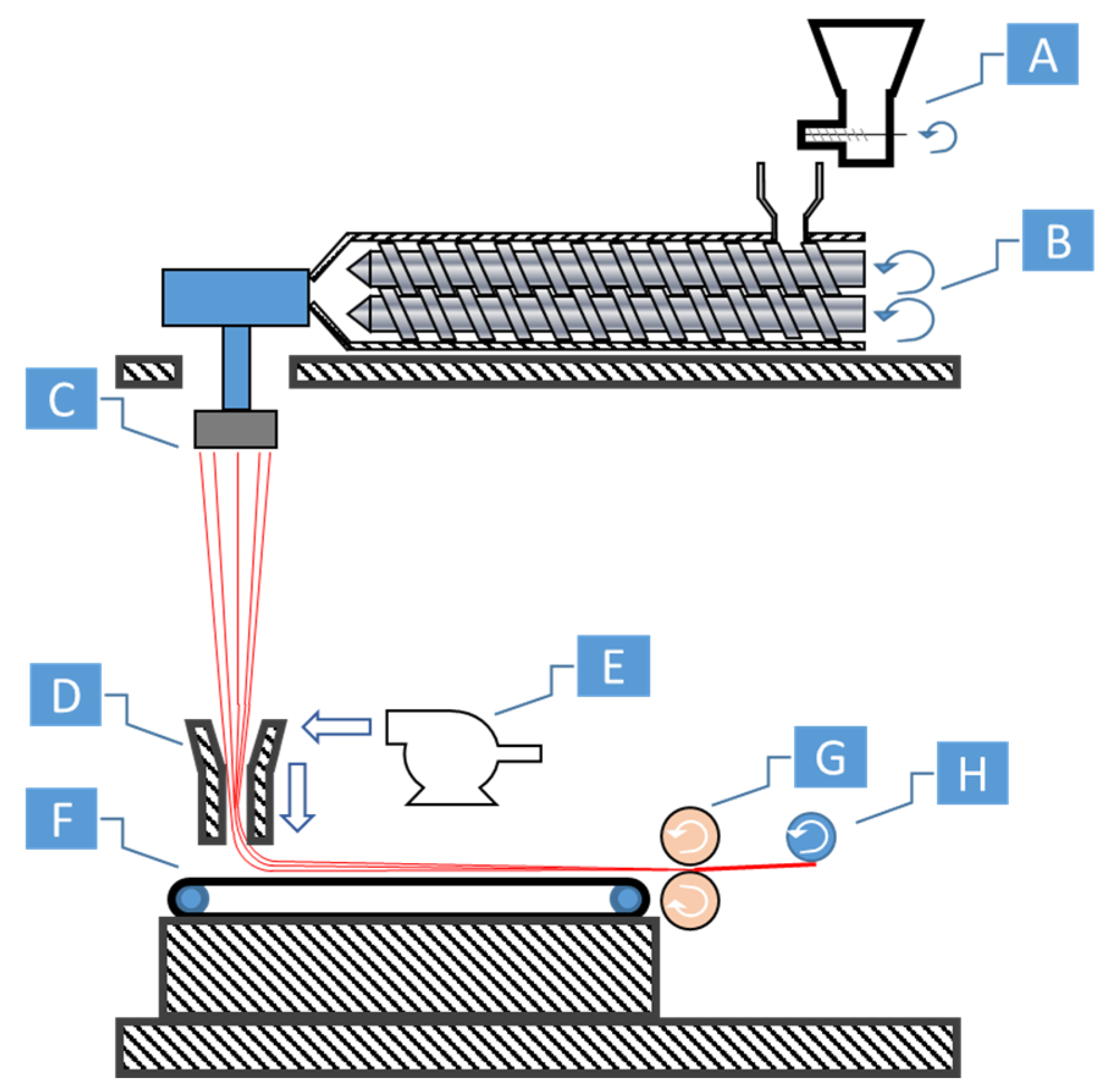
To fibrillate the reinforcement, the sea-island blend undergoes a strong bulk deformation, for which typically uniaxial extensional stresses are used. Uniaxial stress is among the easiest, simplest, and most effective methods to transform the blends into fibers. While experiencing the extensional stress, the matrix becomes deformed and elongated; within the matrix, the stresses are also transmitted to the domains. As time passes, the domains become deformed in the stress direction and small tendrils appear from the domains. With sufficient time, the domains are fibrillated and can coalesce together with their neighbors to create longer fibrils. Melt spinning [
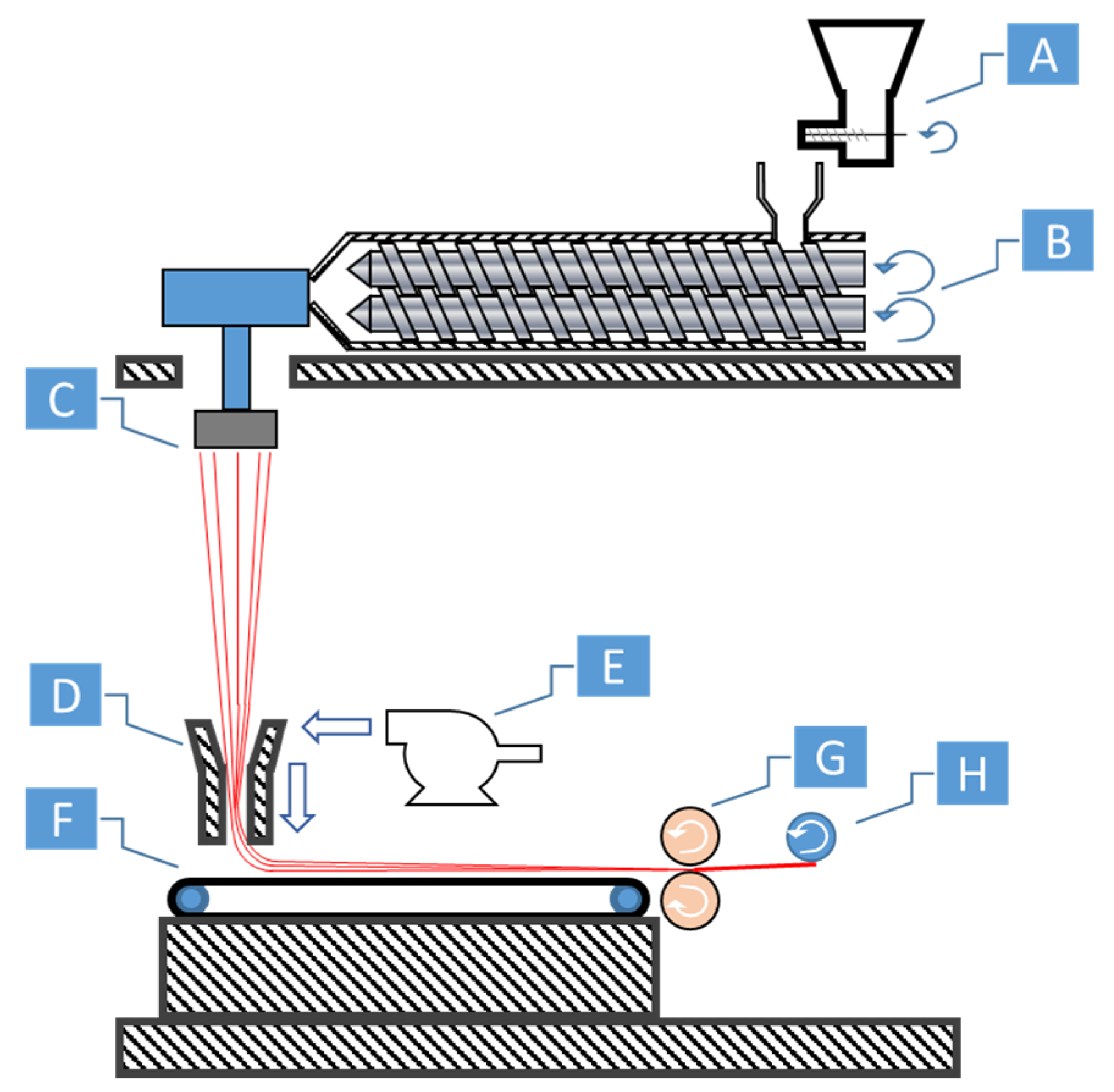
17], spunbonding [
18], melt blowing [
19], and extrusion drawing [
20,
21] are common processes for fibrillation. Extrusion drawing is commonly used among research settings as the additional infrastructure or machine requirements are lower. Melt spinning, spunbonding, and melt blowing are typical processes used for creating different forms of yarns or non-woven textile fabrics. Each process integrates uniaxial extensional forces in their processes, which makes them ideal candidates for creating in situ fibrillated fibers.
Isotropization is one of the last stages of fibrillated composite creation; it serves to randomly orient fibrils and decrease anisotropy. During the process, the temperature is lower than the T
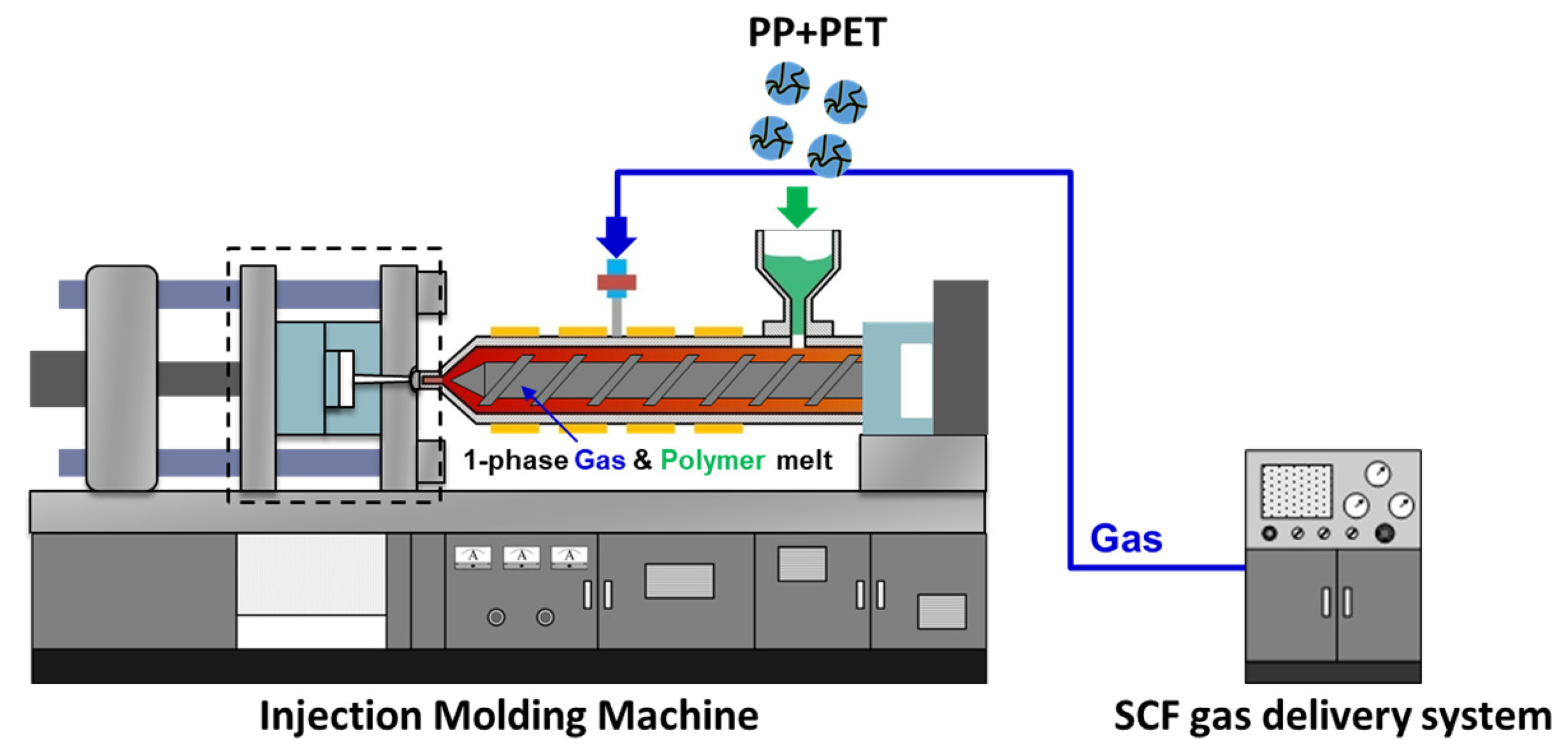
m,Rei to avoid melting or otherwise damaging the fibrils. In laboratory/research settings, hot compression molding is often used to create test specimens. However, to apply this type of material to a more industrial environment, the materials need to be reconfigured into pellets for continuous processes such as extrusion [
2] and injection molding [
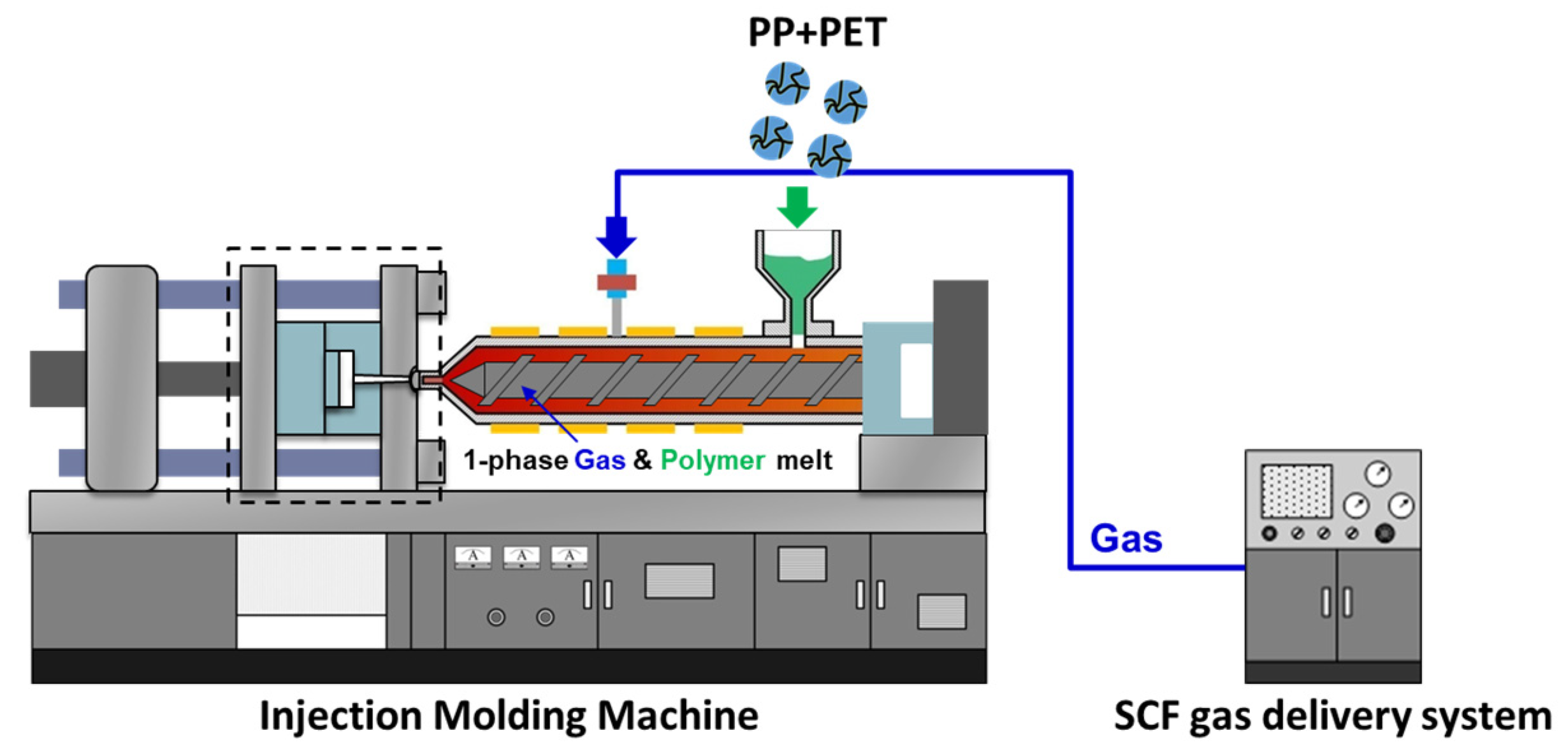
22]. Some reinforced fibers can also be used as-is, skipping a reprocessing stage before isotropization. The textile materials produced via filaments from melt spinning, or via nonwoven fabrics from melt blowing and spunbonding can potentially be directly fed into a machine. After spunbonding, the fibers are collected and roll calandered together to create non-woven materials. The calandered materials have sufficient tear strength to be collected as a spool on a core. This spool can be used to feed the injection molding machine, avoiding the need to have to pelletize the material.
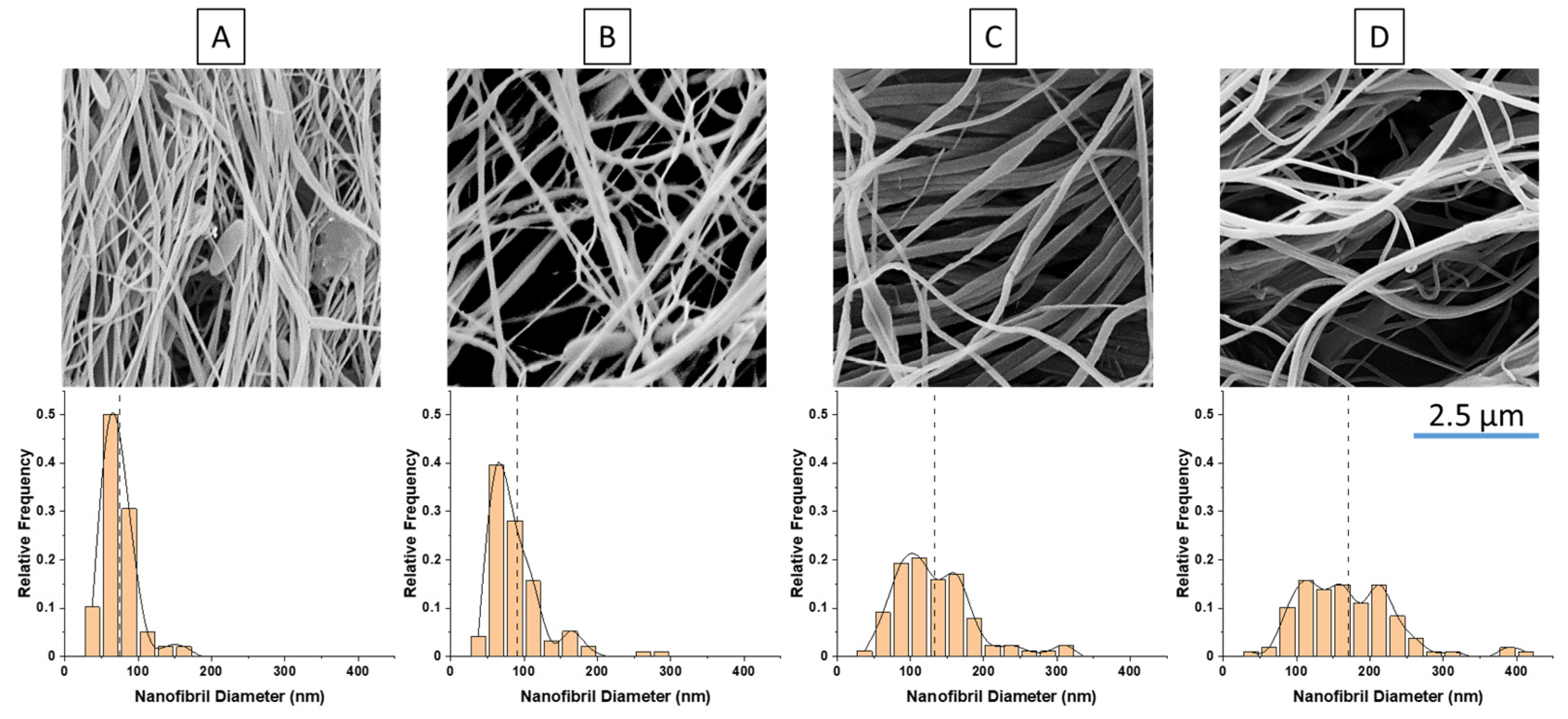
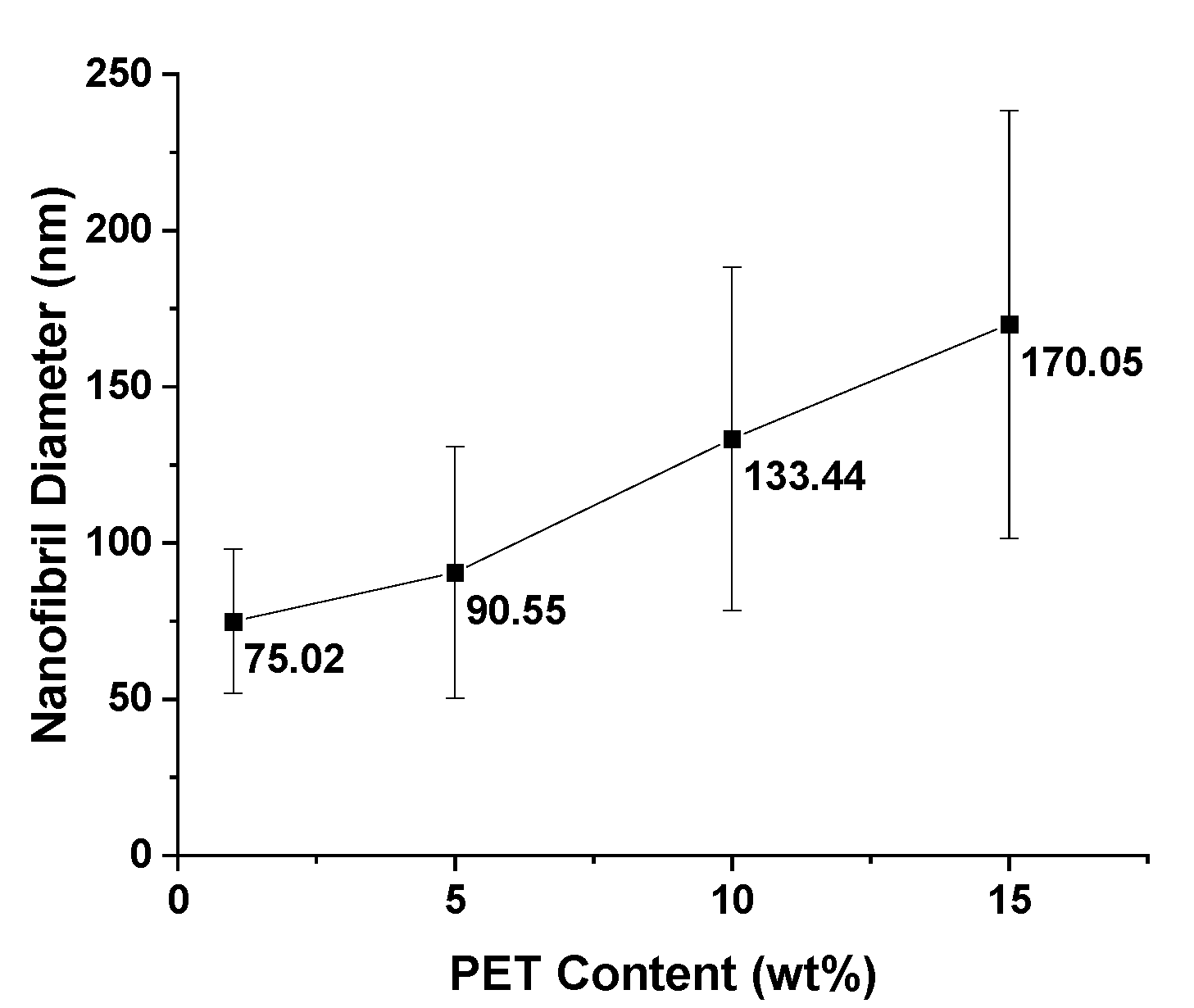
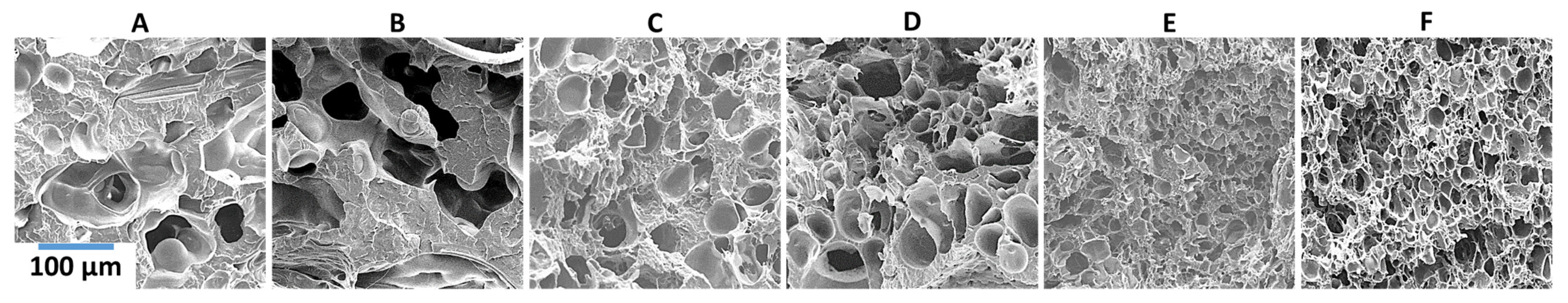
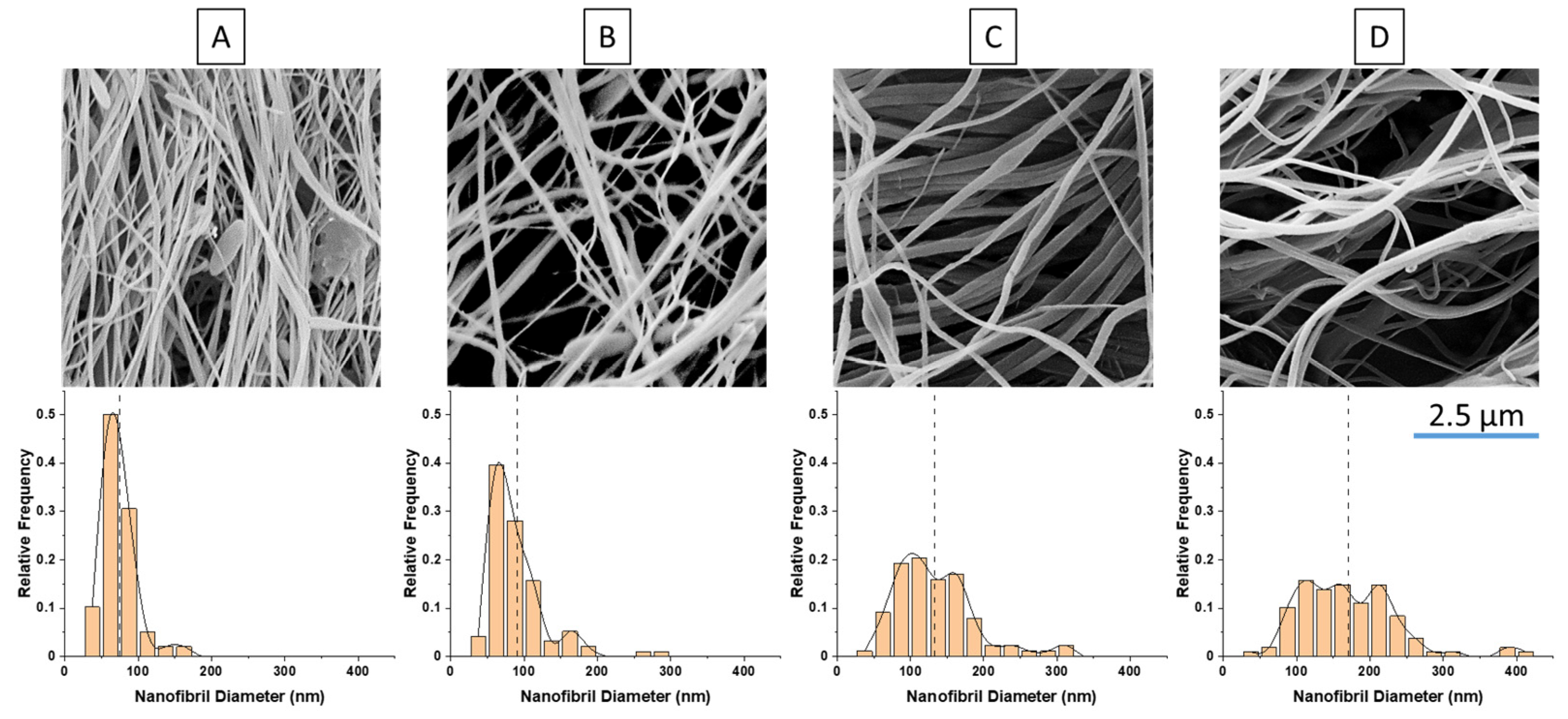
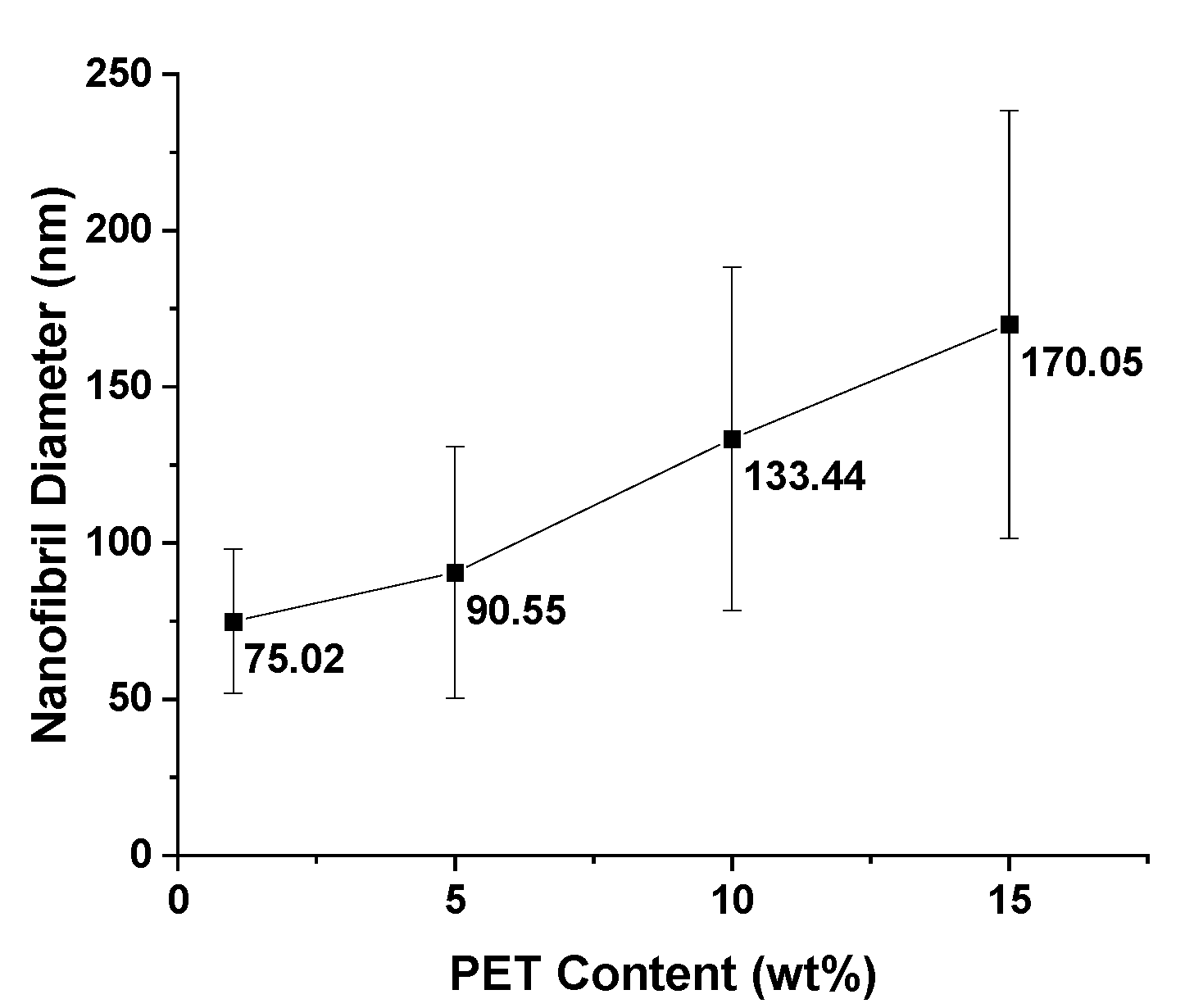
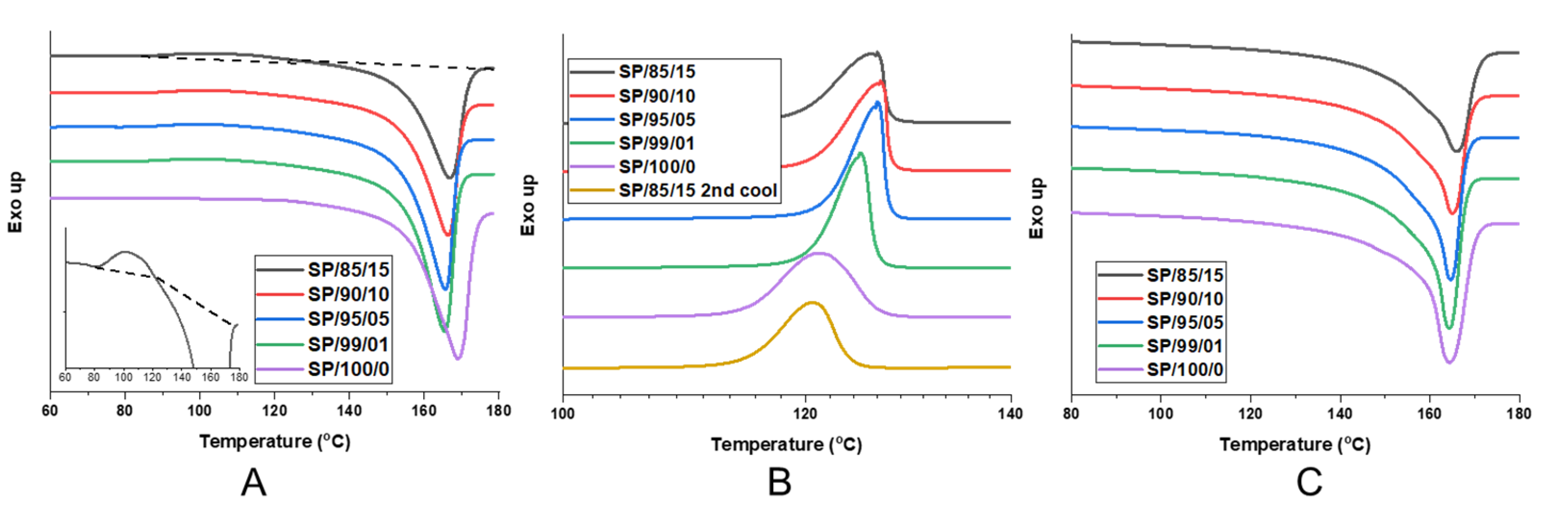
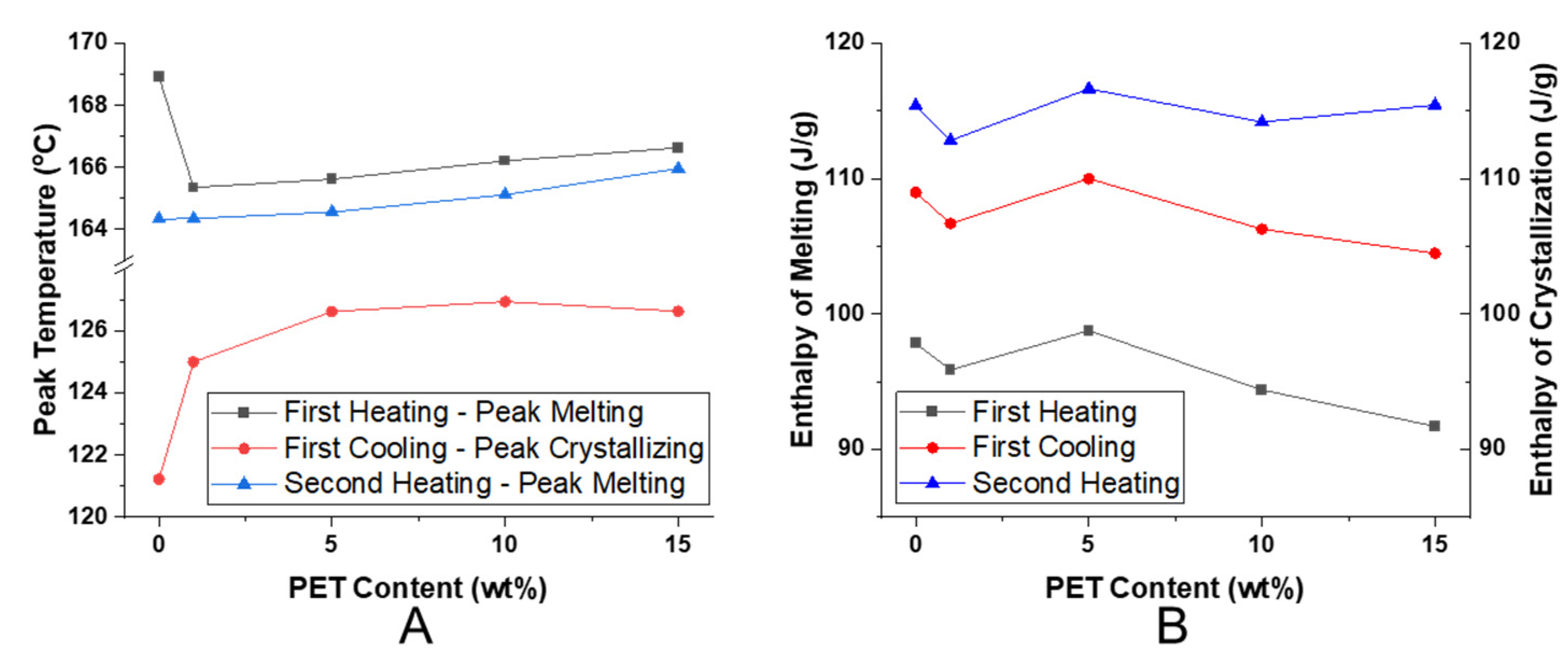
As previously mentioned, for nanofibril composites, the two polymeric components were selected to have distinct and separate melting temperatures. Being able to later reprocess the composite while still maintaining the nanofibril morphology is vital to ensuring high reinforcement properties. Earlier internal testing showed that during isotropization, a high processing temperature or high processing rates can degrade the fibrils. As the fibrils are heated during subsequent processing, they are subjected to excessive heat and stress, which can allow them to soften, relax, and distort. When degraded, the fibrils can shrink back from their elongated morphology, resulting in a low-aspect ratio, larger diameter domains that can be fibrillar or ellipsoidal.
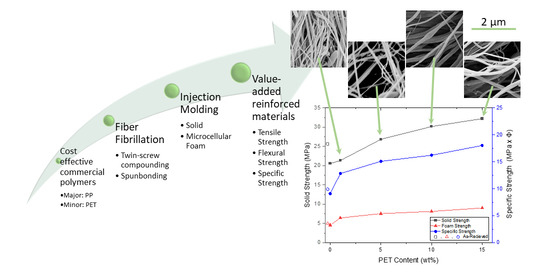
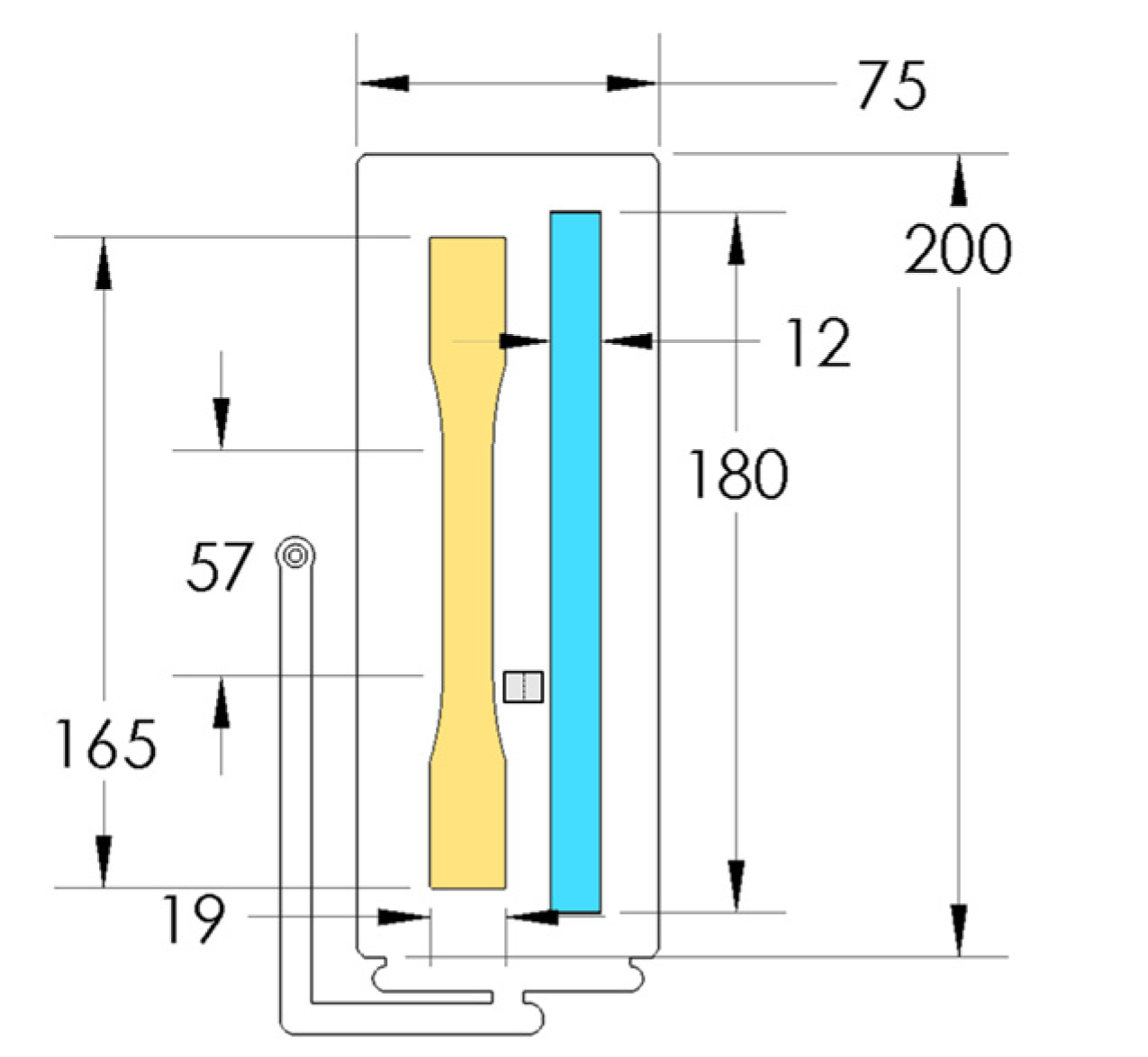
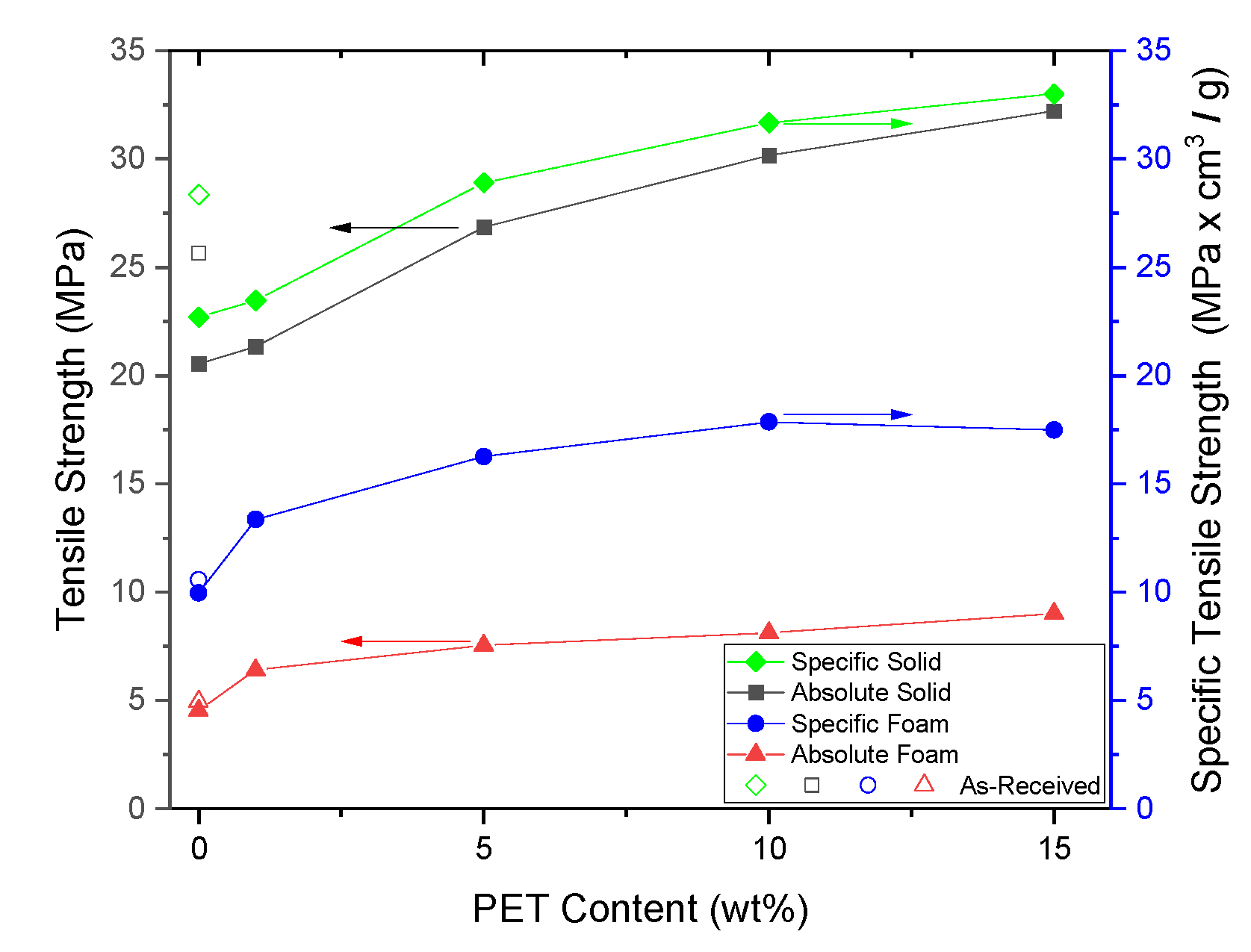
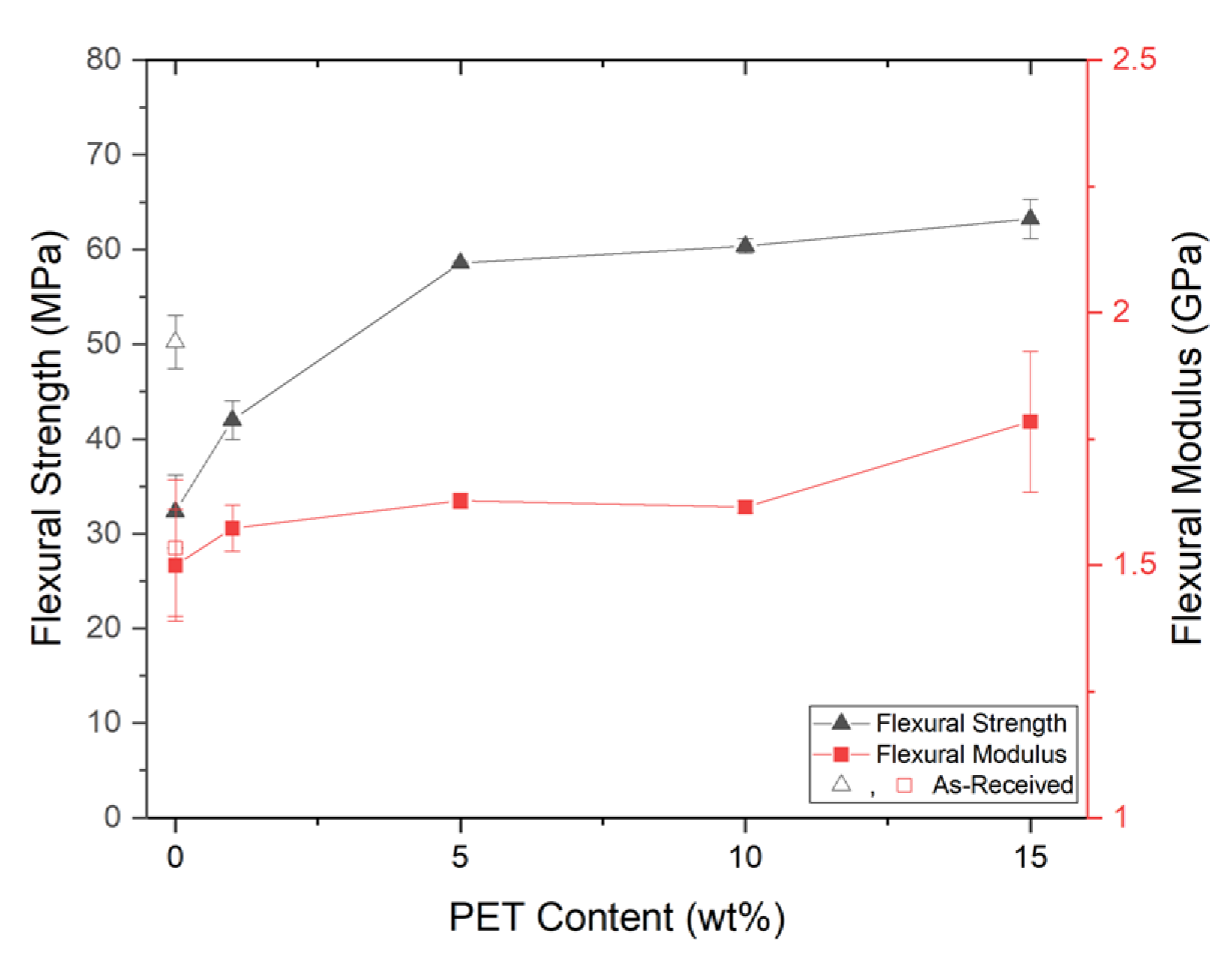
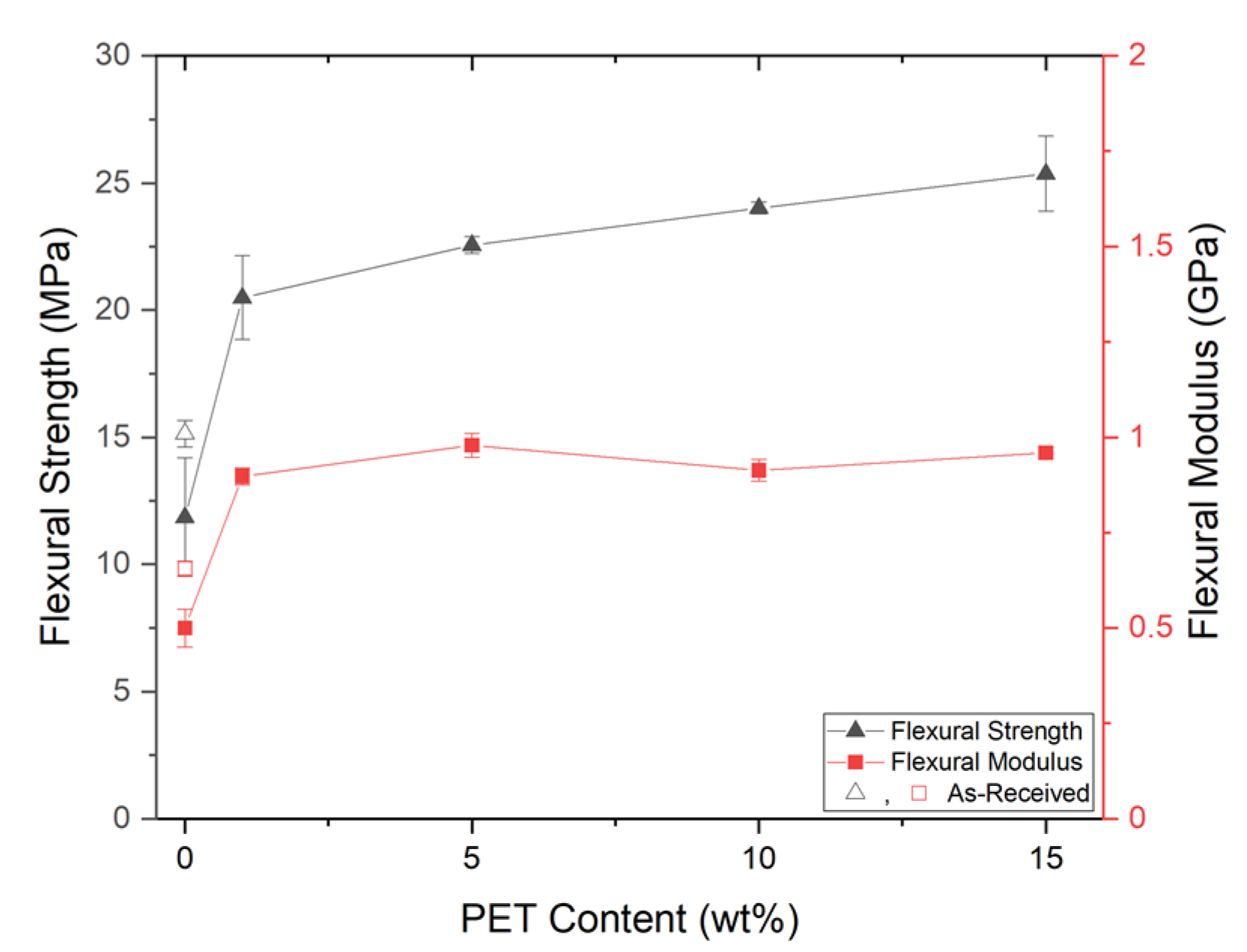
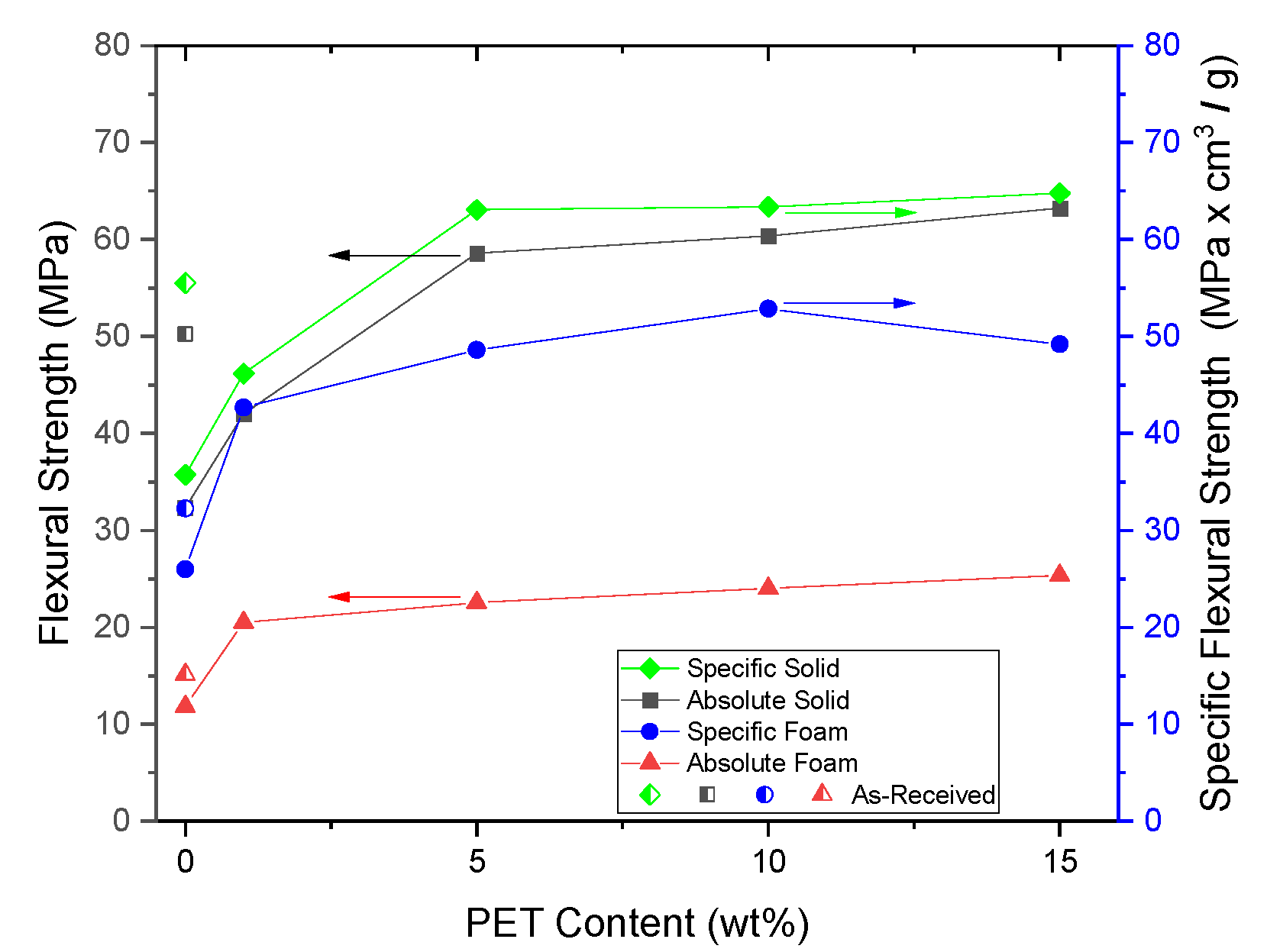
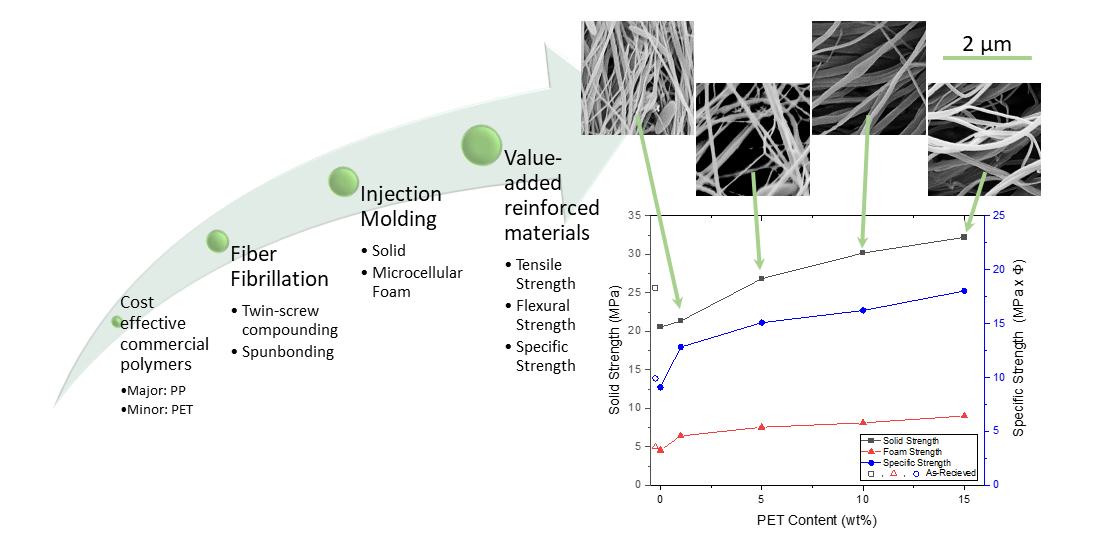
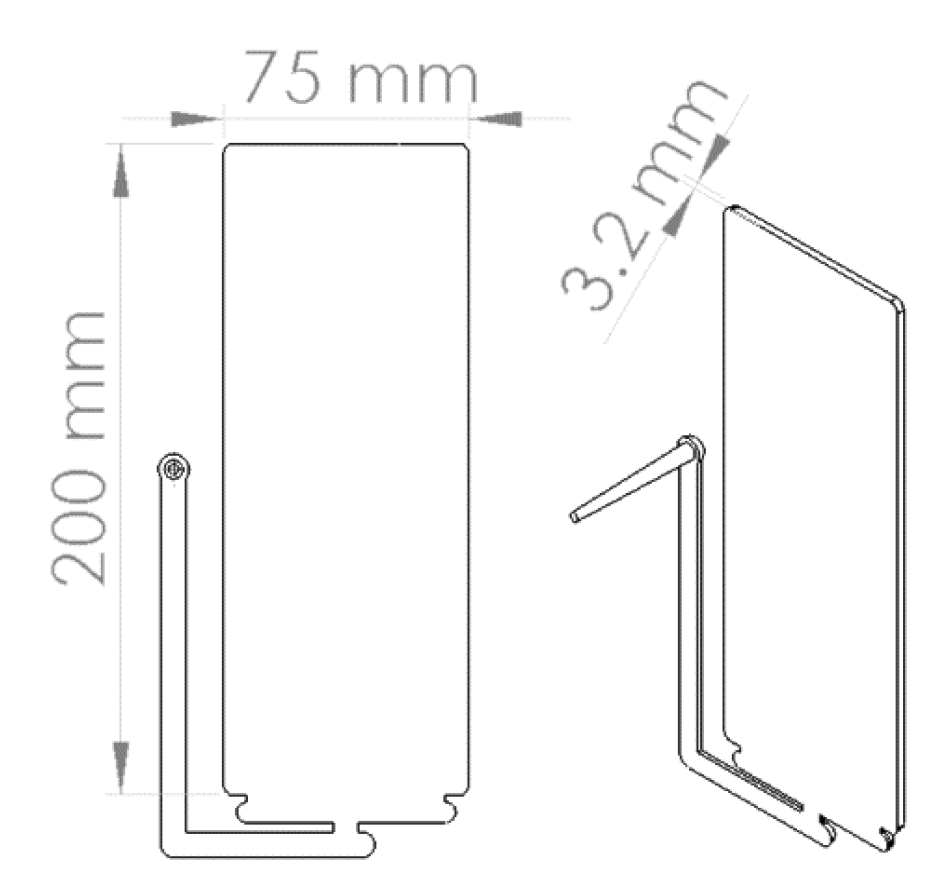
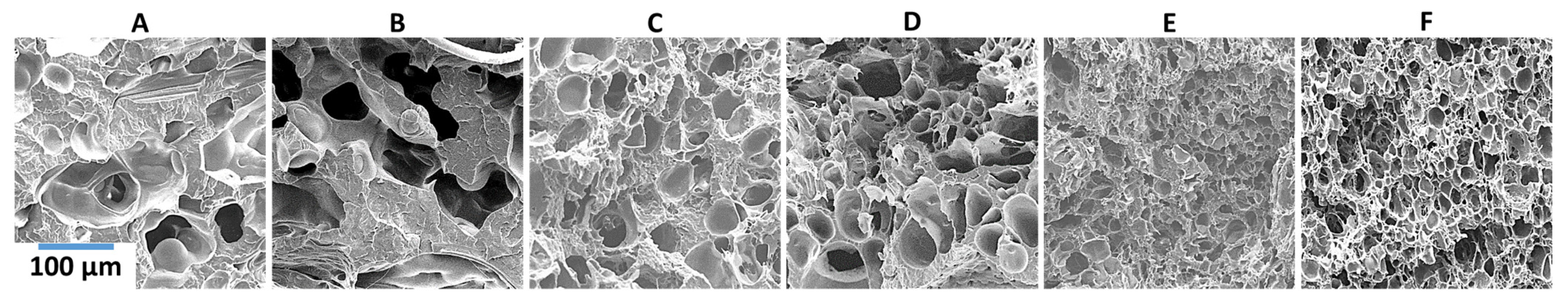
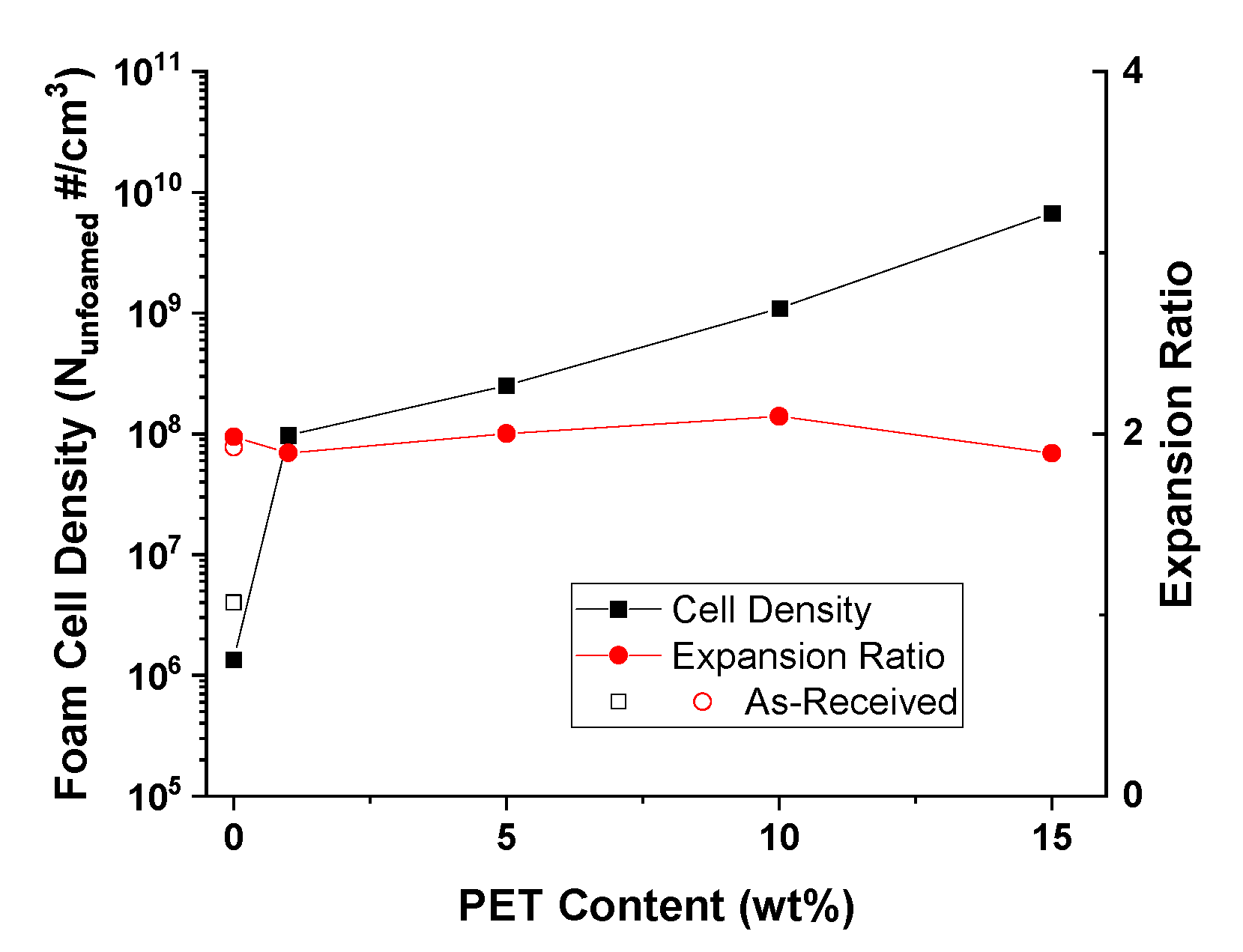
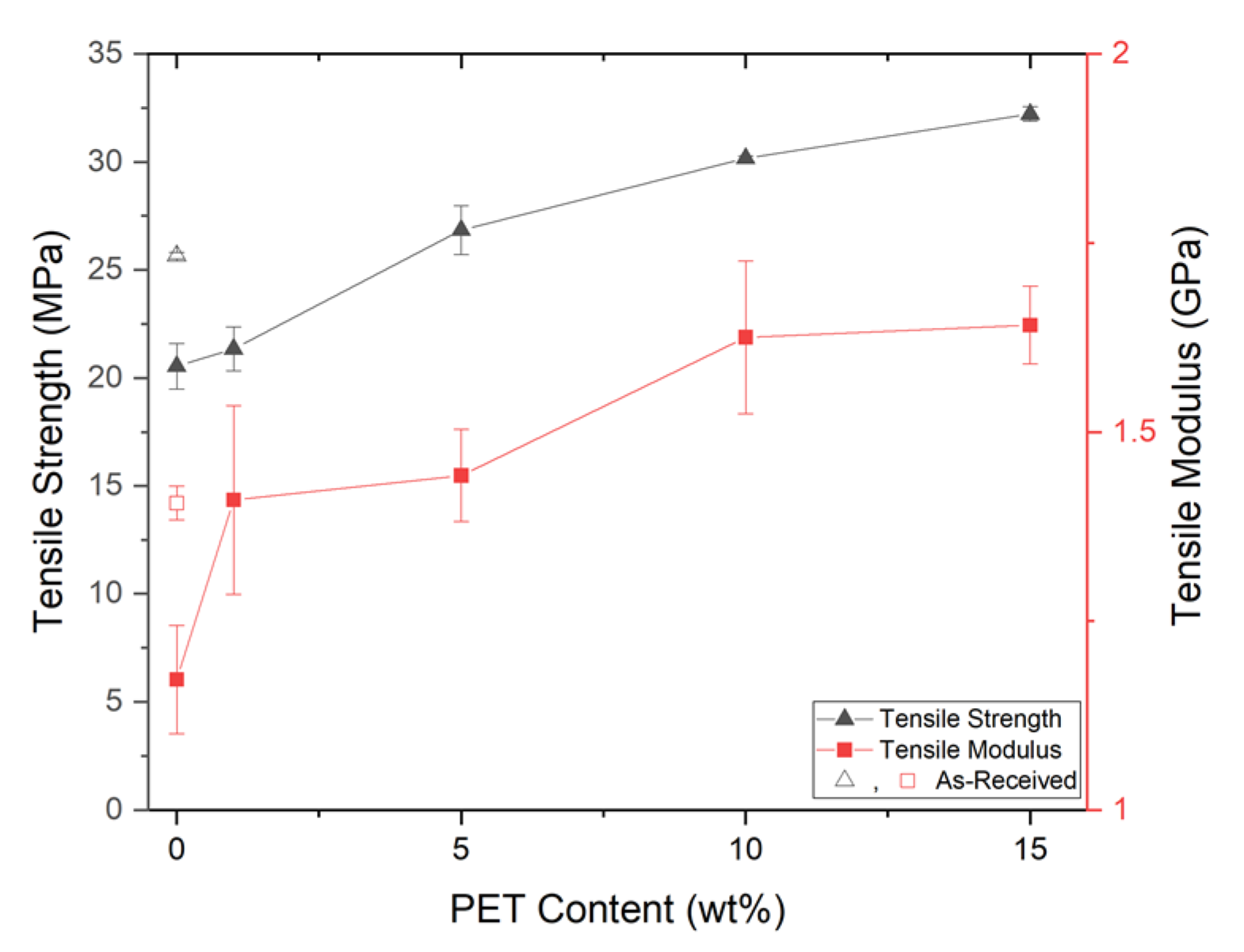
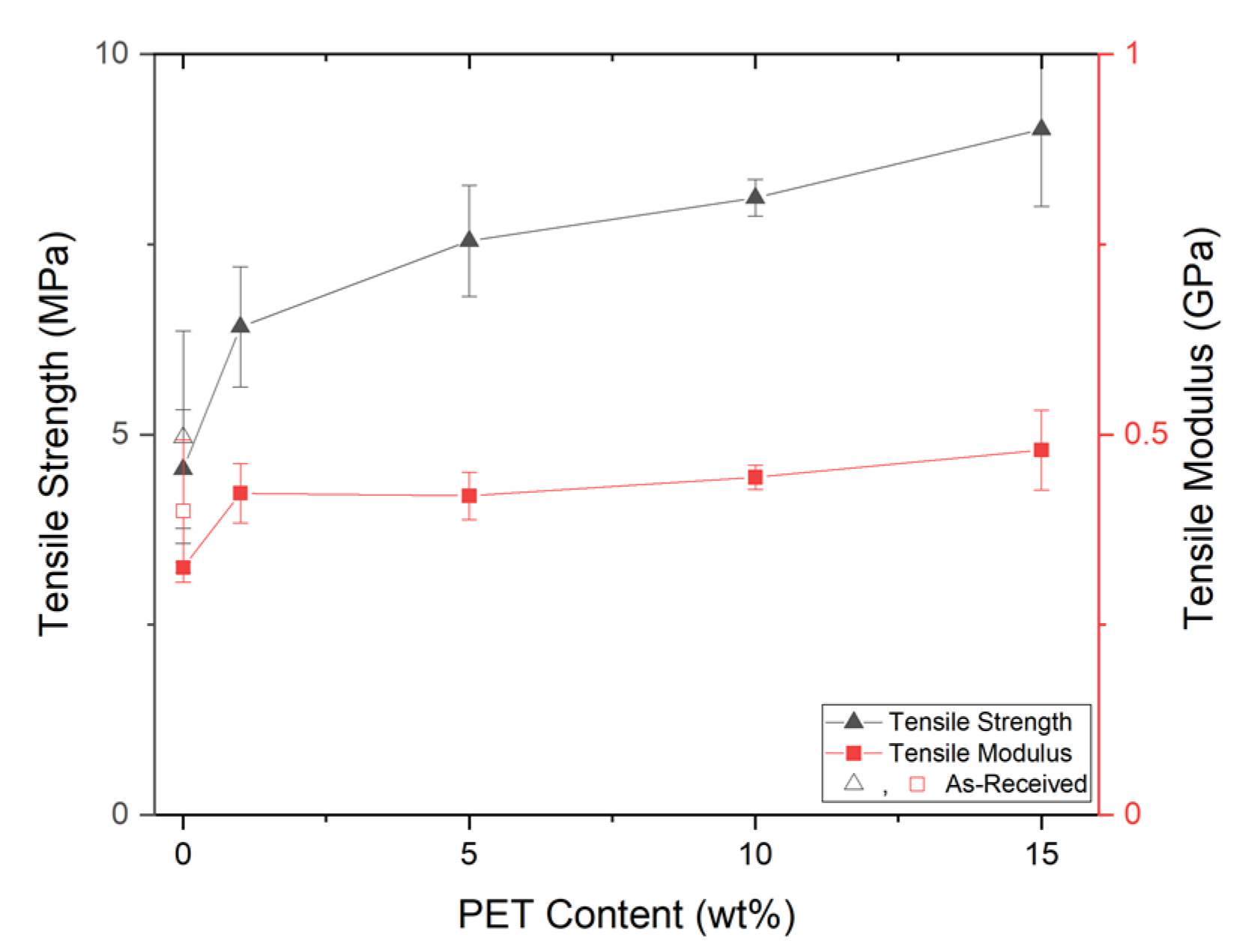
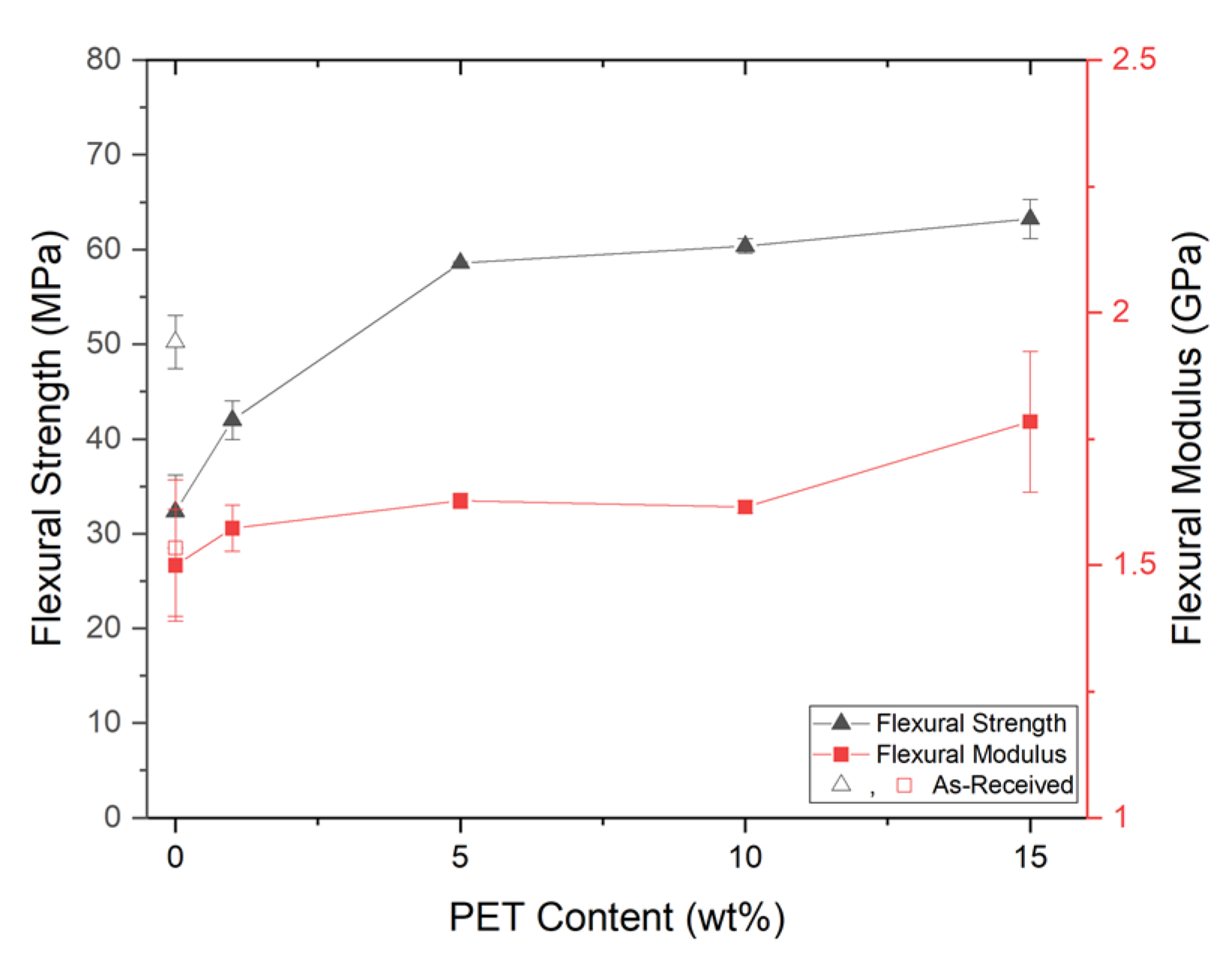
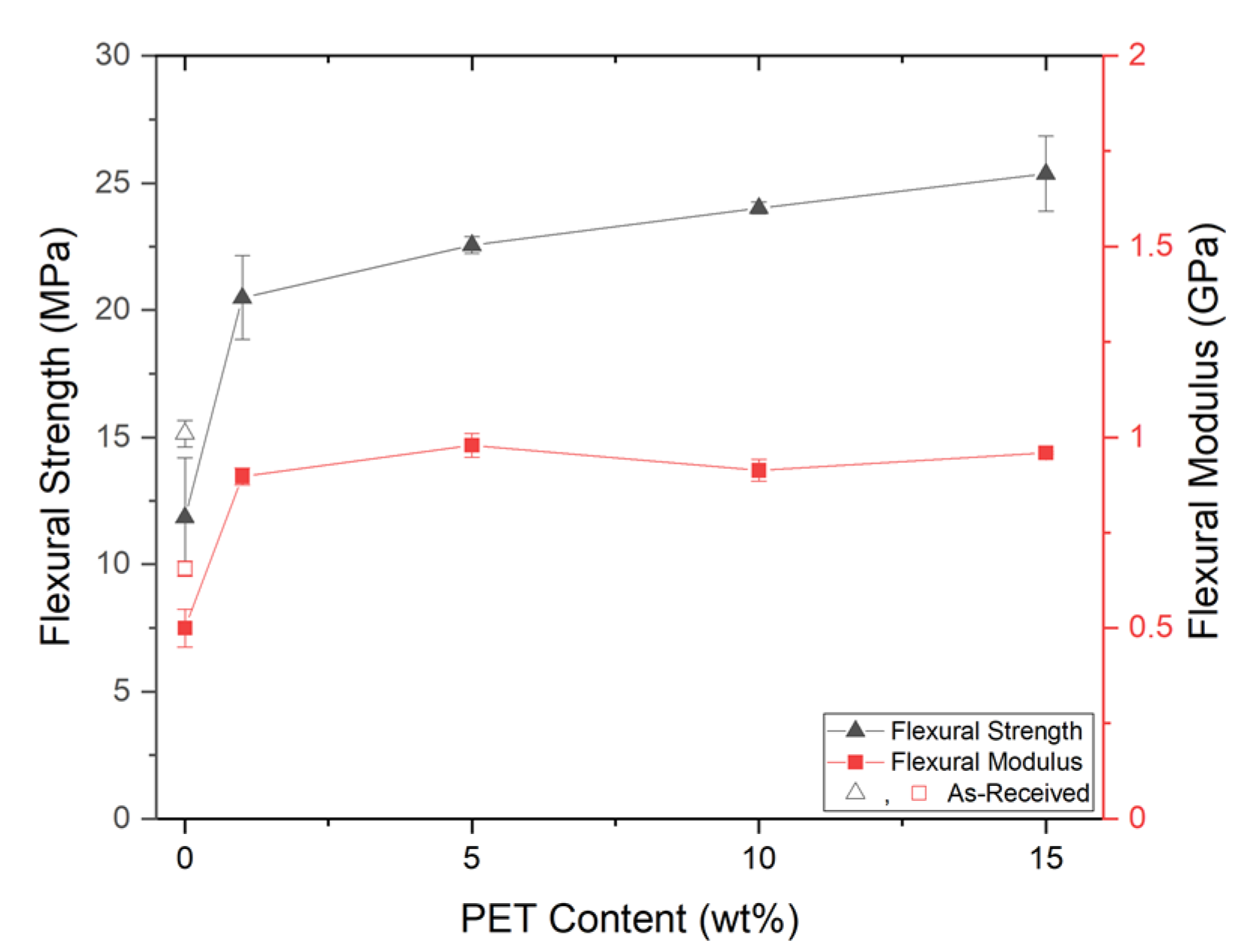
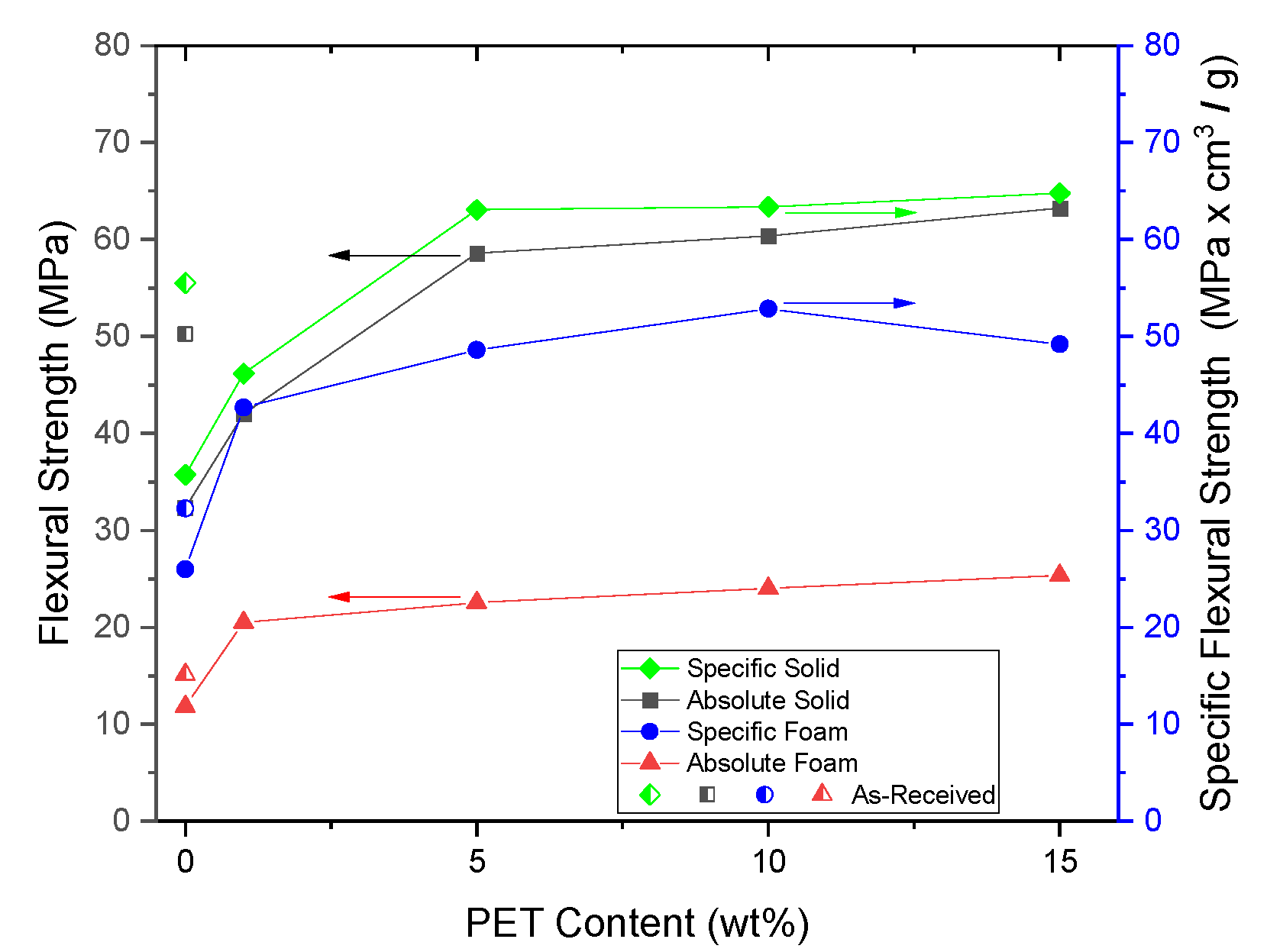
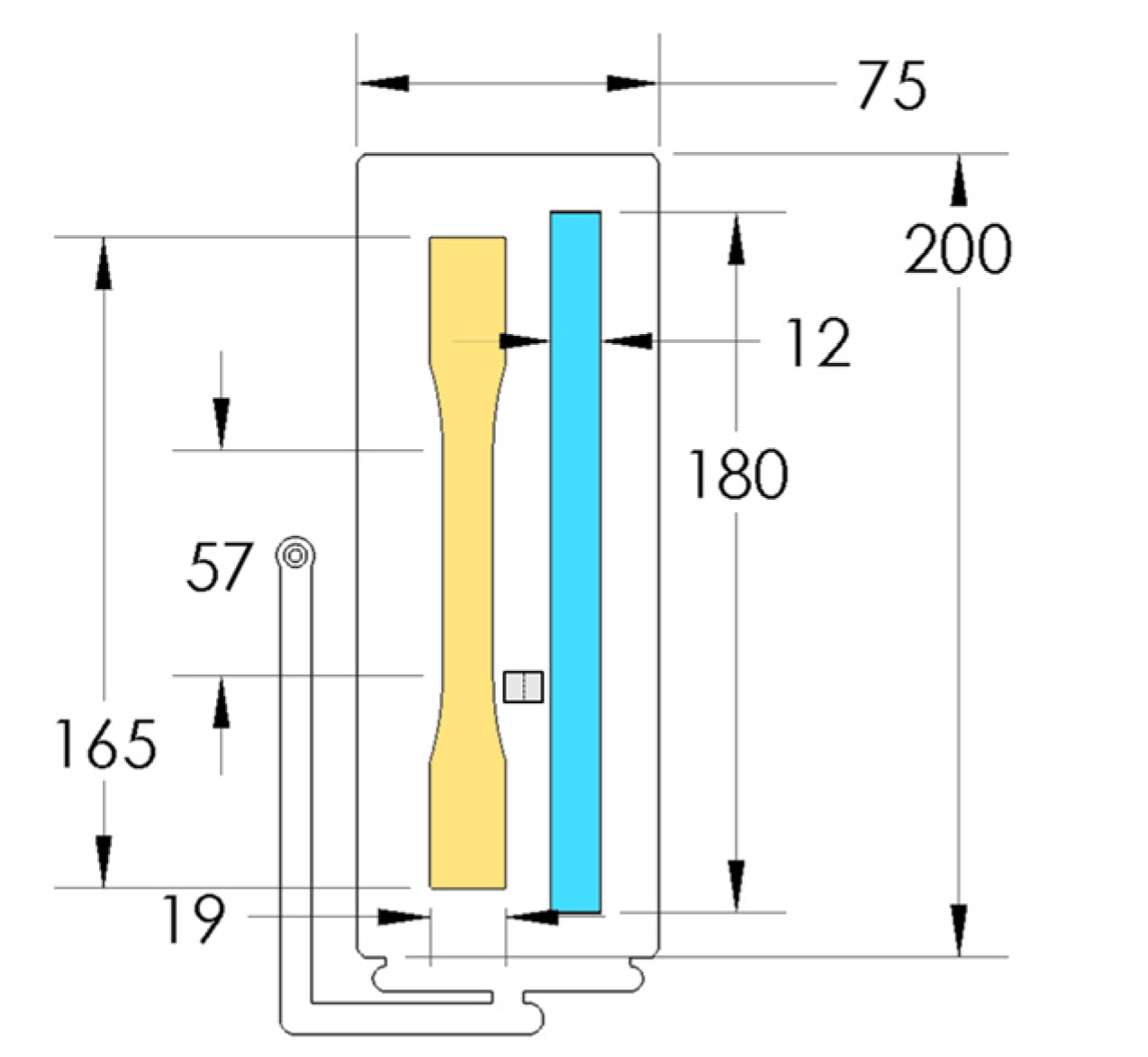
PP composites reinforced with nanofibril PET have been produced using a variety of methods in lab scale settings. However, their application in continuous pilot or industrial processes has not been extensively investigated. Therefore, it is of interest to find methods and strategies to easily create these nanofibrillated materials. In this work, spunbonding was implemented to generate fibrillated composites. The spunbond method has several advantages including the ease of production with a high drawing ratio/extensional force using a vacuum attenuator. This attenuation provides an extra quenching process that allows for faster solidification of the fiber and an increase in the degree of molecular orientation. In this work, we demonstrated a method to easily create PET nanofibril composites and then applied them for solid and foam injection molding. The tensile and flexural properties of both the solid and foam composites were tested.




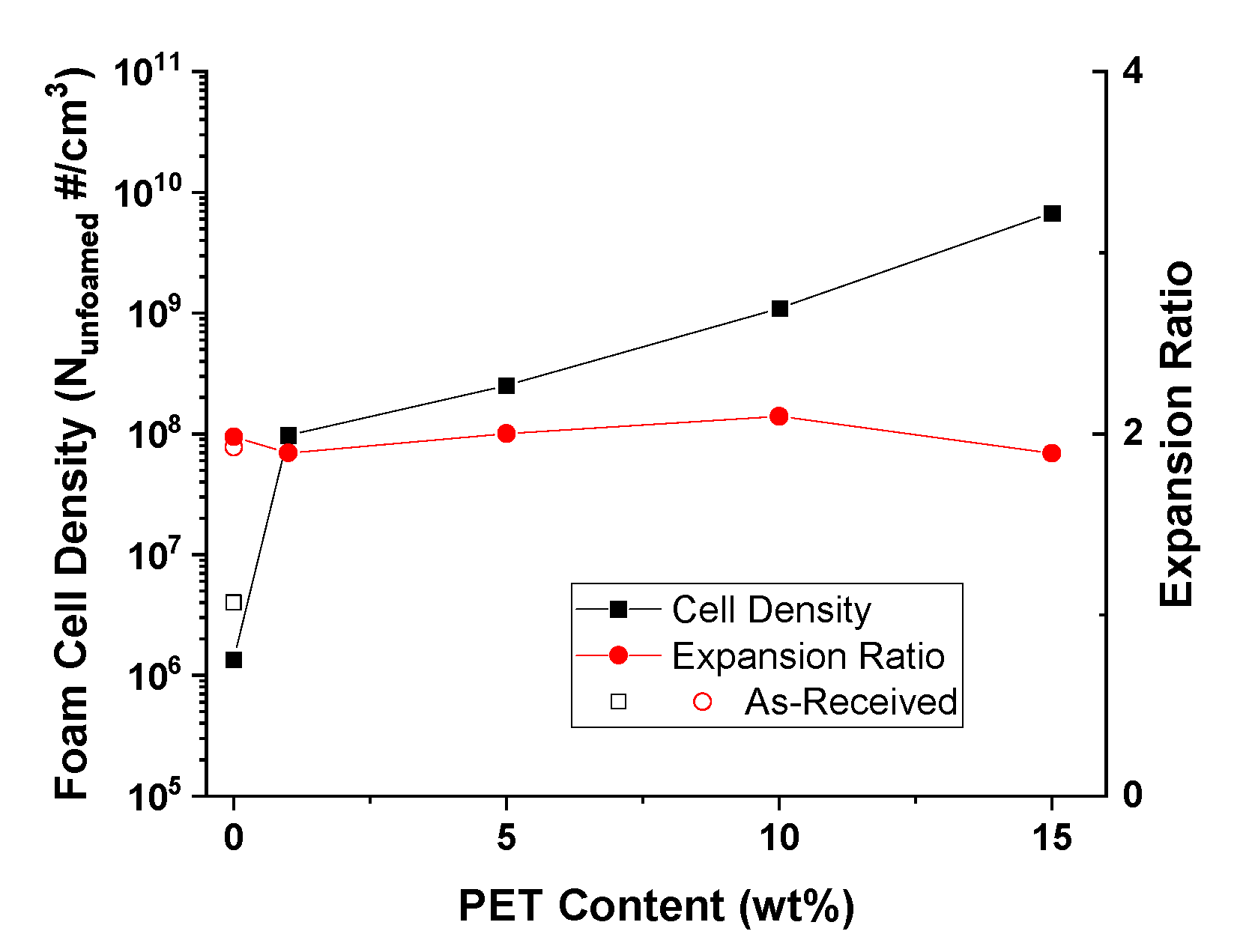
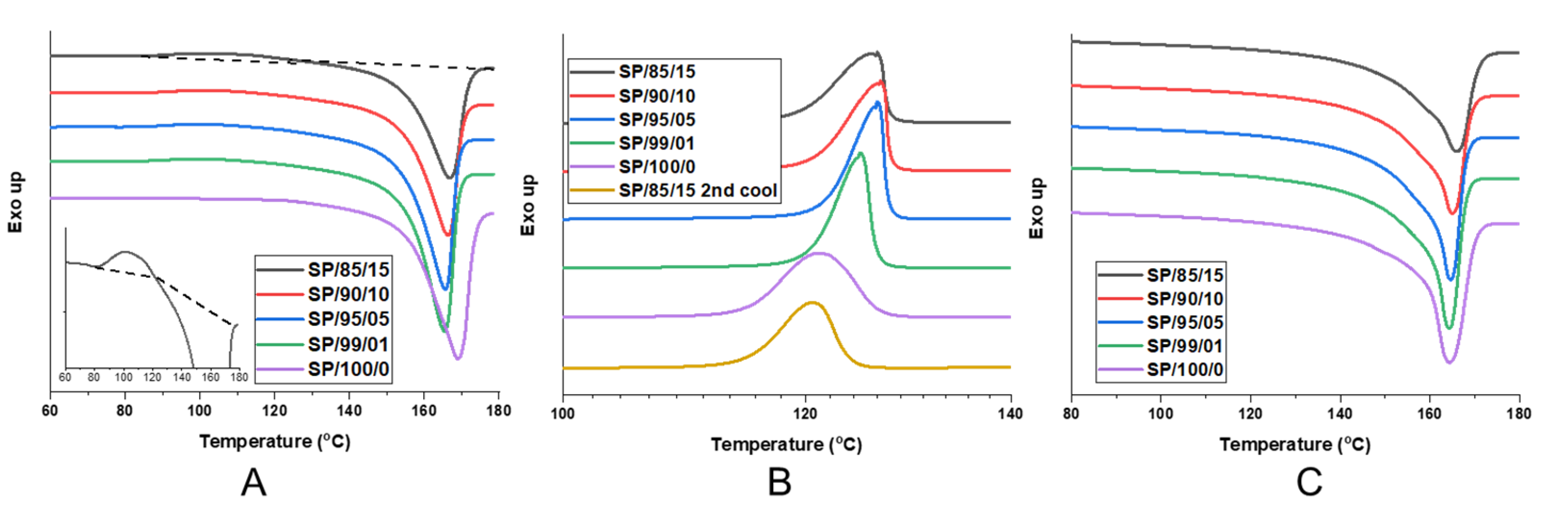
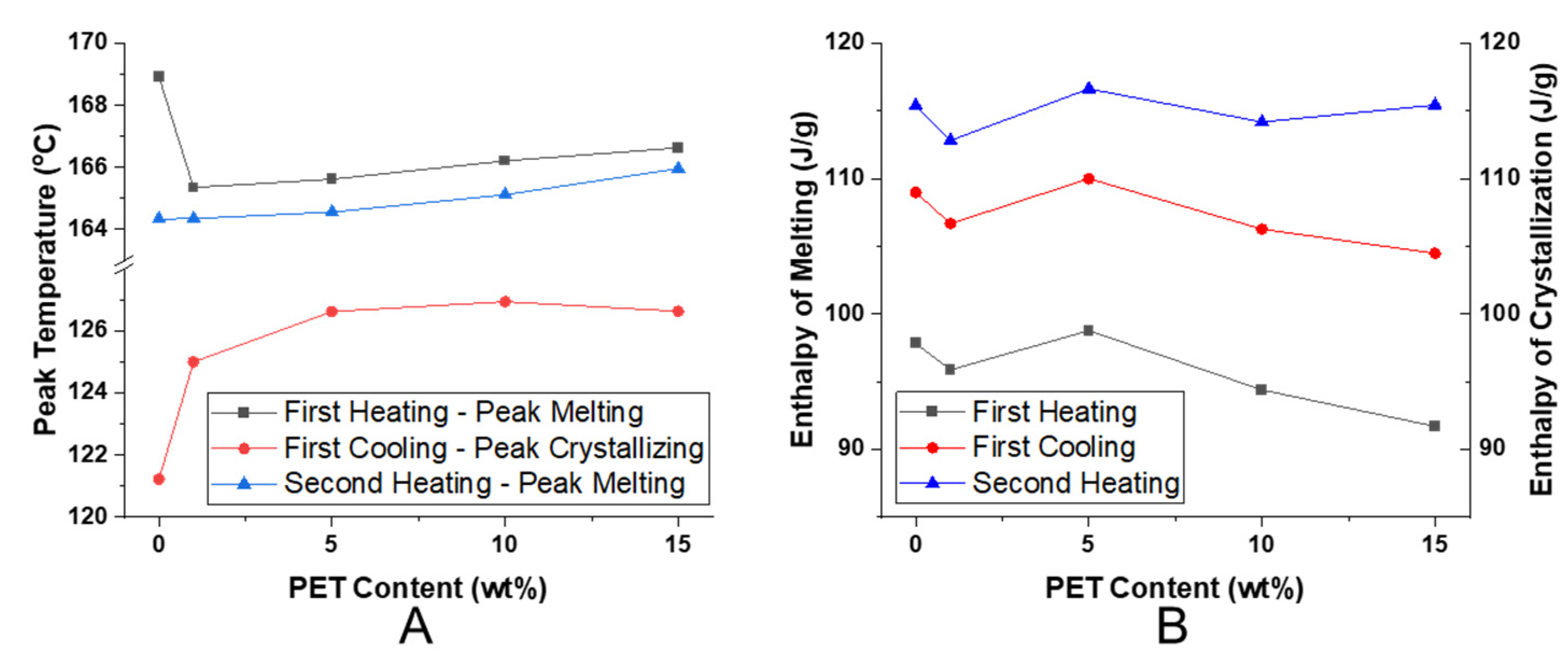

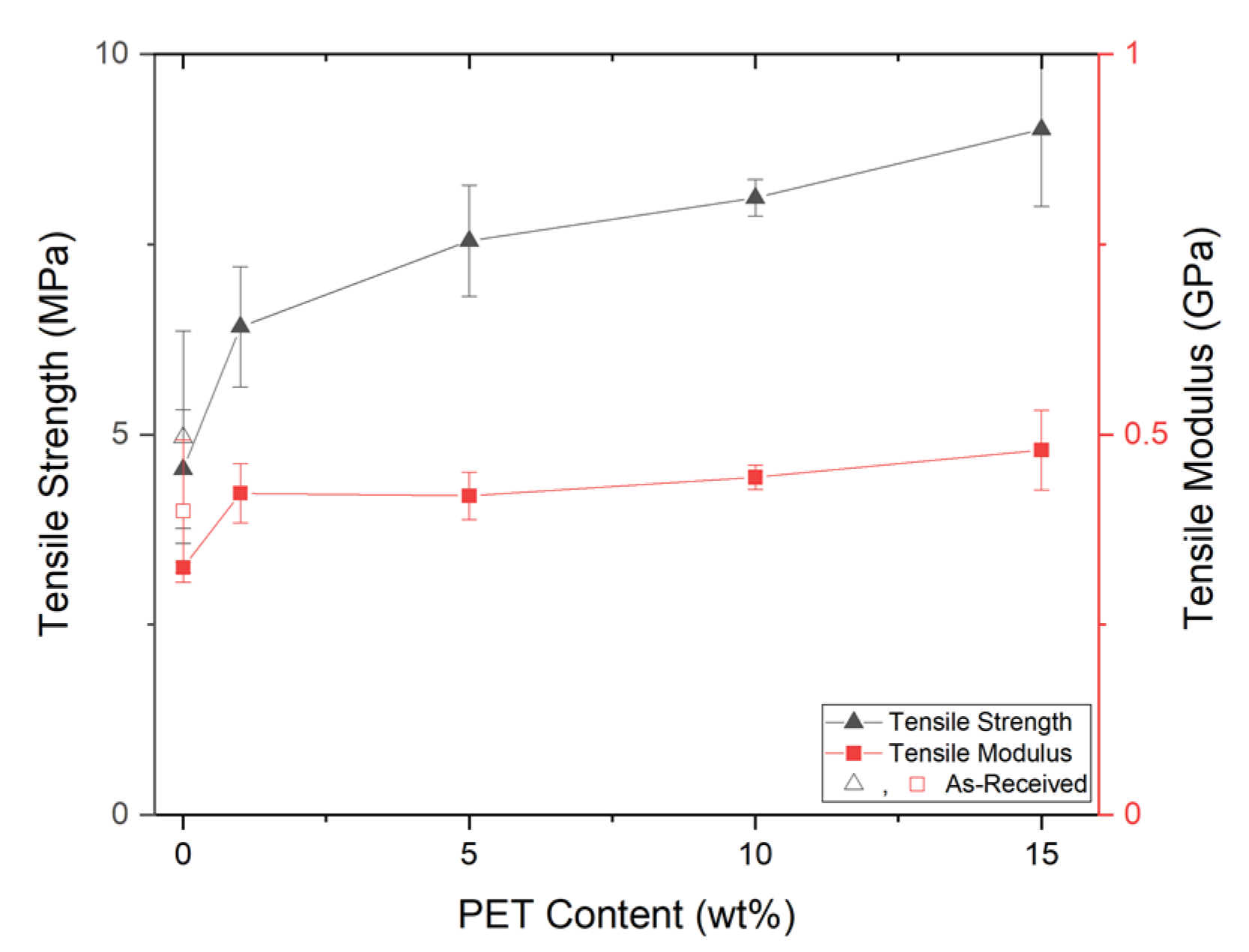

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}