
Intrinsically Stretchable Poly(3,4-ethylenedioxythiophene) Conducting Polymer Film for Flexible Electronics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
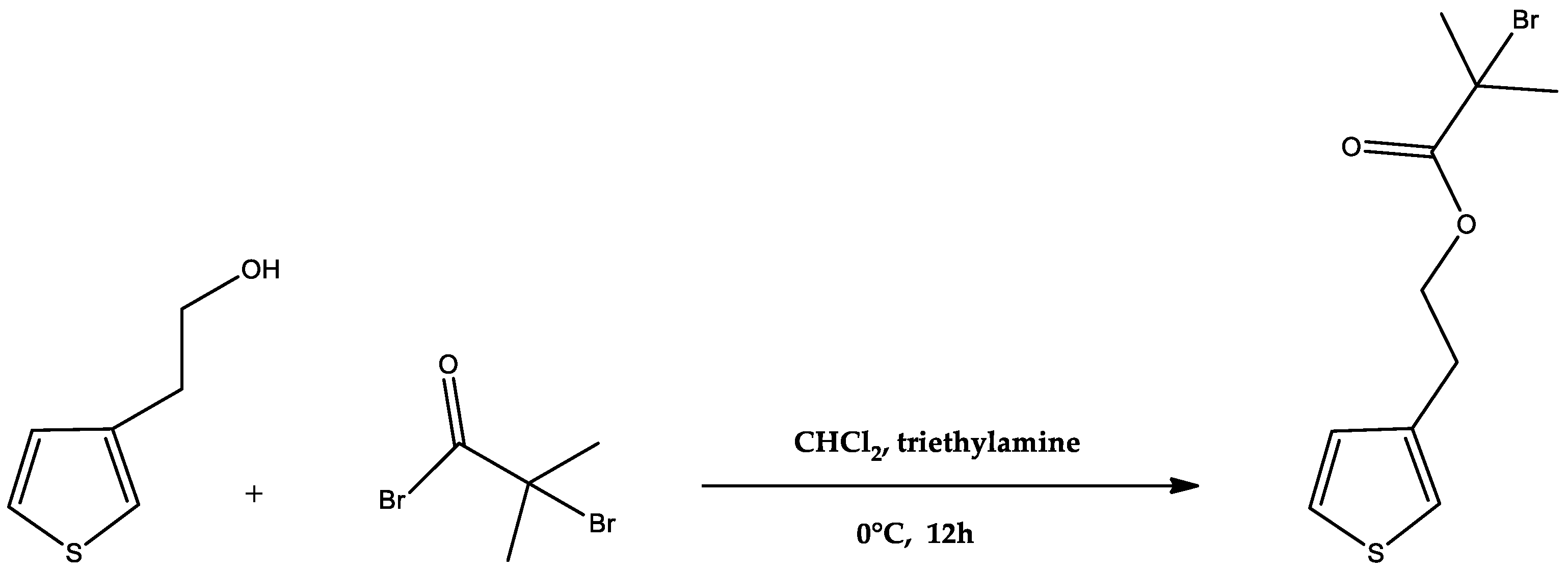
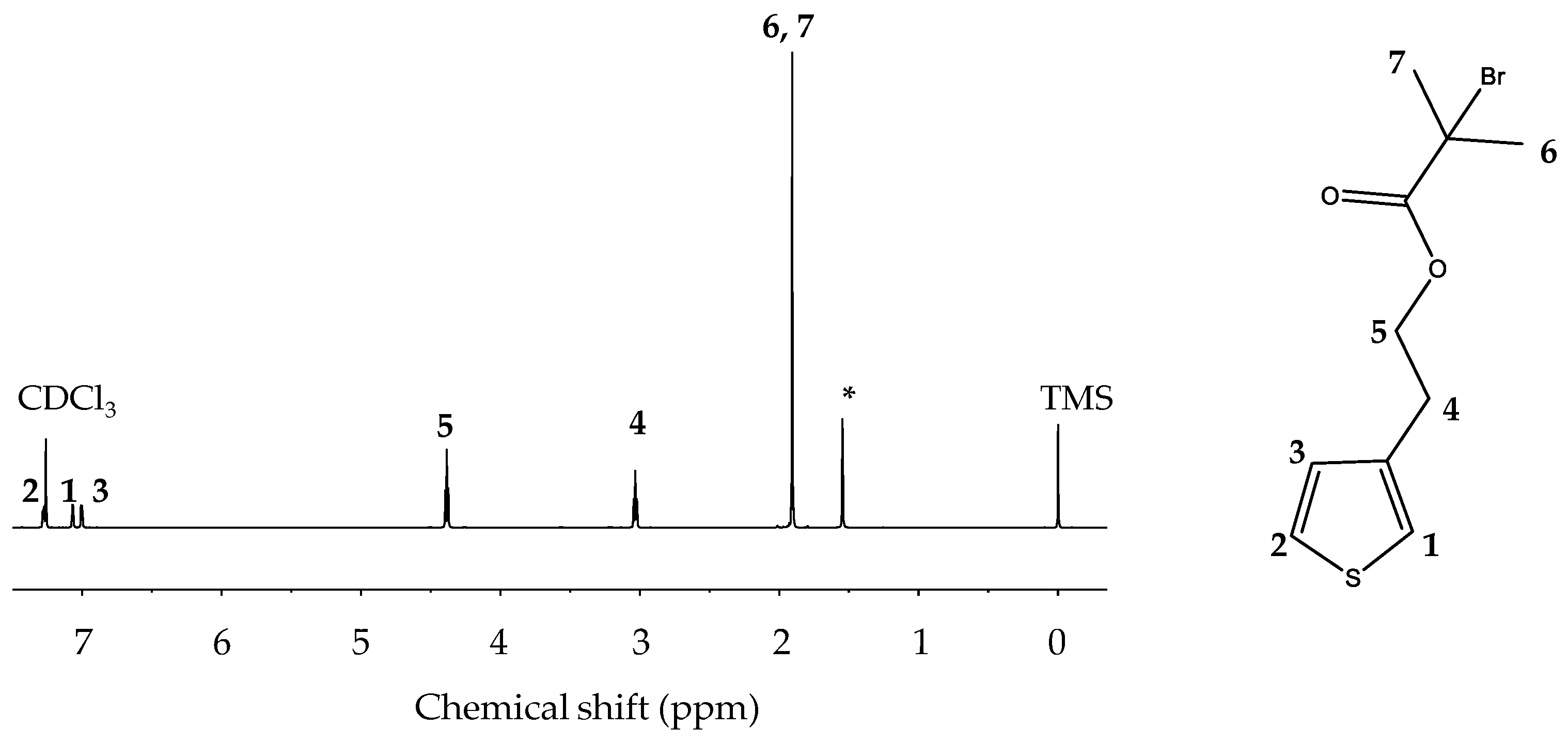
2.2.1. Synthesis of Monomer 2-(Thiophene-3-yl)ethyl 2-bromo-2-methylpropanoate (ThBr)
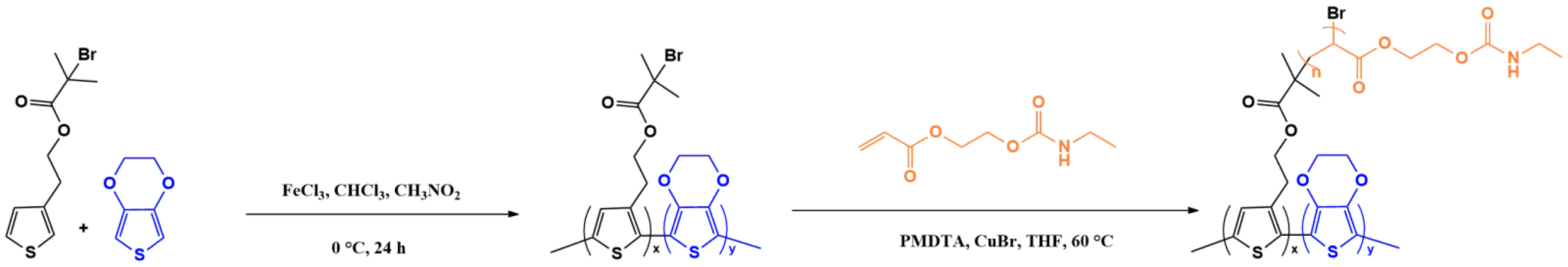
2.2.2. Synthesis of Poly(3,4-ethylenedioxithiophene-co-2-(thiophene-3-yl) ethyl 2-bromo-2-methylpropanoate) ((poly(EDOT-co-ThBr), termed PEDOT-Br)
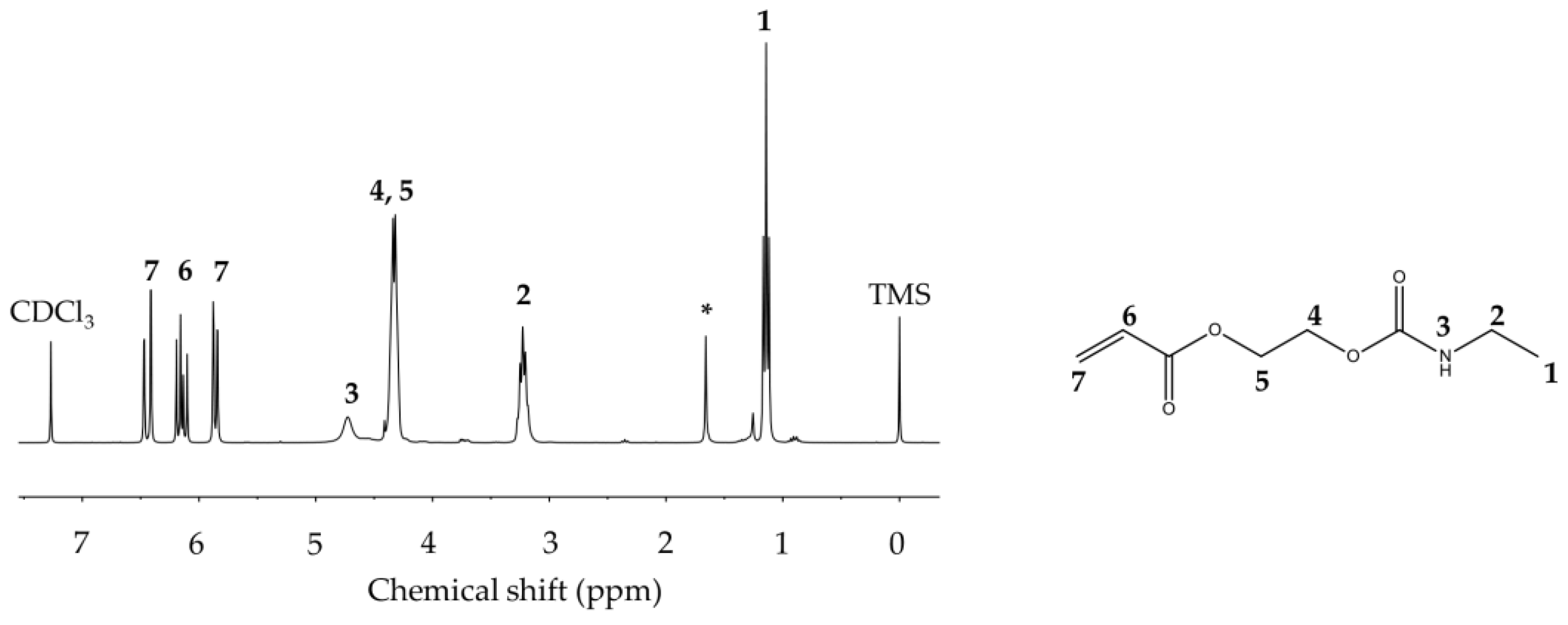
2.2.3. Synthesis of Acrylate Urethane (AU) Monomer
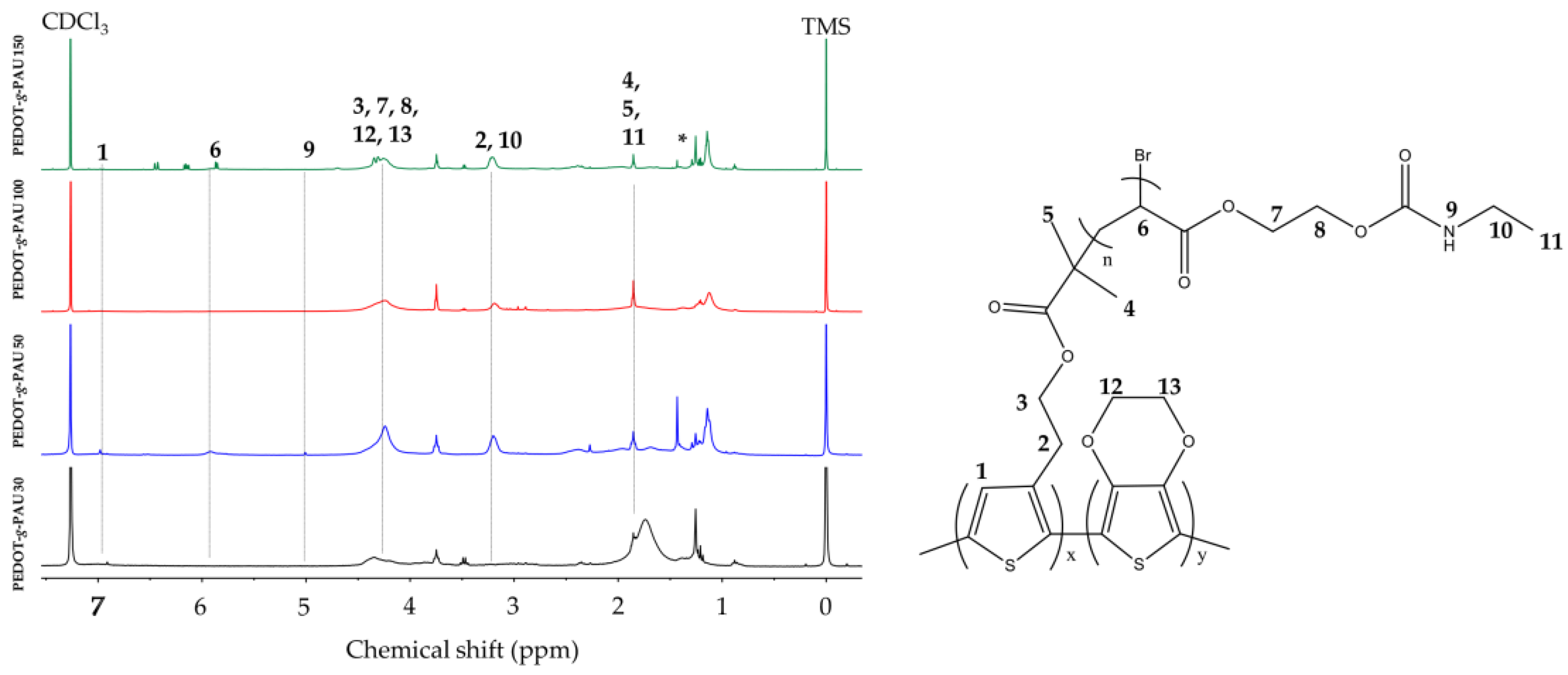
2.2.4. Graft Copolymerization of Poly(3,4-ethylenedioxithiophene-co-2-(thiophene-3-yl) ethyl 2-bromo-2-methylpropanoate)-graft-poly(acrylate-urethane) ((PEDOT-Br-g-PAU), termed PEDOT-g-PAU)
2.3. Characterizations and Instruments
2.3.1. Fourier-Transform Infrared Spectroscopy (FTIR)
2.3.2. Nuclear Magnetic Resonances (NMR)
2.3.3. Gel Permeation Chromatography (GPC)
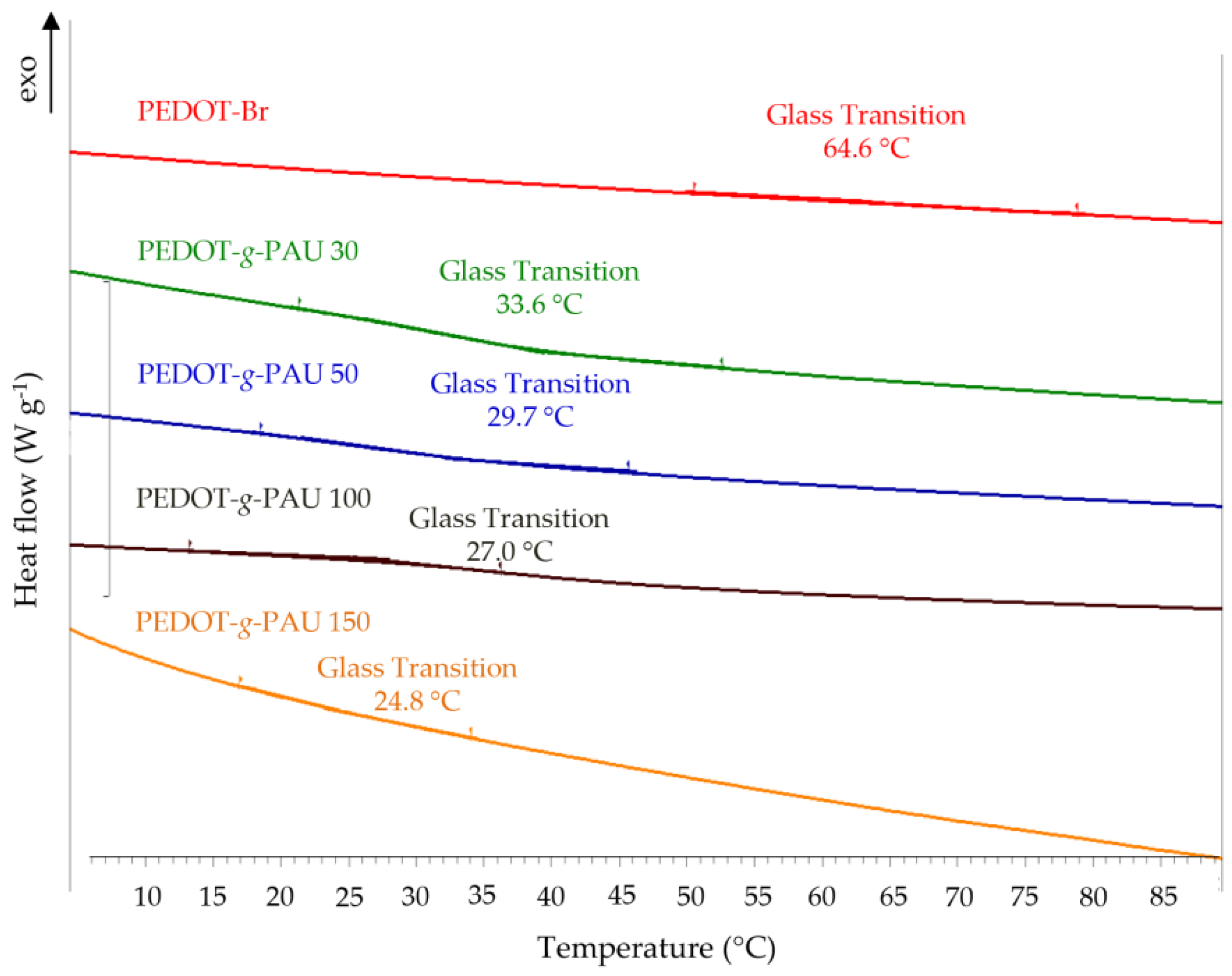
2.3.4. Differential Scanning Calorimetry (DSC)
2.3.5. Thermogravimetric Analysis (TGA)
2.3.6. Four-Point Probe Method (4PP)
2.3.7. Tensile Test
3. Results and Discussion
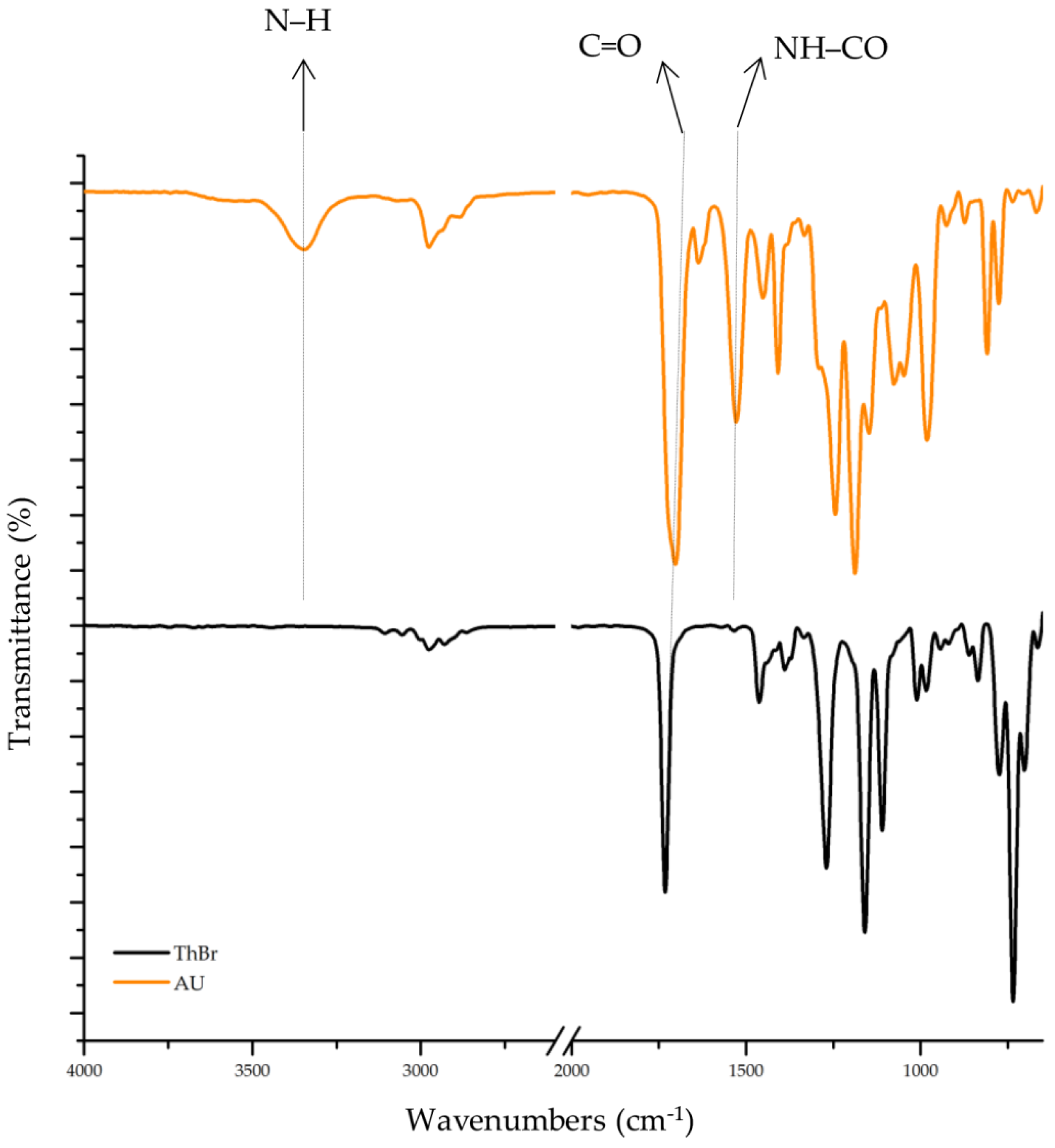
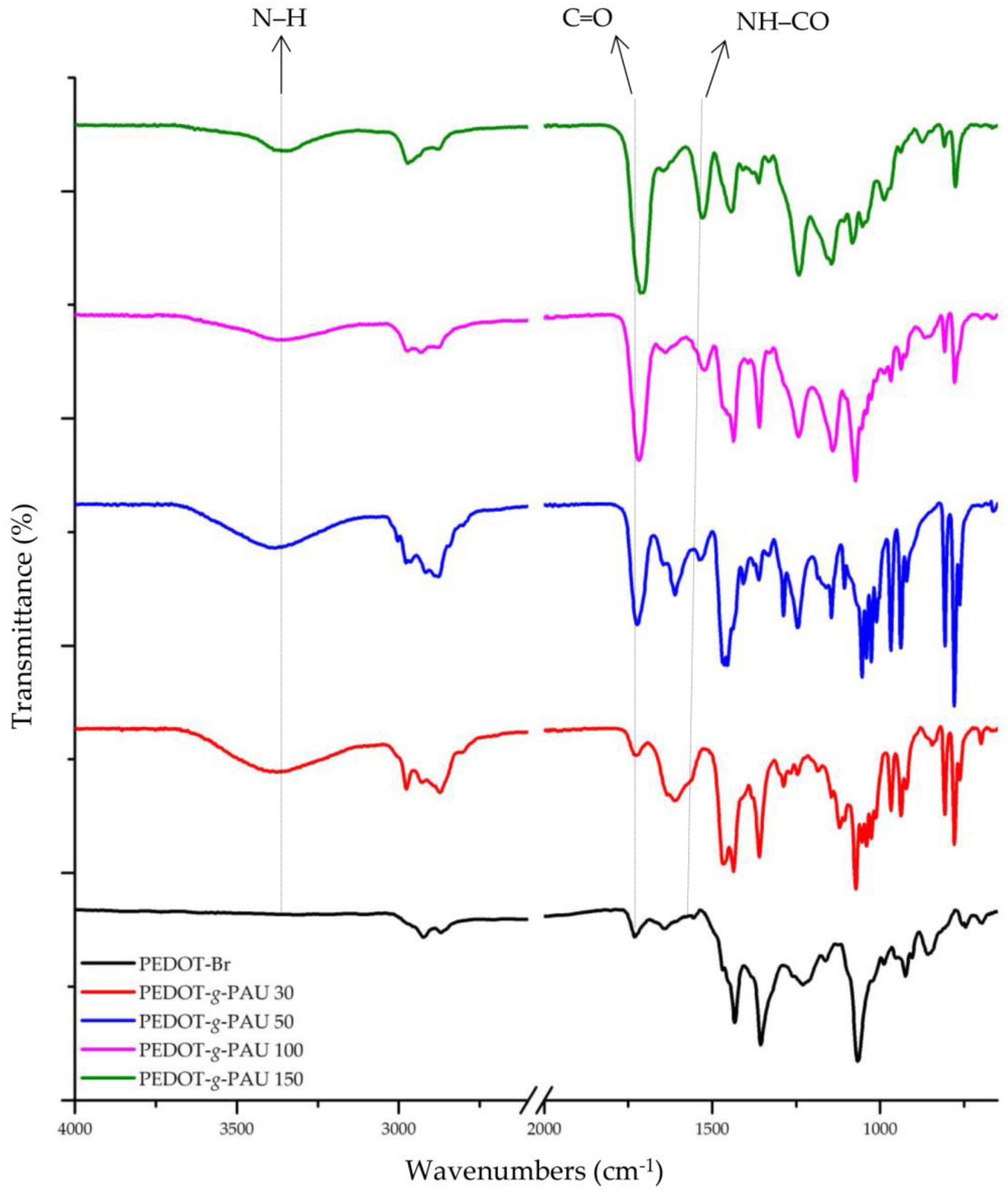
3.1. Fourier-Transform Infrared Spectroscopy (FTIR)
3.2. Nuclear Magnetic Resonances (NMR)
3.3. Gel Permeation Chromatography (GPC)
3.4. Differential Scanning Calorimetry (DSC)
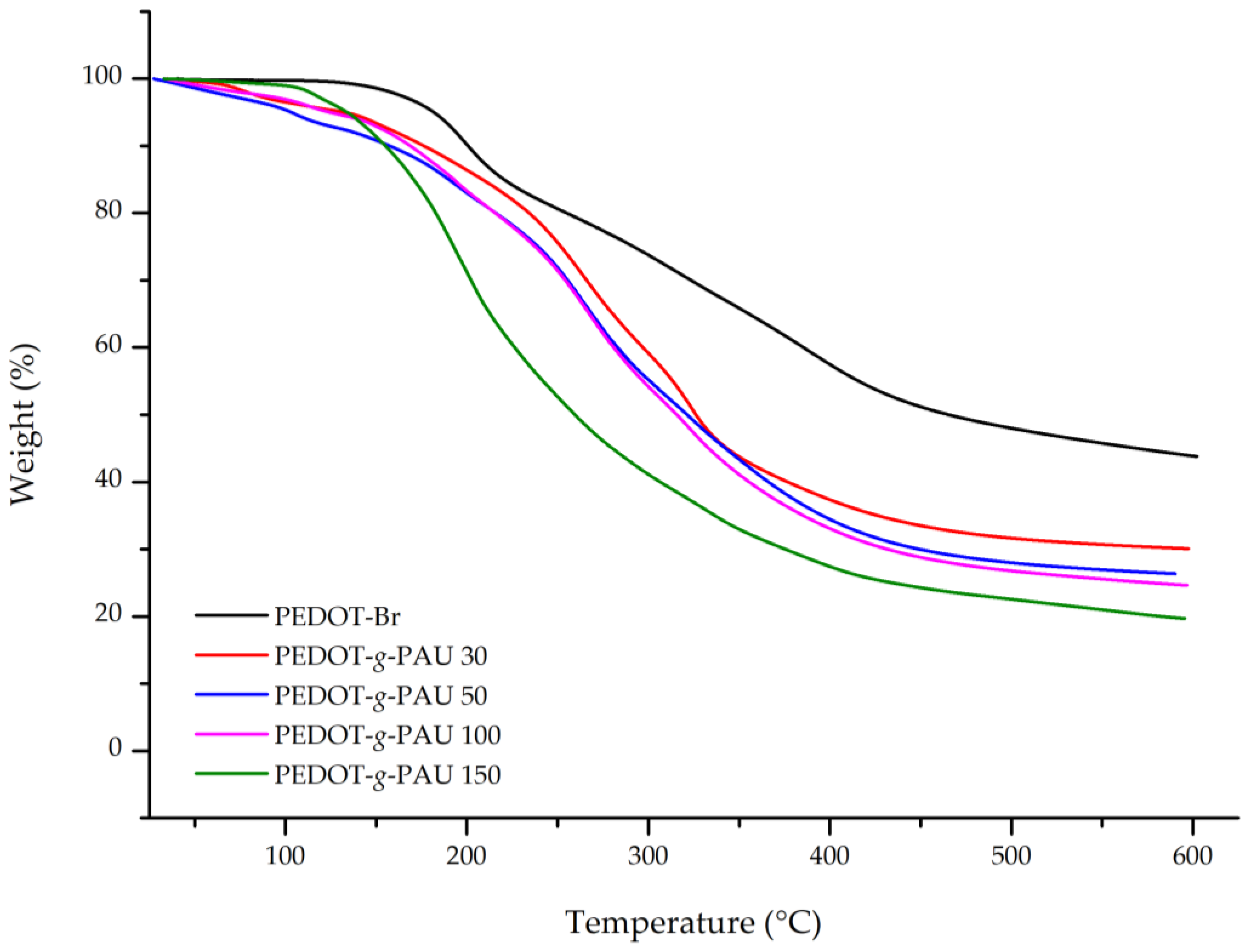
3.5. Thermogravimetric Analysis (TGA)
3.6. Four-Point Probe Method (4PP)
3.7. Tensile Test
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spanu, A.; Casula, G.; Cosseddu, P.; Lai, S.; Martines, L.; Pani, D.; Bonfiglio, A. 20—Flexible and wearable monitoring systems for biomedical applications in organic flexible electronics: Fundamentals, devices, and applications. In Organic Flexible Electronics, 1st ed.; Cosseddu, P., Caironi, M., Eds.; Woodhead Publishing: Cambridge, UK, 2021; pp. 599–625. [Google Scholar] [CrossRef]
- Zhou, J.; Men, D.; Zhang, X.-E. Progress in wearable sweat sensors and their applications. Chin. J. Anal. Chem. 2022, 50, 87–96. [Google Scholar] [CrossRef]
- Won, P.; Jeong, S.; Majidi, C.; Ko, S.H. Recent advances in liquid-metal-based wearable electronics and materials. iScience 2021, 24, 102698. [Google Scholar] [CrossRef]
- Park, M.; Park, J.; Jeong, U. Design of conductive composite elastomers for stretchable electronics. Nano Today 2014, 9, 244–260. [Google Scholar] [CrossRef]
- Xu, K.; Lu, Y.; Takei, K. Multifunctional Skin-Inspired Flexible Sensor Systems for Wearable Electronics. Adv. Mater. Technol. 2019, 4, 1800628. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, H.; Louis, E.J.; MacDiarmid, A.G.; Chiang, C.K.; Heeger, A.J. Synthesis of electrically conducting organic polymers: Halogen derivatives of polyacetylene, (CH)x. J. Chem. Soc. Chem. Commun. 1977, 16, 578–580. [Google Scholar] [CrossRef]
- Mokhtar, S.M.A.; Alvarez de Eulate, E.; Yamada, M.; Prow, T.W.; Evans, D.R. Conducting polymers in wearable devices. Med. Devices Sens. 2021, 4, e10160. [Google Scholar] [CrossRef]
- Chapman, C.A.R.; Cuttaz, E.A.; Goding, J.A.; Green, R.A. Actively controlled local drug delivery using conductive polymer-based devices. Appl. Phys. Lett. 2020, 116, 010501. [Google Scholar] [CrossRef] [Green Version]
- Talikowska, M.; Fu, X.; Lisak, G. Application of conducting polymers to wound care and skin tissue engineering: A review. Biosens. Bioelectron. 2019, 135, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Puiggalí-Jou, A.; del Valle, L.J.; Alemán, C. Drug delivery systems based on intrinsically conducting polymers. J. Control. Release 2019, 309, 244–264. [Google Scholar] [CrossRef]
- Park, Y.; Jung, J.; Chang, M. Research Progress on Conducting Polymer-Based Biomedical Applications. Appl. Sci. 2019, 9, 1070. [Google Scholar] [CrossRef] [Green Version]
- Nezakati, T.; Seifalian, A.; Tan, A.; Seifalian, A.M. Conductive Polymers: Opportunities and Challenges in Biomedical Applications. Chem. Rev. 2018, 118, 6766–6843. [Google Scholar] [CrossRef] [PubMed]
- Tajik, S.; Beitollahi, H.; Nejad, F.G.; Shoaie, I.S.; Khalilzadeh, M.A.; Asl, M.S.; Le, Q.V.; Zhang, K.; Jang, H.W.; Shokouhimehr, M. Recent developments in conducting polymers: Applications for electrochemistry. RSC Adv. 2020, 10, 37834–37856. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L.A.; Hillier, J.K.; Burchell, M.J.; Armes, S.P. Space science applications for conducting polymer particles: Synthetic mimics for cosmic dust and micrometeorites. Chem. Commun. 2015, 51, 16886–16899. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.; Park, K.S.; Baek, J.; Oh, H.S.; Lee, Y.E.K.; Sung, M.M. Single-crystal poly(3,4-ethylenedioxythiophene) nanowires with ultrahigh conductivity. Nano Lett. 2014, 14, 3321–3327. [Google Scholar] [CrossRef]
- Shi, H.; Liu, C.; Jiang, Q.; Xu, J. Effective Approaches to Improve the Electrical Conductivity of PEDOT:PSS: A Review. Adv. Electron. Mater. 2015, 1, 1500017. [Google Scholar] [CrossRef]
- Kee, S.; Kim, N.; Kim, B.S.; Park, S.; Jang, Y.H.; Lee, S.H.; Kim, J.; Kim, J.; Kwon, S.; Lee, K. Controlling Molecular Ordering in Aqueous Conducting Polymers Using Ionic Liquids. Adv. Mater. 2016, 28, 8625–8631. [Google Scholar] [CrossRef]
- Marina, S.; Mantione, D.; ManojKumar, K.; Kari, V.; Gutierrez, J.; Tercjak, A.; Sanchez-Sanchez, A.; David Mecerreyes, D. New electroactive macromonomers and multi-responsive PEDOT graft copolymers. Polym. Chem. 2018, 9, 3780–3790. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Li, M.; Yang, K.; Xiong, Z.; Yang, B.; Wang, M.; Tang, X.; Zang, Z.; Liu, X.; Li, B.; et al. PEDOT:PSS monolayers to enhance the hole extraction and stability of perovskite solar cells. J. Mater. Chem. A 2018, 6, 16583–16589. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, N.; Lv, T.; Yao, Y.; Peng, H.; Shi, J.; Cao, S.; Chen, T. Ag-Doped PEDOT:PSS/CNT composites for thin-film all-solid-state supercapacitors with a stretchability of 480%. J. Mater. Chem. A 2018, 6, 941–947. [Google Scholar] [CrossRef]
- Liang, A.; Li, D.; Zhou, W.; Wu, Y.; Ye, G.; Wu, J.; Chang, Y.; Wang, R.; Xu, J.; Nie, G.; et al. Robust flexible WS2/PEDOT:PSS film for use in high-performance miniature supercapacitors. J. Electroanal. Chem 2018, 824, 136–146. [Google Scholar] [CrossRef]
- Xu, S.; Hong, M.; Shi, X.-L.; Wang, Y.; Ge, L.; Bai, Y.; Wang, L.; Dargusch, M.; Zou, J.; Chen, Z.-G. High-Performance PEDOT:PSS Flexible Thermoelectric Materials and Their Devices by Triple Post-Treatments. Chem. Mater. 2019, 31, 5238–5244. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.H.; Wang, G.J.N.; Zhu, C.X.; Luo, S.C.; Jin, L.H.; Gu, X.D.; Chen, S.C.; Feig, V.R.; To, J.W.F.; et al. Highly stretchable polymer semiconductor films through the nanoconfinement effect. Science 2017, 355, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, T.; Niittynen, J.; Kankkunen, T.; Kraft, T.M.; Mäntysalo, M. Inkjet-printed graphene/PEDOT:PSS temperature sensors on a skin-conformable polyurethane substrate. Sci. Rep. 2016, 6, 1500017. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Swetha, P.; Zhu, Y. Nanomaterial-Enabled Wearable Sensors for Healthcare. Adv. Healthc. Mater. 2018, 7, 1700889. [Google Scholar] [CrossRef]
- Çetin, M.Z.; Camurlu, P. An amperometric glucose biosensor based on PEDOT nanofibers. RSC Adv. 2018, 8, 19724–19731. [Google Scholar] [CrossRef] [Green Version]
- Savagatrup, S.; Printz, A.D.; O’Connor, T.F.; Zaretski, A.V.; Lipomi, D.J. Molecularly stretchable electronics. Chem. Mater. 2014, 26, 3028–3041. [Google Scholar] [CrossRef]
- Savagatrup, S.; Printz, A.D.; Rodriquez, D.; Lipomi, D.J. Best of both worlds: Conjugated polymers exhibiting good photovoltaic behavior and high tensile elasticity. Macromolecules 2014, 47, 1981–1992. [Google Scholar] [CrossRef]
- Müller, C.; Goffri, S.; Breiby, D.W.; Andreasen, J.W.; Chanzy, H.D.; Janssen, R.A.J.; Nielsen, M.M.; Radano, C.P.; Sirringhaus, H.; Smith, P.; et al. Tough, semiconducting polyethylene-poly(3-hexylthiophene) diblock copolymers. Adv. Funct. Mater. 2007, 17, 2674–2679. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [Green Version]
- Yuk, H.; Zhang, T.; Lin, S.; Parada, G.A.; Zhao, X. Tough bonding of hydrogels to diverse non-porous surfaces. Nat. Mater. 2016, 15, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Xu, W.; Wang, W.; Fong, H.; Zhu, Z. Scalable and Facile Preparation of Highly Stretchable Electrospun PEDOT:PSS@PU Fibrous Nonwovens toward Wearable Conductive Textile Applications. ACS Appl. Mater. Interfaces 2017, 9, 30014–30023. [Google Scholar] [CrossRef]
- Taroni, P.J.; Santagiuliana, G.; Wan, K.; Calado, P.; Qiu, M.; Zhang, H.; Pugno, N.M.; Palma, M.; Stingelin-Stutzman, N.; Heeney, M.; et al. Toward Stretchable Self-Powered Sensors Based on the Thermoelectric Response of PEDOT:PSS/Polyurethane Blends. Adv. Funct. Mater. 2018, 28, 1704285. [Google Scholar] [CrossRef]
- Baek, P.; Aydemir, N.; An, Y.; Chan, E.W.C.; Sokolova, A.; Nelson, A.; Mata, J.P.; McGillivray, D.; Barker, D.; Travas-Sejdic, J. Molecularly Engineered Intrinsically Healable and Stretchable Conducting Polymers. Chem. Mater. 2017, 29, 8850–8858. [Google Scholar] [CrossRef]
- Ramírez-Gómez, M.A.; Guzmán-Rabadán, K.K.; González-Juárez, E.; Güizado-Rodríguez, M.; Ramos-Ortiz, G.; Alba-Rosales, J.E.; Panzo-Medrano, H.; Barba, V.; Rodríguez, M.; Maldonado, J.L.; et al. Physicochemical and Luminescent Properties of Copolymers Composed of Three Monomers: Polythiophenes Based on 3-Hexylthiophene and 3,4-Ethylenedioxythiophene. Int. J. Polym. Sci. 2017, 2017, 1918602. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, H.; Lin, Y.; Xu, Y.; Ruan, Y.; Weng, W.; Xia, H. A simple and versatile approach to self-healing polymers and electrically conductive composites. RSC Adv. 2015, 5, 13261–13269. [Google Scholar] [CrossRef]
- Wang, M.; Kee, S.; Barker, D.; Travas-Sejdic, J. Highly stretchable, solution-processable, and crosslinkable poly(3,4-ethylenedioxithiophene)-based conjugated polymers. Eur. Polym. J. 2020, 125, 109508. [Google Scholar] [CrossRef]
- Raic, M.; Sačer, D.; Rokovic, M.K. Structural and capacitive properties of graphene obtained by a green method of graphene oxide reduction. Chem. Biochem. Eng. Q. 2019, 33, 385–393. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to read and interpret ftir spectroscope of organic material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom transfer radical polymerization: From mechanisms to applications. Isr. J. Chem. 2012, 52, 206–220. [Google Scholar] [CrossRef]
- Zhang, H.R.; Pang, H.; Zhang, L.; Chen, X.; Liao, B. Biodegradability of Polyurethane Foam from Liquefied Wood Based Polyols. J. Polym. Environ. 2013, 21, 329–334. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assignment (cm−1) | NH | C–H | C=O | NH–CO | C–C, C=C Thiophene Ring | C–N | C–O | C–S |
|---|---|---|---|---|---|---|---|---|
| ThBr | − | 2975, 2864 | 1732 | − | 1462, 1389 | − | 1160 | 983 |
| PEDOT-Br | − | 2924, 2869 | 1730 | − | 1433, 1355 | − | 1163 | 988 |
| AU | 3346 | 2976, 2882 | 1703 | 1528 | − | 1243 | 1187 | − |
| PEDOT-g-PAU 30 | 3374 | 2977, 2870 | 1726 | 1564 | 1436, 1360 | 1246 | 1145 | 988 |
| PEDOT-g-PAU 50 | 3381 | 2971, 2875 | 1723 | 1533 | 1464, 1361 | 1245 | 1145 | 986 |
| PEDOT-g-PAU 100 | 3360 | 2975, 2872 | 1716 | 1521 | 1436, 1360 | 1243 | 1140 | 985 |
| PEDOT-g-PAU 150 | 3360 | 2972, 2876 | 1709 | 1529 | 1442, 1362 | 1241 | 1144 | 988 |
| Sample | Mn | Mw | Đ |
|---|---|---|---|
| PEDOT-Br | 84 477 ± 1151 | 129 250 ± 3484 | 1.53 ± 0.03 |
| PEDOT-g-PAU 30 | 116 434 ± 1541 | 140 885 ± 983 | 1.21 ± 0.01 |
| PEDOT-g-PAU 50 | 131 770 ± 2606 | 162 077 ± 6120 | 1.23 ± 0.03 |
| PEDOT-g-PAU 100 | 175 758 ± 3740 | 227 020 ± 5941 | 1.29 ± 0.04 |
| PEDOT-g-PAU 150 | 193 655 ± 5477 | 263 370 ± 6178 | 1.36 ± 0.01 |
| Sample | T90 (°C) | T50 (°C) | Residue (%) |
|---|---|---|---|
| PEDOT-Br | 200.7 | 464.9 | 41.4 |
| PEDOT-g-PAU 30 | 176.3 | 326.3 | 29.0 |
| PEDOT-g-PAU 50 | 157.2 | 320.8 | 25.2 |
| PEDOT-g-PAU 100 | 168.9 | 315.3 | 23.3 |
| PEDOT-g-PAU 150 | 153.0 | 259.6 | 19.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiket, L.; Božičević, M.; Brkić, L.; Žagar, P.; Horvat, A.; Katančić, Z. Intrinsically Stretchable Poly(3,4-ethylenedioxythiophene) Conducting Polymer Film for Flexible Electronics. Polymers 2022, 14, 2340. https://doi.org/10.3390/polym14122340
Fiket L, Božičević M, Brkić L, Žagar P, Horvat A, Katančić Z. Intrinsically Stretchable Poly(3,4-ethylenedioxythiophene) Conducting Polymer Film for Flexible Electronics. Polymers. 2022; 14(12):2340. https://doi.org/10.3390/polym14122340
Chicago/Turabian StyleFiket, Lucija, Marin Božičević, Lana Brkić, Patricia Žagar, Anamarija Horvat, and Zvonimir Katančić. 2022. "Intrinsically Stretchable Poly(3,4-ethylenedioxythiophene) Conducting Polymer Film for Flexible Electronics" Polymers 14, no. 12: 2340. https://doi.org/10.3390/polym14122340
APA StyleFiket, L., Božičević, M., Brkić, L., Žagar, P., Horvat, A., & Katančić, Z. (2022). Intrinsically Stretchable Poly(3,4-ethylenedioxythiophene) Conducting Polymer Film for Flexible Electronics. Polymers, 14(12), 2340. https://doi.org/10.3390/polym14122340