
Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study


Abstract
1. Introduction
2. Method
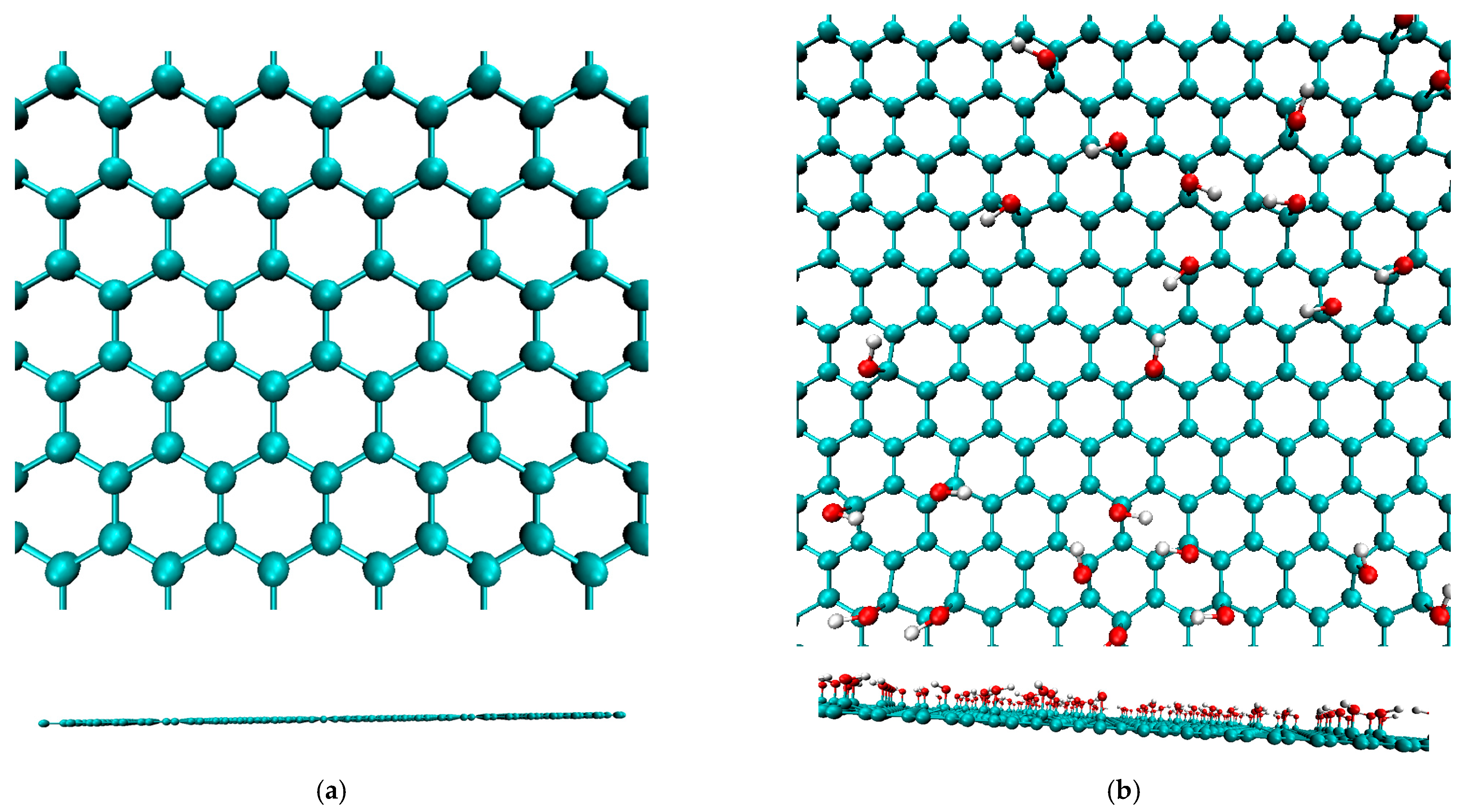
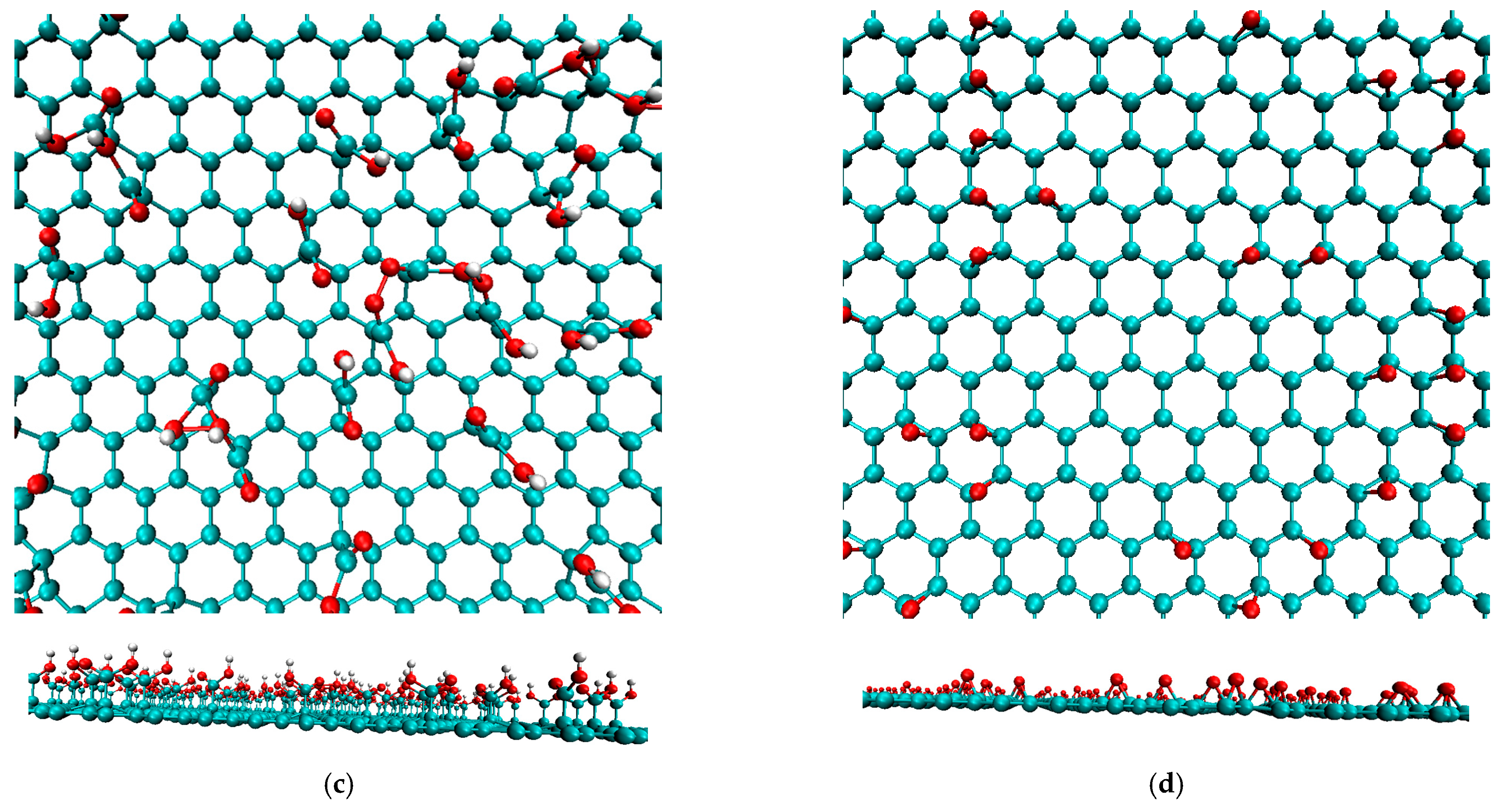
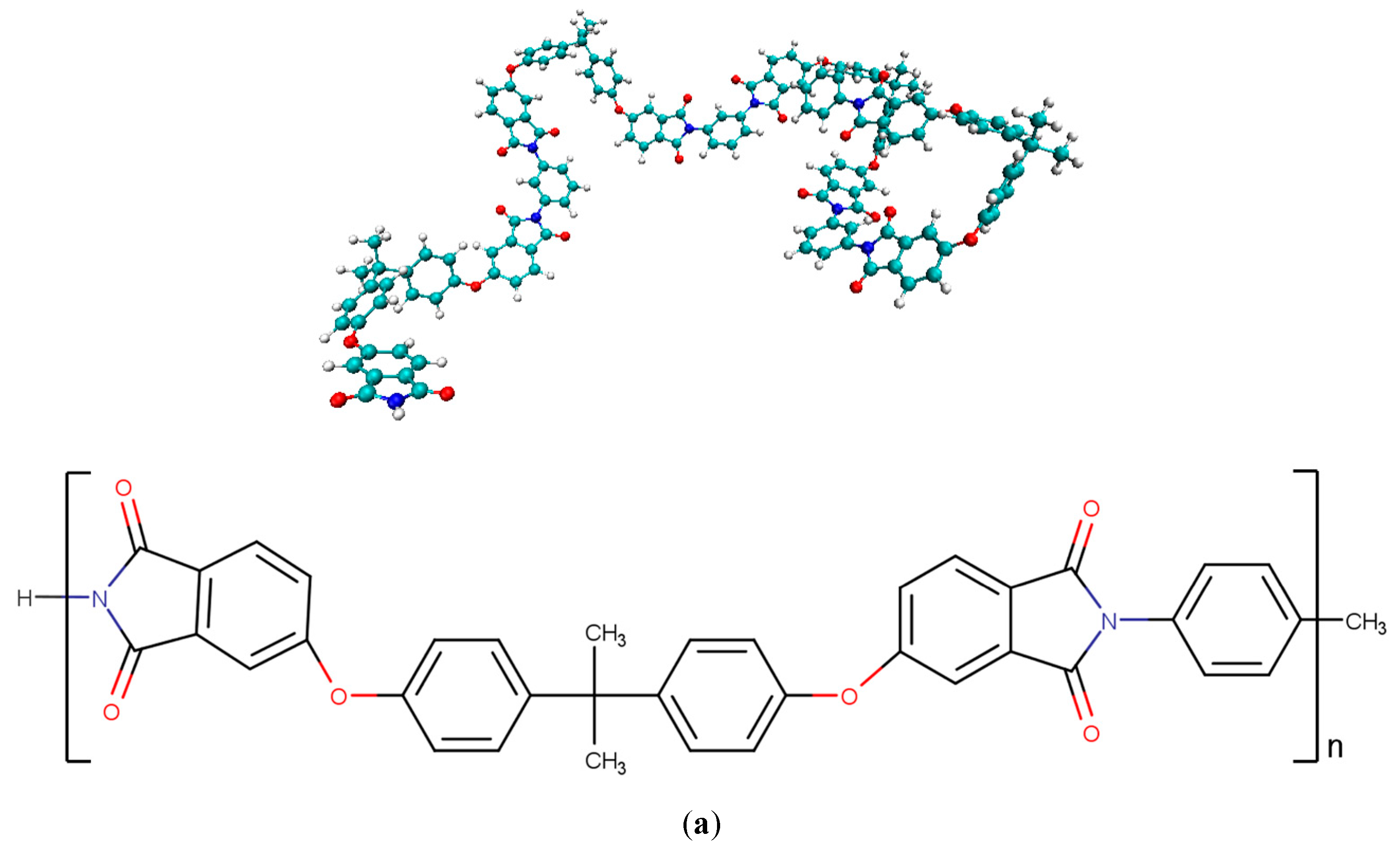
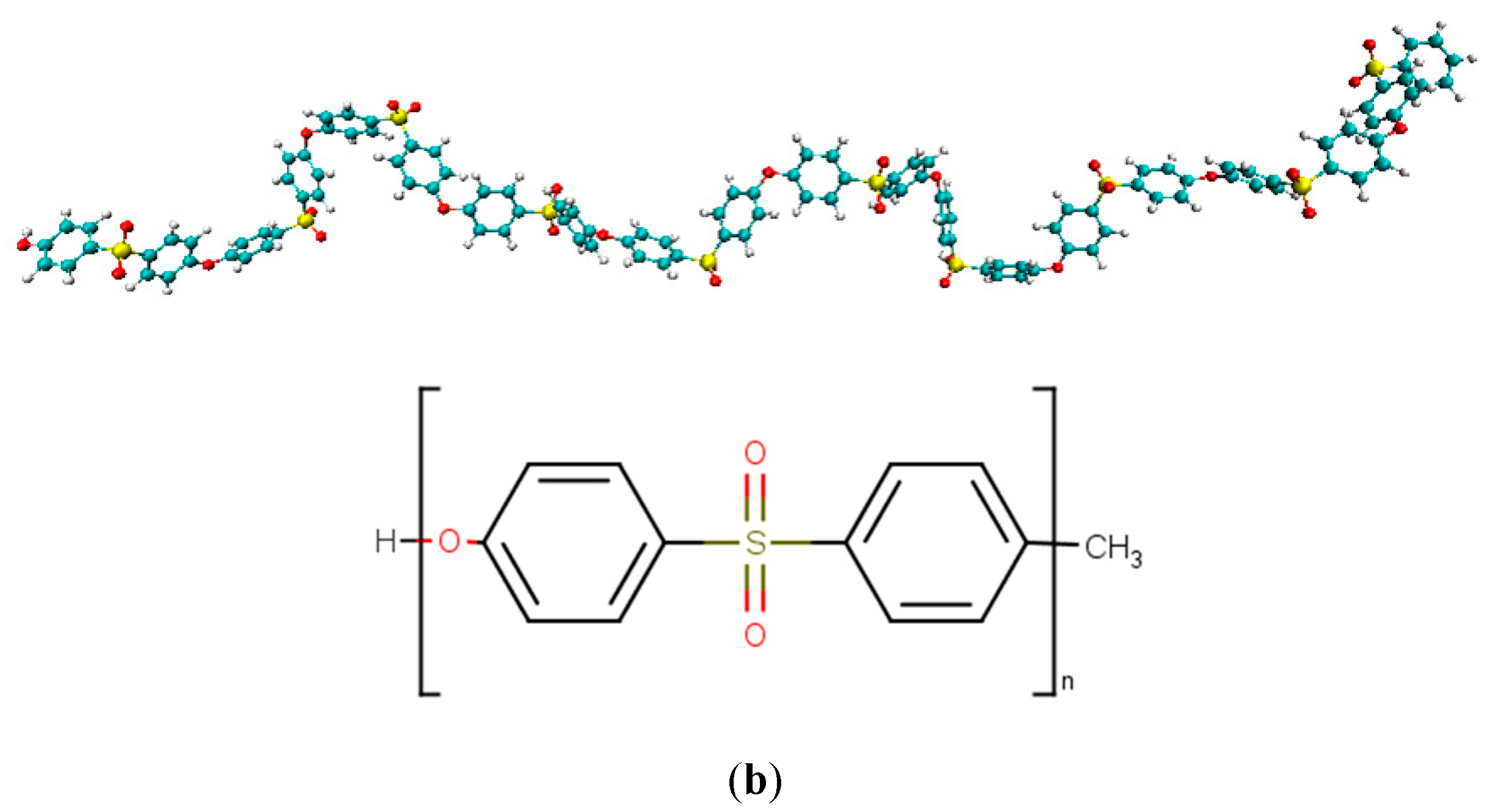
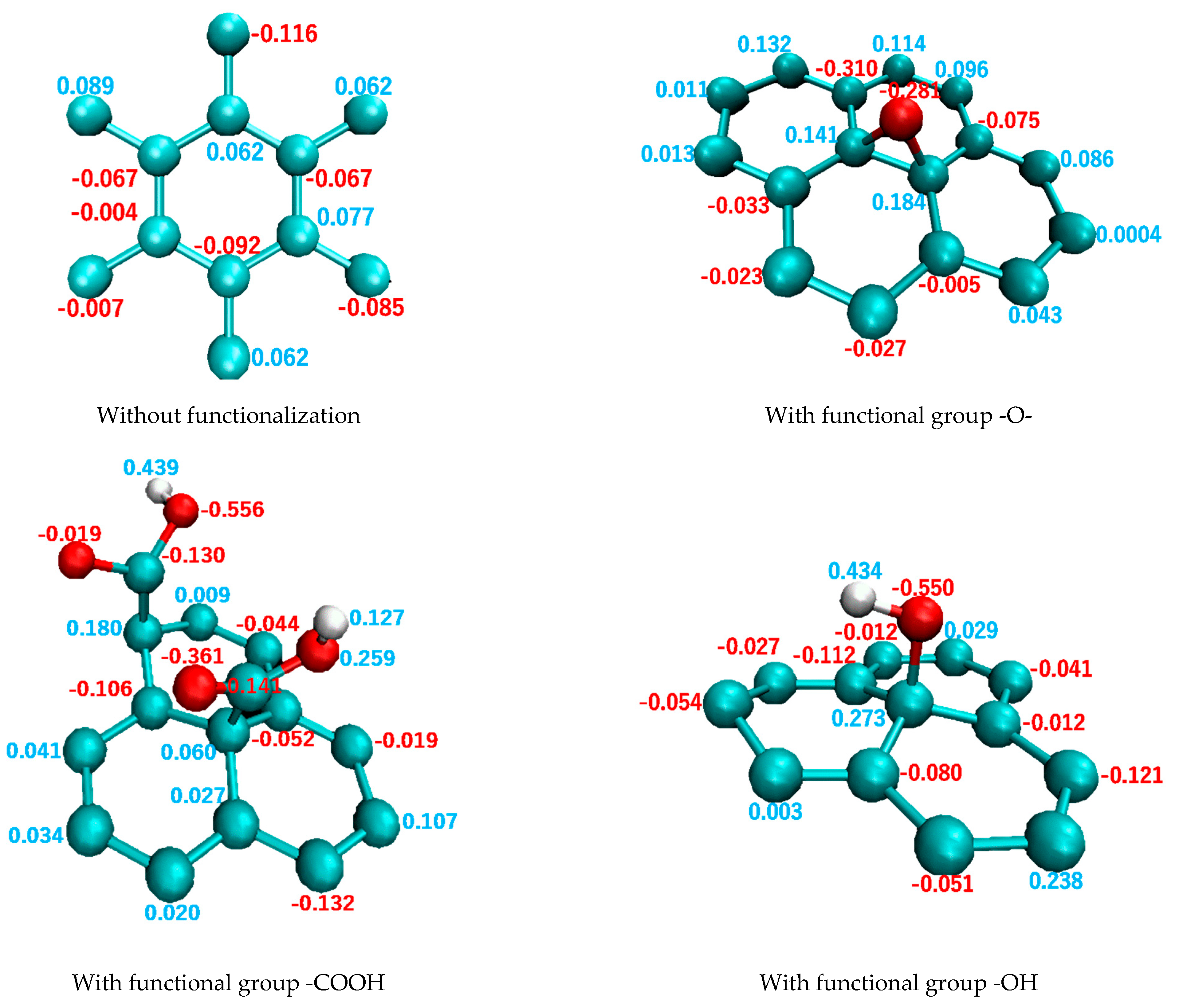
2.1. Creating Composite Models
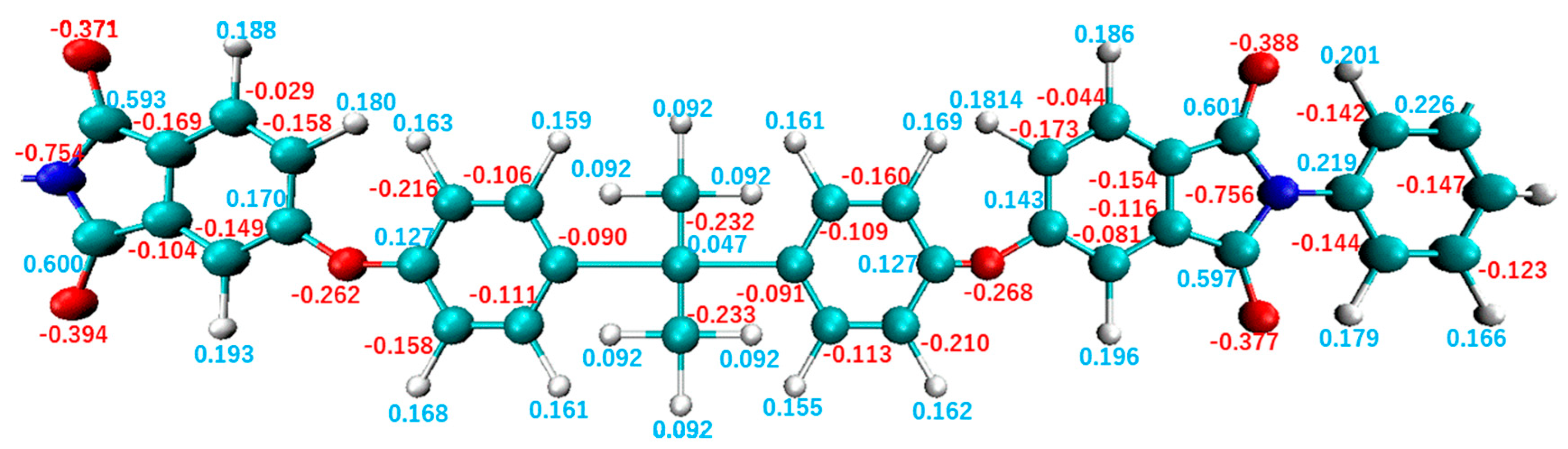
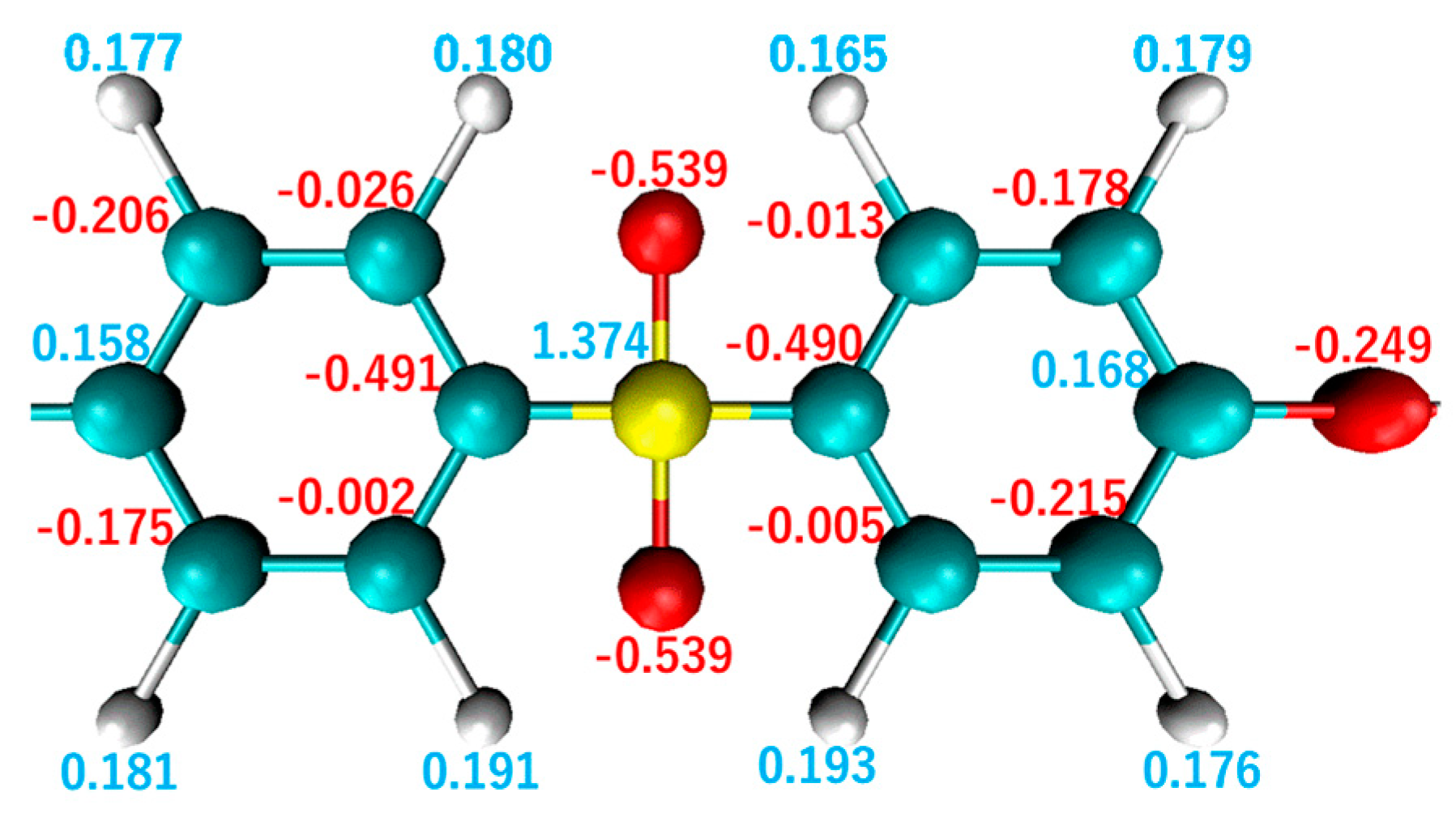
2.2. Relaxation Calculation
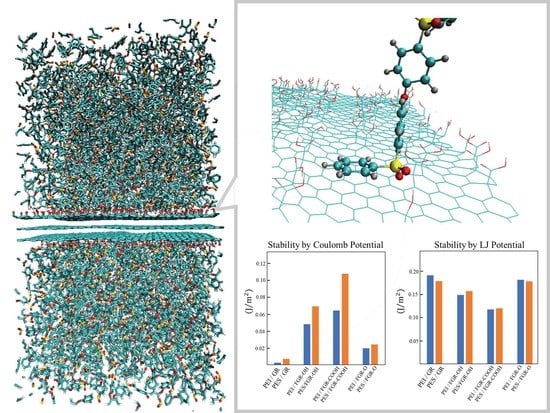
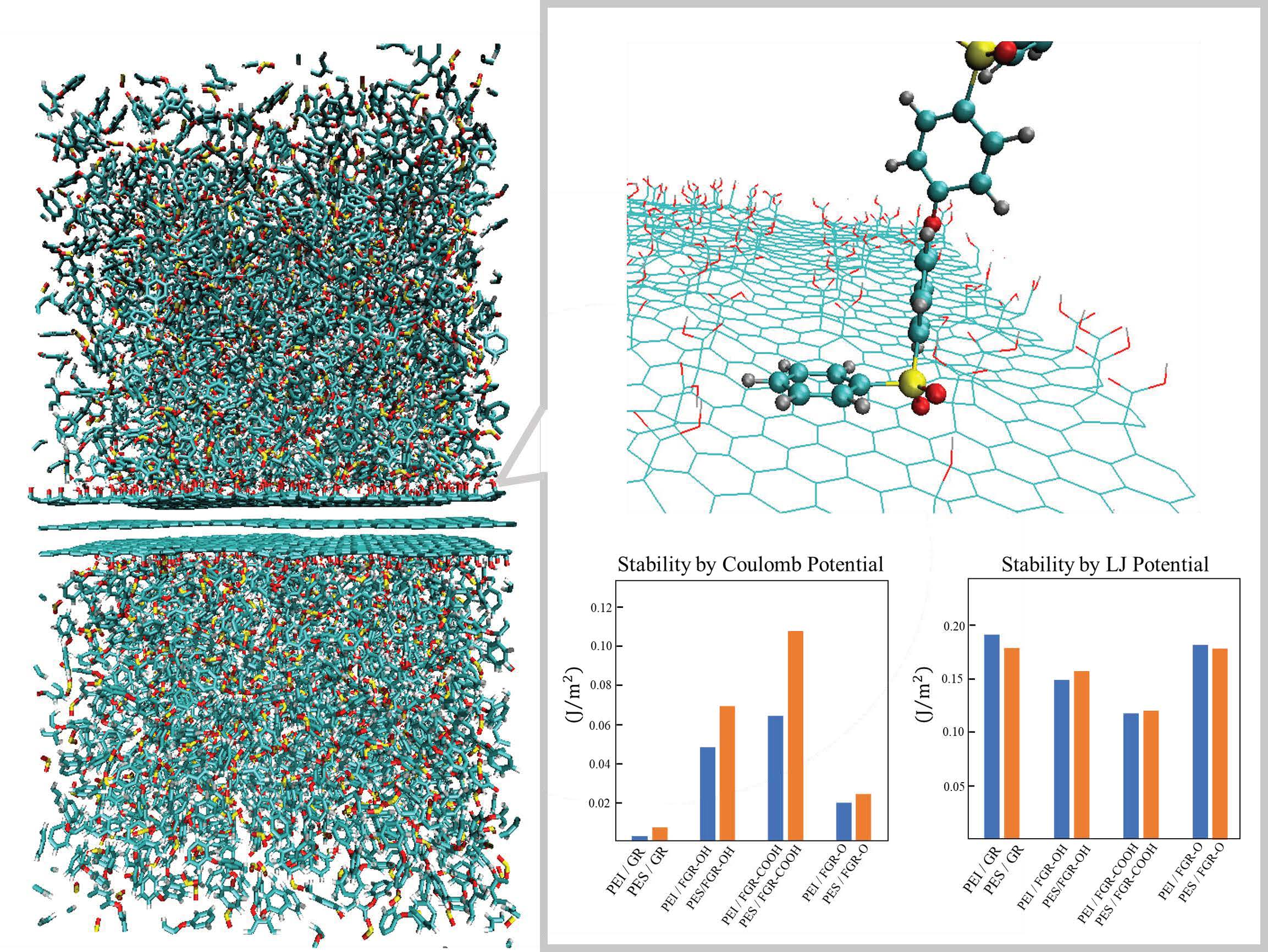
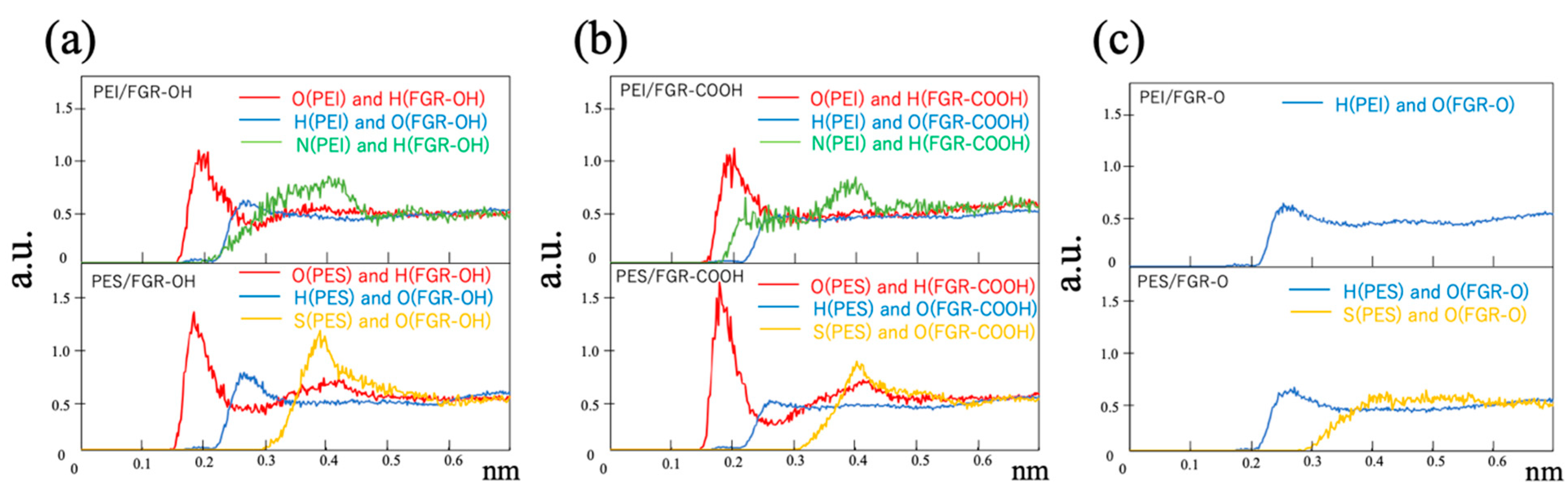
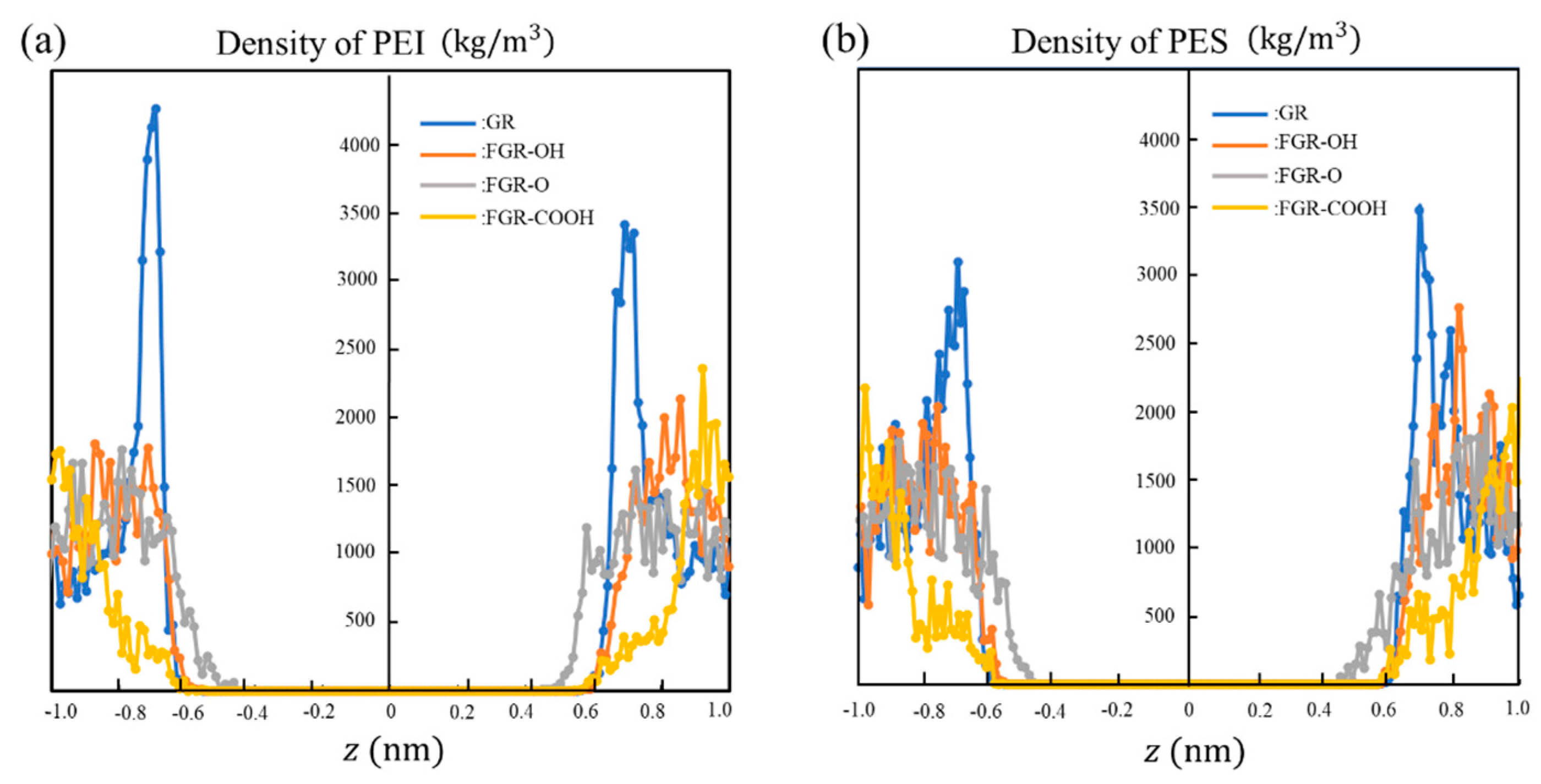
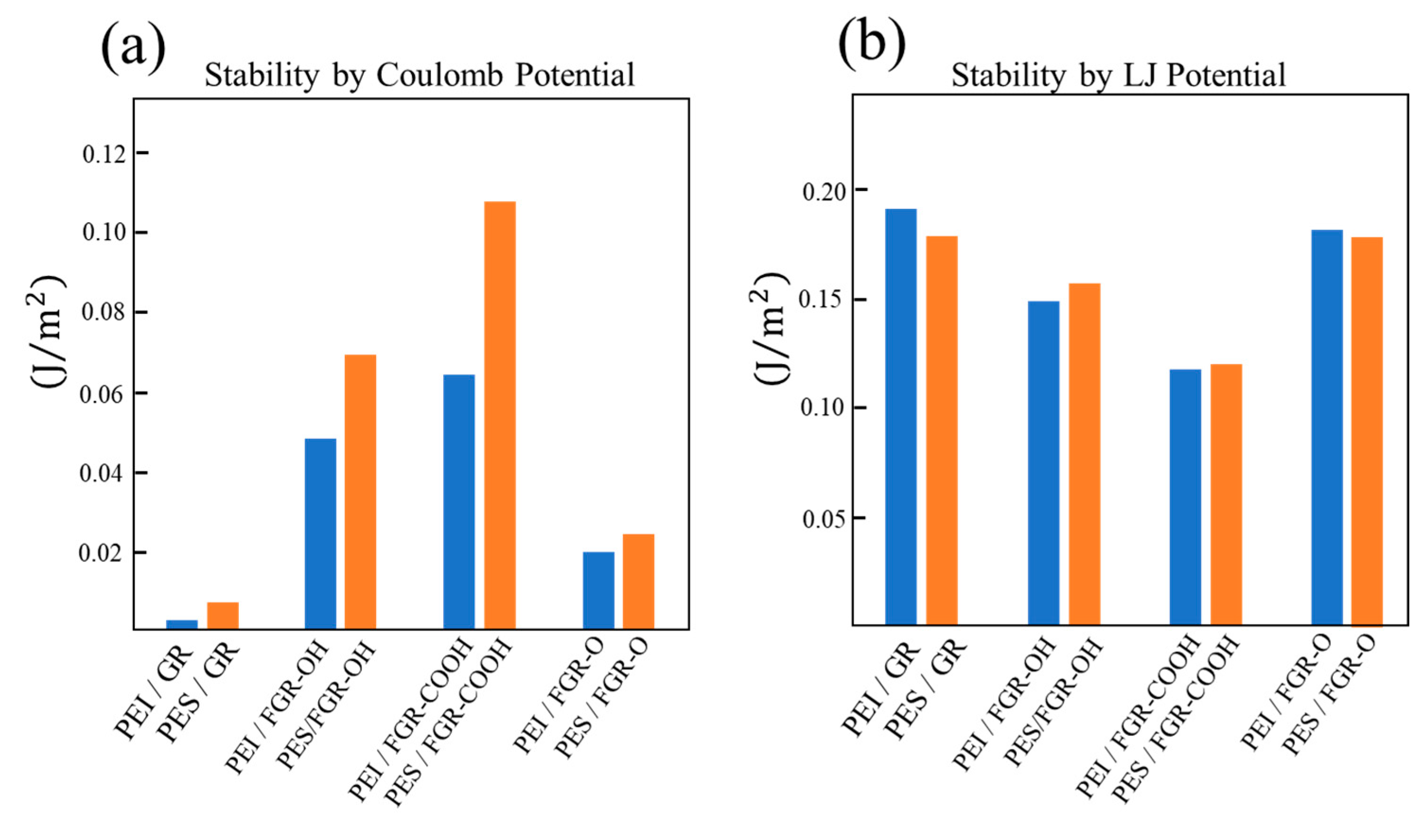
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alshammari, B.A.; Alsuhybani, M.S.; Almushaikeh, A.M.; Altotaibi, B.M.; Alenad, A.M.; Alqahtani, N.B.; Alharbi, A.G. Comprehensive review of the properties and modifications of carbon fiber-reinforced thermoplastic composites. Polymers 2021, 13, 2474. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.S.; Jin, F.L.; Rhee, K.Y.; Hui, D.; Park, S.J. Recent advances in carbon-fiber-reinforced thermoplastic composites: A review. Compos. Part B 2018, 142, 241–250. [Google Scholar] [CrossRef]
- Ning, F.; Cong, W.; Qiu, J.; Wei, J.; Wang, S. Additive manufacturing of carbon fiber reinforced thermoplastic composites using fused deposition modeling. Compos. Part B 2015, 80, 369–378. [Google Scholar] [CrossRef]
- Jiang, B.; Chen, Q.; Yang, J. Advances in joining technology of carbon fiber-reinforced thermoplastic composite materials and aluminum alloys. Int. J. Adv. Manufuct. Technol. 2020, 110, 2631–2649. [Google Scholar] [CrossRef]
- Wang, H.; Jin, K.; Tao, J. Improving the interfacial shear strength of carbon fibre and epoxy via mechanical interlocking effect. Compos. Sci. Technol. 2020, 200, 108423. [Google Scholar] [CrossRef]
- Jiao, W.; Zheng, T.; Liu, W.; Jiao, W.; Wang, R. Molecular dynamics simulations of the effect of sizing agent on the interface property in carbon fiber reinforced vinyl ester resin composite. Appl. Surf. Sci. 2019, 479, 1192–1199. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Lv, Y.; Huang, L.; Zhang, X. Influence of Humidity on Fatigue Performance of CFRP: A Molecular Simulation. Polymers 2020, 13, 140. [Google Scholar] [CrossRef]
- Takamura, M.; Uehara, K.; Koyanagi, J.; Takeda, S. Multi-Timescale Simulations of Temperature Elevation for Ultrasonic Welding of CFRP with Energy Director. J. Multiscale Modell. 2021, 12, 2143003. [Google Scholar] [CrossRef]
- Koyanagi, J.; Takase, N.; Mori, K.; Sakai, T. Molecular dynamics simulation for the quantitative prediction of experimental tensile strength of a polymer material. Compos. Part C 2020, 2, 100041. [Google Scholar] [CrossRef]
- Johnston, J.P.; Koo, B.; Subramanian, N.; Chattopadhyay, A. Modeling the molecular structure of the carbon fiber/polymer interphase for multiscale analysis of composites. Compos. Part B Eng. 2017, 111, 27–36. [Google Scholar] [CrossRef]
- Jiao, W.; Liu, W.; Yang, F.; Jiang, L.; Jiao, W.; Wang, R. Improving the interfacial strength of carbon fiber/vinyl ester resin composite by self-migration of acrylamide: A molecular dynamics simulation. Appl. Surf. Sci. 2018, 454, 74–81. [Google Scholar] [CrossRef]
- Xiao, Y.; Xian, G. Effects of moisture ingress on the bond between carbon fiber and epoxy resin investigated with molecular dynamics simulation. Polym. Compos. 2018, 39, E2074–E2083. [Google Scholar] [CrossRef]
- Alizadeh, A.S.; Mokarizadeh, A.H.; George, D.; Rodrigue, D.; Baniassadi, M.; Foroutan, M. Insights into interphase thickness characterization for graphene/epoxy nanocomposites: A molecular dynamics simulation. Phys. Chem. Chem. Phys. 2019, 21, 19890–19903. [Google Scholar] [CrossRef]
- Kasahara, S.; Koyanagi, J.; Mori, K.; Yabe, M. Evaluation of interface properties of carbon fiber/resin using the full atomistic model considering the electric charge state. Adv. Compos. Mater. 2020, 30, 164–175. [Google Scholar] [CrossRef]
- Wang, H.; Jin, K.; Tao, J. Design and Experiment of Polyimide/Ti Wire Composite with the Interface Chemically Grafted through Carbon Nanotubes. Adv. Mater. Interfaces 2020, 8, 2001863. [Google Scholar] [CrossRef]
- Frankland, S.J.; Caglar, A.; Brenner, D.W.; Griebel, M. Molecular Simulation of the Influence of Chemical Cross-Links on the Shear Strength of Carbon Nanotube-Polymer Interfaces. J. Phys. Chem. B 2002, 106, 3046–3048. [Google Scholar] [CrossRef]
- Fan, J.; Yang, J.; Wang, L.; Li, H.; Tian, J.; Ye, J.; Zhao, Y. Enhanced mechanical properties of epoxy nanocomposites with mildly surface-functionalized graphene oxide by tuned amine species. Appl. Surf. Sci. 2021, 558, 149964. [Google Scholar] [CrossRef]
- Sun, Y.; Tang, X.; Bao, H.; Yang, Z.; Ma, F. The effects of hydroxide and epoxide functional groups on the mechanical properties of graphene oxide and its failure mechanism by molecular dynamics simulations. RSC Adv. 2020, 10, 29610–29617. [Google Scholar] [CrossRef]
- Wang, H.; Jin, K.; Wang, C.; Guo, X.; Chen, Z.; Tao, J. Effect of fiber surface functionalization on shear behavior at carbon fiber/epoxy interface through molecular dynamics analysis. Compos. Part A 2019, 126, 105611. [Google Scholar] [CrossRef]
- Sun, S.; Chen, S.; Weng, X.; Shan, F.; Hu, S. Effect of Carbon Nanotube Addition on the Interfacial Adhesion between Graphene and Epoxy: A Molecular Dynamics Simulation. Polymers 2019, 11, 121. [Google Scholar] [CrossRef]
- Takasa, N.; Koyanagi, J.; Mori, K.; Sakai, T. Molecular dynamis simulation for evaluating fracture entropy of a polymer material under various combined stress states. Materials 2021, 14, 1884. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Takasa, N.; Oya, Y.; Koyanagi, J. A possibility for quantitative detection of mechanically-induced invisible damage by thermal property measurement via entropy generation for a polymer material. Materials 2022, 15, 737. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Chen, H.; Luo, Y.; Li, Y.; Cheng, Y.; Liu, Y. Toughning mechanism based on the physical entanglement of branched epoxy resin in the non-phase-separated inhomogeneous crosslinking network: An experimental and molecular dynamics simulation study. Polymer 2022, 247, 124754. [Google Scholar] [CrossRef]
- Okabe, T.; Oya, Y.; Tanabe, K.; Kikugawa, G.; Yoshioka, K. Molecular dynamics simulation of crosslinked epoxy resins: Curing and mechanical properties. Eur. Polym. J. 2016, 80, 78. [Google Scholar] [CrossRef]
- Hassanloo, H.; Sadeghzadeh, S.; Ahmadi, R. Reactive molecular dynamics simulation of thermo-physicochemical properties of non-covalent functionalized graphene nanofluids. Mater. Today Commun. 2022, 32, 103869. [Google Scholar] [CrossRef]
- Shirasu, K.; Kitayama, S.; Liu, F.; Yamamoto, G.; Hashida, T. Molecular Dynamics Simulations and theoretical model for engineering tensile properties of single-and multi-walled carbon nanotubes. Nanomaterials 2021, 11, 795. [Google Scholar] [CrossRef]
- Park, C.; Yun, G.J. Characterization of Interfacial Properties of Graphene-Reinforced Polymer Nanocomposites by Molecular Dynamics-Shear Deformation Model. J. Appl. Mech. 2018, 85, 091007. [Google Scholar] [CrossRef]
- Alian, A.R.; Meguid, S.A. Molecular dynamics simulations of the effect of waviness and agglomeration of CNTs on interface strength of thermoset nanocomposites. Phys. Chem. Chem. Phys. 2017, 19, 4426–4434. [Google Scholar] [CrossRef]
- Mahmud, H.A.; Radue, M.S.; Chinkanjanarot, S.; Pisani, W.A.; Gowtham, S.; Odegard, G.M. Multiscale modeling of carbon fiber- graphene nanoplatelet-epoxy hybrid composites using a reactive force field. Compos. Part B Eng. 2019, 172, 628–635. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, L.; Cui, L.; Zhang, Y.; Du, X. Molecular dynamics simulation of cross-linked epoxy resin and its interaction energy with graphene under two typical force fields. Comput. Mater. Sci. 2018, 143, 240–247. [Google Scholar] [CrossRef]
- Salahshoor, H.; Rahbar, N. Nano-scale fracture toughness and behavior of graphene/epoxy interface. J. Appl. Phys. 2012, 112, 023510. [Google Scholar] [CrossRef]
- Oya, Y.; Inuyama, K.; Okabe, T. Analysis of structure characteristics in laminated graphene oxide nanocomposites using molecular dynamics simulation. Adv. Compos. Mater. 2018, 27, 427–438. [Google Scholar] [CrossRef]
- Shen, J.; Li, X.; Zhang, L.; Lin, X.; Li, H.; Shen, X.; Ganesan, V.; Liu, J. Mechanical and Viscoelastic Properties of Polymer-Grafted Nanorod Composites from Molecular Dynamics Simulation. Macromolecules 2018, 51, 2641–2652. [Google Scholar] [CrossRef]
- Yang, M.; Koutsos, V.; Zaiser, M. Interactions between Polymers and Carbon Nanotubes: A Molecular Dynamics Study. J. Phys. Chem. B 2005, 109, 10009–10014. [Google Scholar] [CrossRef]
- Eslami, H.; Behrouz, M. Molecular Dynamics Simulation of a Polyamide-66/Carbon Nanotube Nanocomposite. J. Phys. Chem. C 2014, 118, 9841–9851. [Google Scholar] [CrossRef]
- Lee, S.; Lyulin, A.V.; Frank, C.W.; Yoon, D.Y. Interface characteristics of polystyrene melts in free-standing thin films and on graphite surface from molecular dynamics simulations. Polymer 2017, 116, 540–548. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Wang, Q.; Xing, M. A comparison study on mechanical properties of polymer composites reinforced by carbon nanotubes and graphene sheet. Compos. Part B Eng. 2018, 133, 35–41. [Google Scholar] [CrossRef]
- Azimi, M.; Mirjavadi, S.S.; Hamouda, A.M.S.; Makki, H. Heterogeneities in Polymer Structural and Dynamic Properties in Graphene and Graphene Oxide Nanocomposites: Molecular Dynamics Simulations. Macromol. Theory Simul. 2017, 26, 1600086. [Google Scholar] [CrossRef]
- Duan, K.; Li, L.; Wang, F.; Meng, W.; Hu, Y.; Wang, X. Importance of interface in the coarse-grained model of CNT/epoxy nanocomposites. Nanomaterials 2019, 9, 1479. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Karatrantos, A.V.; Kroger, M. Ionic Polymer Nanocomposites subjected to uniaxial extension: A nonequilibrium molecular dynamics study. Polymer 2021, 13, 4001. [Google Scholar] [CrossRef]
- Karatasos, K.; Kritikos, G. Characterization of a graphene oxide/poly(acrylic acid) nanocomposite by means of molecular dynamics simulations. RSC Adv. 2016, 6, 109267. [Google Scholar] [CrossRef]
- Zhang, X.; Nguyen, H.; Daly, M.; Nguyen, S.T.; Espinosa, H.D. Nanoscale toughning of ultrathin graphene oxide-polymer composites: Mechanochemical insights into hydrogen-bonding/ vander Waals interactions, polymer chain alignment, and steric parameters. Nanoscale 2019, 11, 12305. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Wang, H.; Tao, J.; Zhang, X. Interface strengthening mechanisms of Ti/CFRP fiber metal laminate after adding MWCNTs to resin matrix. Compos. Part B 2019, 171, 254–263. [Google Scholar] [CrossRef]
- Irisawa, T.; Iwamura, R.; Kozawa, Y.; Kobayashi, S.; Tanabe, Y. Recycling methods for thermoplastic-matrix composites having high thermal stability in focusing on reuse of the carbon fibers. Carbon 2017, 280, 175–181. [Google Scholar] [CrossRef]
- Irisawa, T.; Hashimoto, R.; Arai, M.; Tanabe, Y. The Suitability Evaluation of Aromatic Amorphous Thermoplastics as Matrix Resin for CFRTP Having High Thermal Stability. J. Fiber Sci. Technol. 2017, 73, 61–66. [Google Scholar] [CrossRef][Green Version]
- PolyParGen. Available online: http://polypargen.com/ (accessed on 24 May 2022).
- Yabe, I.; Mori, K.; Koyanagi, J. Development of PolyParGenv2 Software for Determination of Molecular Dynamics Simulation Parameters for Molecules with Crosslinked or Condensed Ring Structures. J. Comput. Chem. Jpn. Int. Ed. 2020, 6, 2019–2031. [Google Scholar] [CrossRef]
- MarvinSketch [Soft]. Available online: chemaxon.com/products/marvin (accessed on 24 May 2022).
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 110, 1657–1666. [Google Scholar] [CrossRef]
- GROMACS [Soft]. Available online: https://www.gromacs.org/ (accessed on 24 May 2022).
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of sovation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, Q.; Wu, X.; Wu, L.; Lin, J.H. Exploring the interfacial phase and pi-pi stacking in aligned carbon nanotube/polyimide nanocomposites. Nanomaterials 2020, 10, 1158. [Google Scholar] [CrossRef]
- Mishra, K.; Bastola, P.B.; Singh, R.P.; Vaidyanathat, R. Effect of graphene oxide on the interlaminar fracture toughness of carbon fiber/epoxy composites. Polym. Eng. Sci. 2019, 59, 1199–1208. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Groups on Graphene | ||||
|---|---|---|---|---|
| None | –OH | –COOH | –O– | |
| PEI | 0.194 (1.72 × 10−3) | 0.197 (2.85 × 10−3) | 0.182 (1.99 × 10−3) | 0.201 (1.68 × 10−3) |
| PES | 0.186 (3.83 × 10−3) | 0.226 (0.51 × 10−3) | 0.228 (0.76 × 10−3) | 0.202 (0.11 × 10−3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morita, M.; Oya, Y.; Kato, N.; Mori, K.; Koyanagi, J. Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study. Polymers 2022, 14, 2579. https://doi.org/10.3390/polym14132579
Morita M, Oya Y, Kato N, Mori K, Koyanagi J. Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study. Polymers. 2022; 14(13):2579. https://doi.org/10.3390/polym14132579
Chicago/Turabian StyleMorita, Mayu, Yutaka Oya, Nobuhiko Kato, Kazuki Mori, and Jun Koyanagi. 2022. "Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study" Polymers 14, no. 13: 2579. https://doi.org/10.3390/polym14132579
APA StyleMorita, M., Oya, Y., Kato, N., Mori, K., & Koyanagi, J. (2022). Effect of Electrostatic Interactions on the Interfacial Energy between Thermoplastic Polymers and Graphene Oxide: A Molecular Dynamics Study. Polymers, 14(13), 2579. https://doi.org/10.3390/polym14132579