
Blood-Vessel-Inspired Hierarchical Trilayer Scaffolds: PCL/Gelatin-Driven Protein Adsorption and Cellular Interaction

 ,
,  ,
,  ,
, 
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PCL Oxidation
2.3. O-PCL Physicochemical Characterization
Biocompatibility
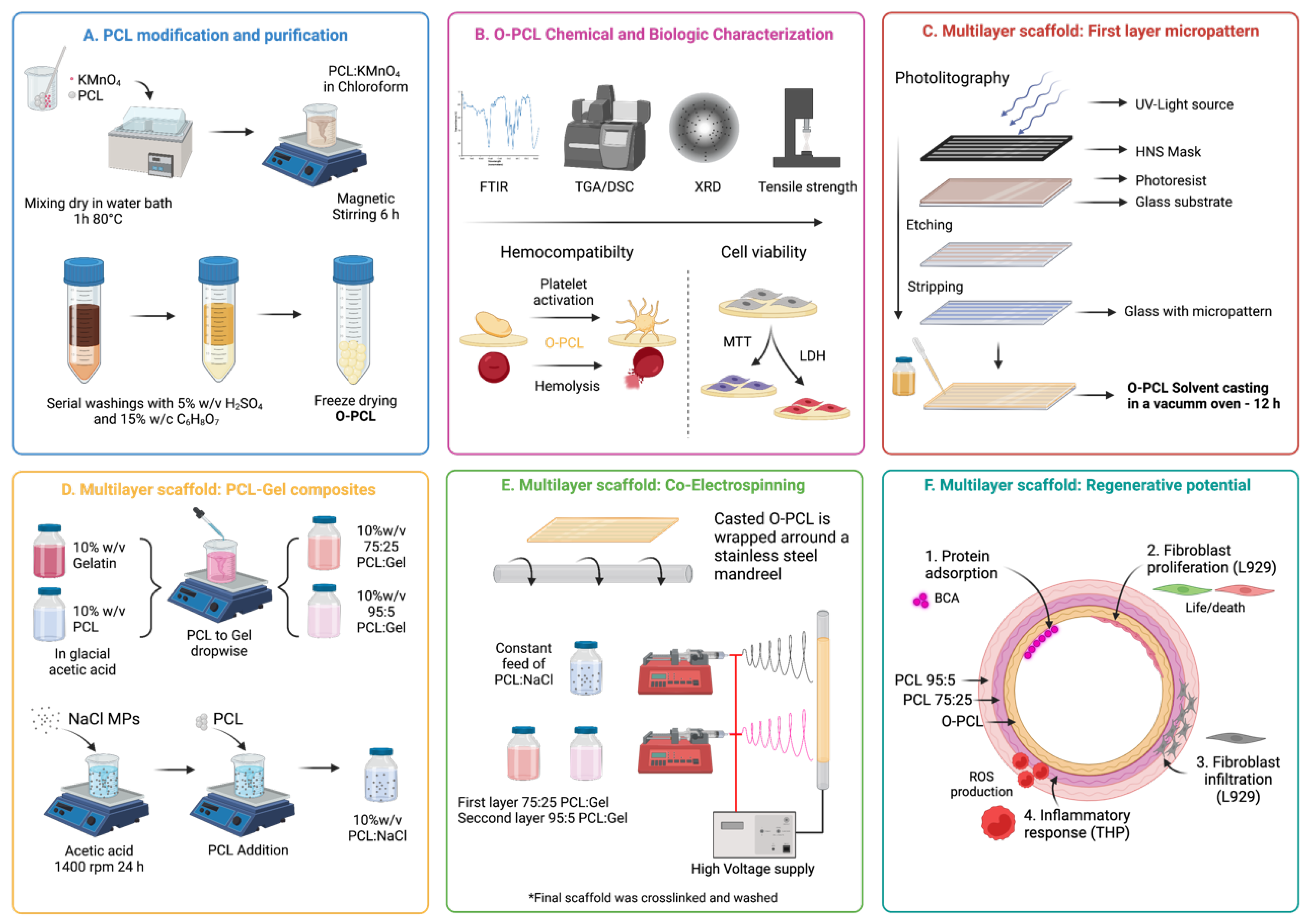
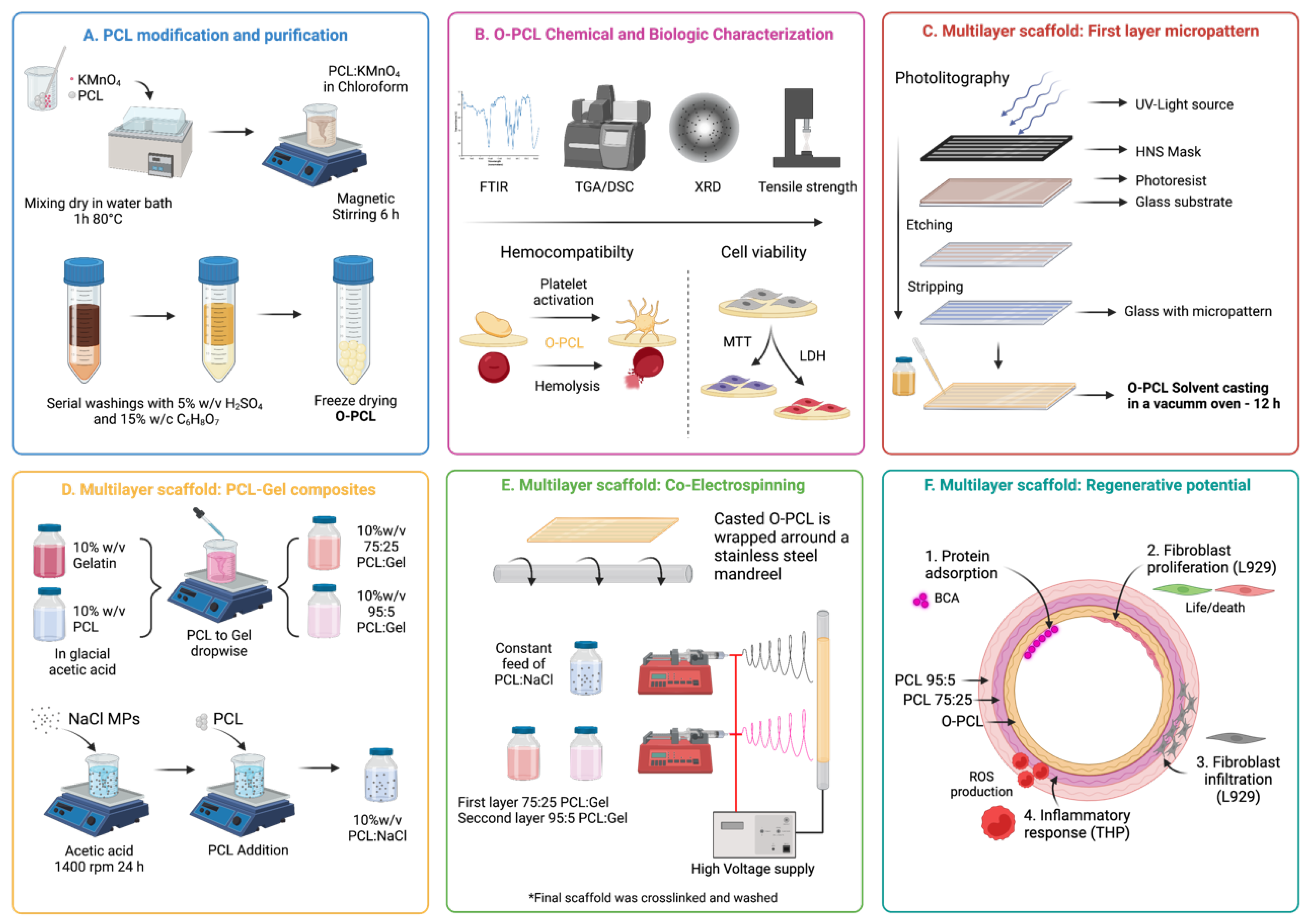
2.4. Multilayer Scaffold Fabrication
2.4.1. Fabrication of Microgrooved First Layer
2.4.2. Blends Preparation
2.4.3. Electrospinning
2.5. Scanning Electron Microscopy
2.6. Protein Adsorption Capacity
2.7. Secondary Conformational Changes of Adsorbed Proteins
2.8. Protein Adsorption Kinetics and Isotherms
2.9. Tensile Strength Test
2.10. In Vitro Cell Morphology and Proliferation
2.11. Cell Viability
2.12. Intracellular Oxidative Stress Levels
2.13. Statistical Analysis
3. Results
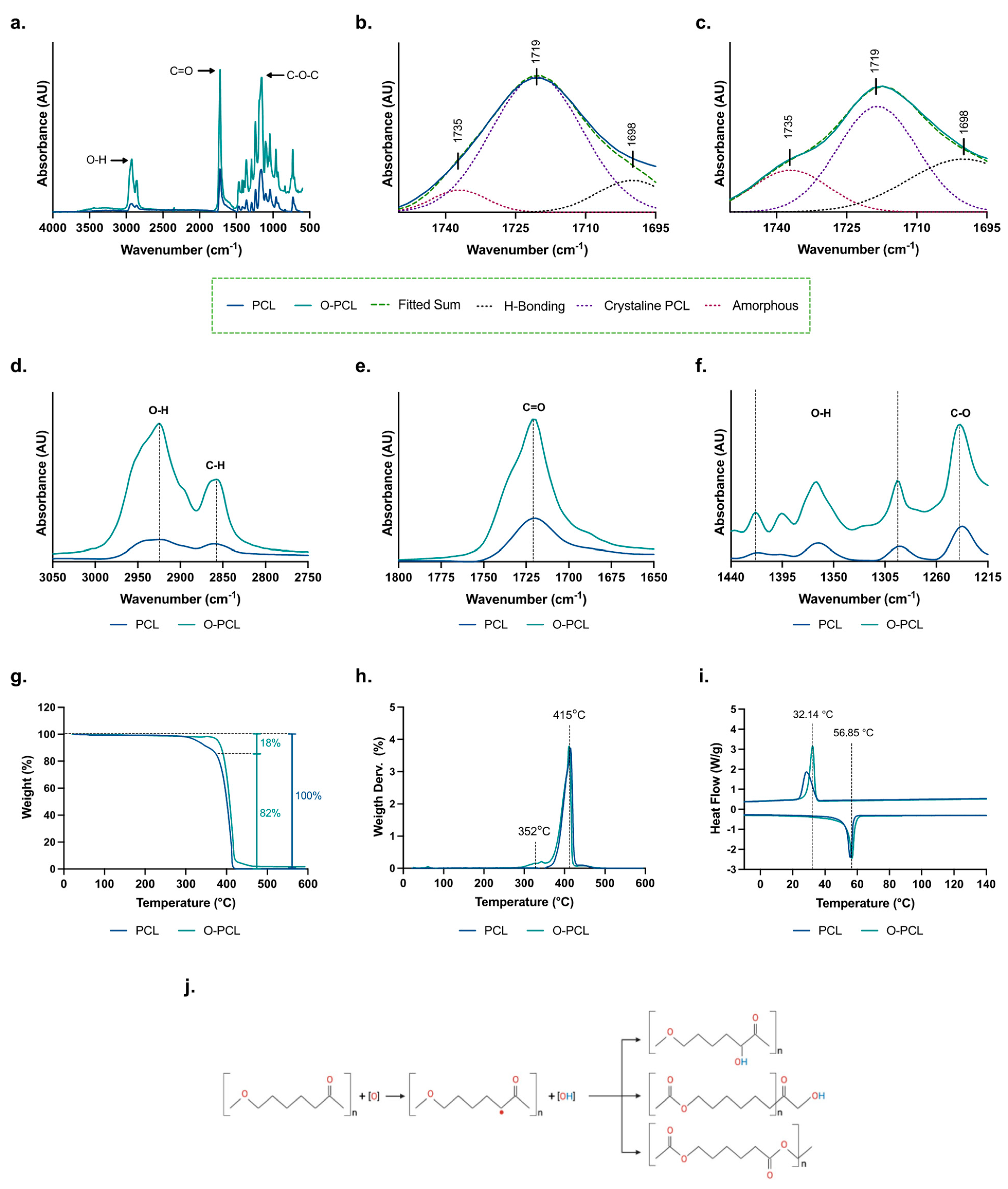
3.1. PCL Oxidation
3.1.1. FTIR Spectroscopy
3.1.2. Thermogravimetric Analysis
3.1.3. X-ray Diffraction
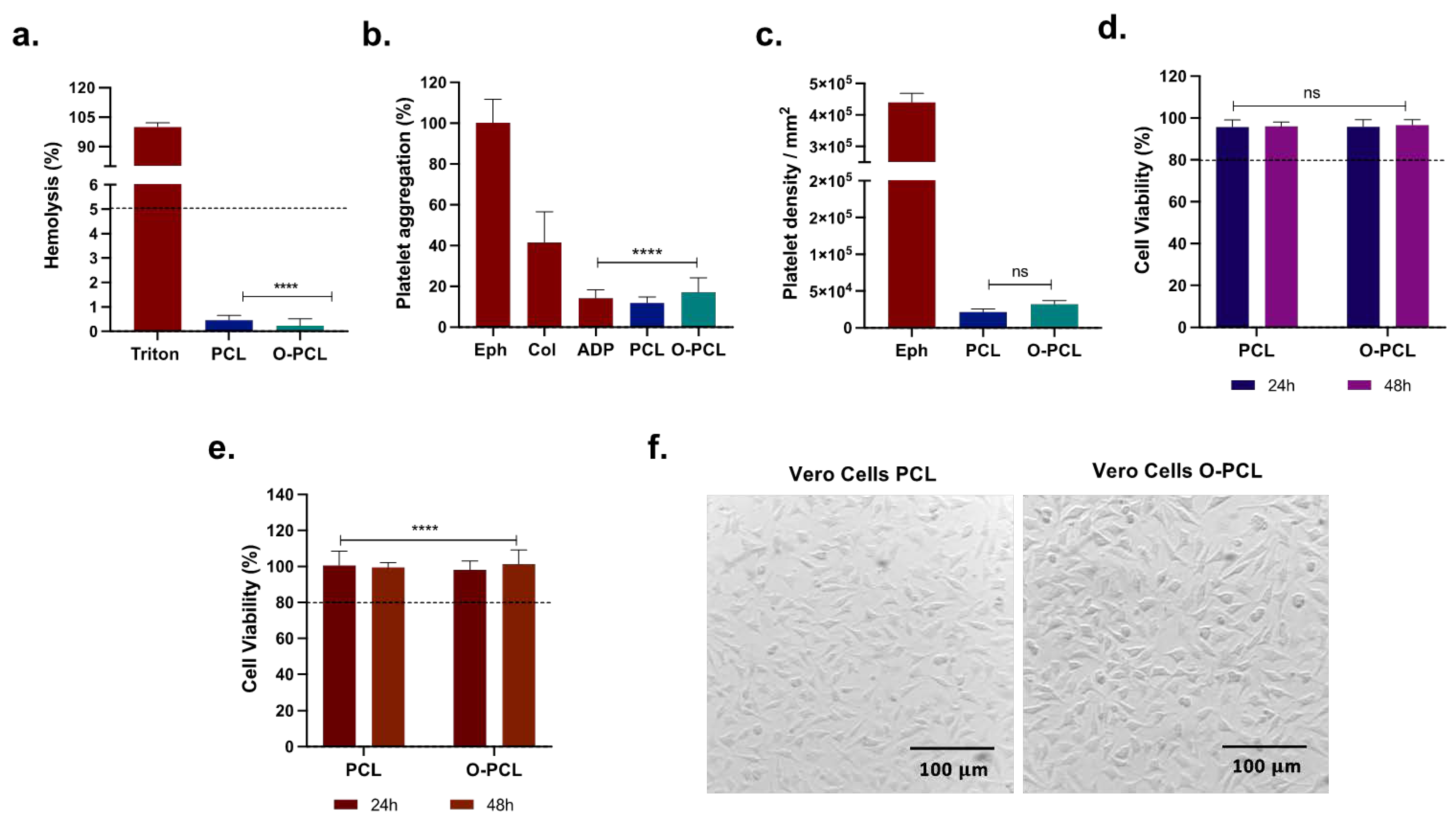
3.1.4. O-PCL Biocompatibility
3.2. Scaffold Characterization
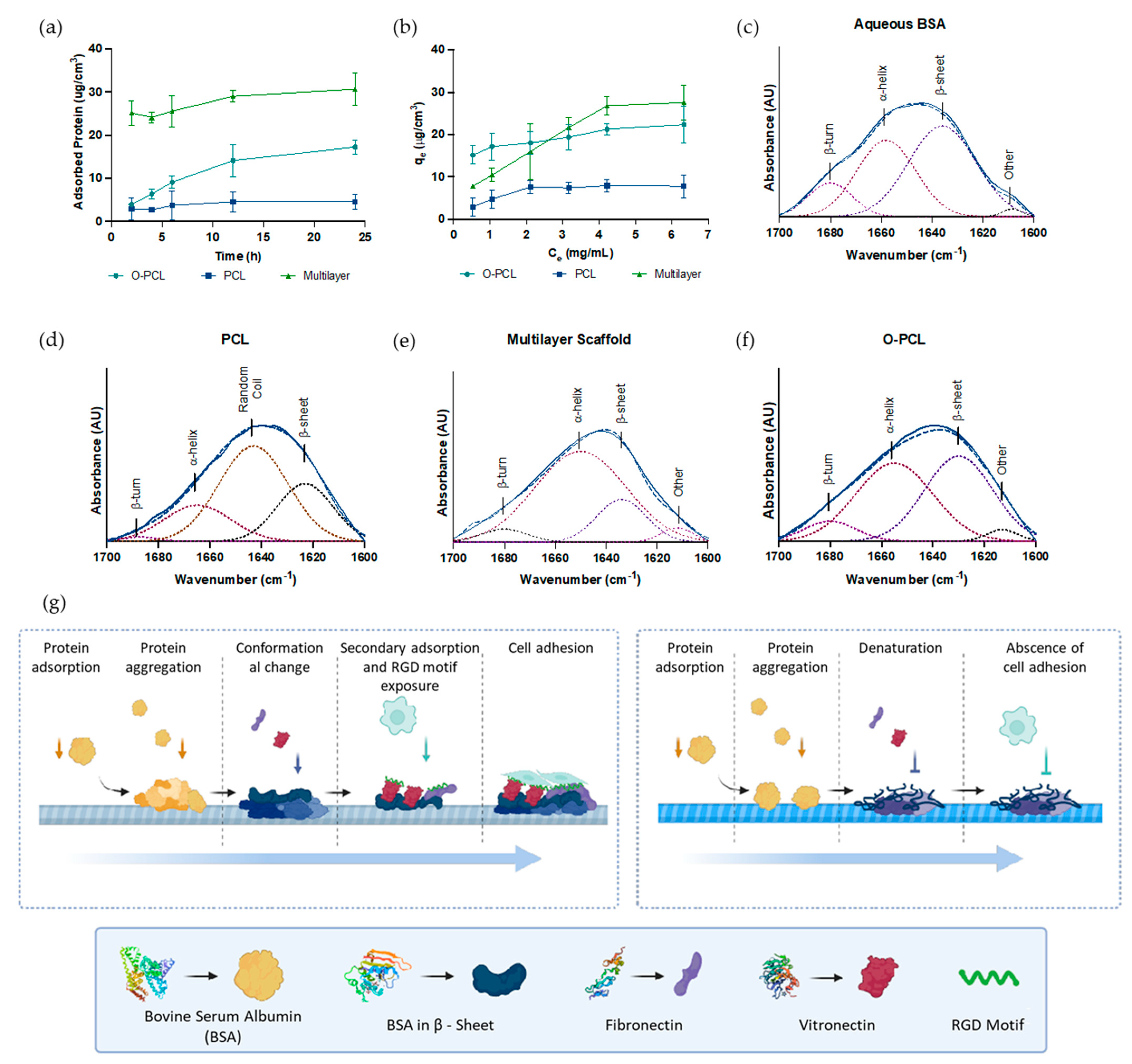
3.2.1. Protein Adsorption Capacity
3.2.2. Protein Adsorption Kinetics and Isotherms
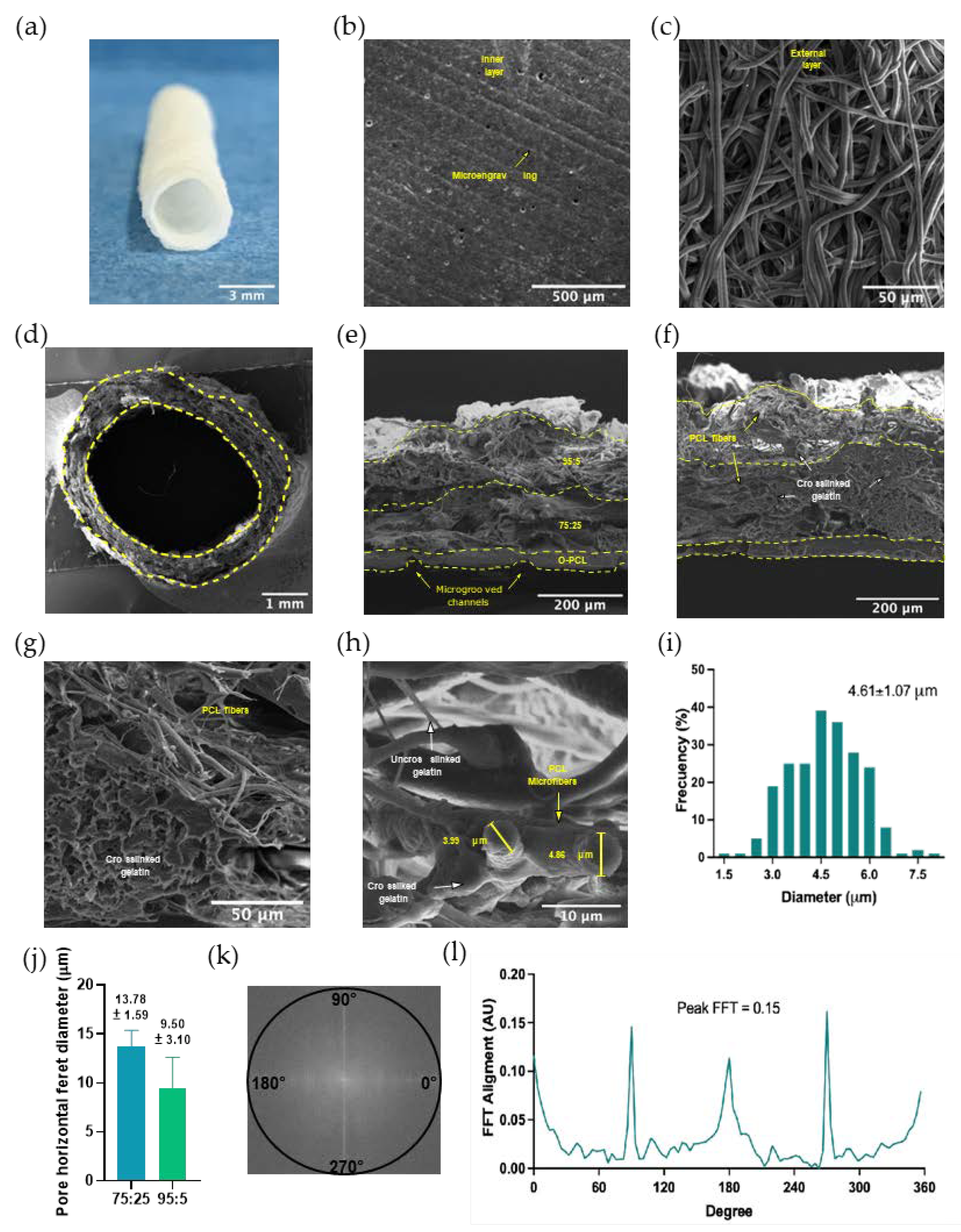
3.2.3. Micropatterning and Fiber Size and Alignment
3.3. Tensile Properties
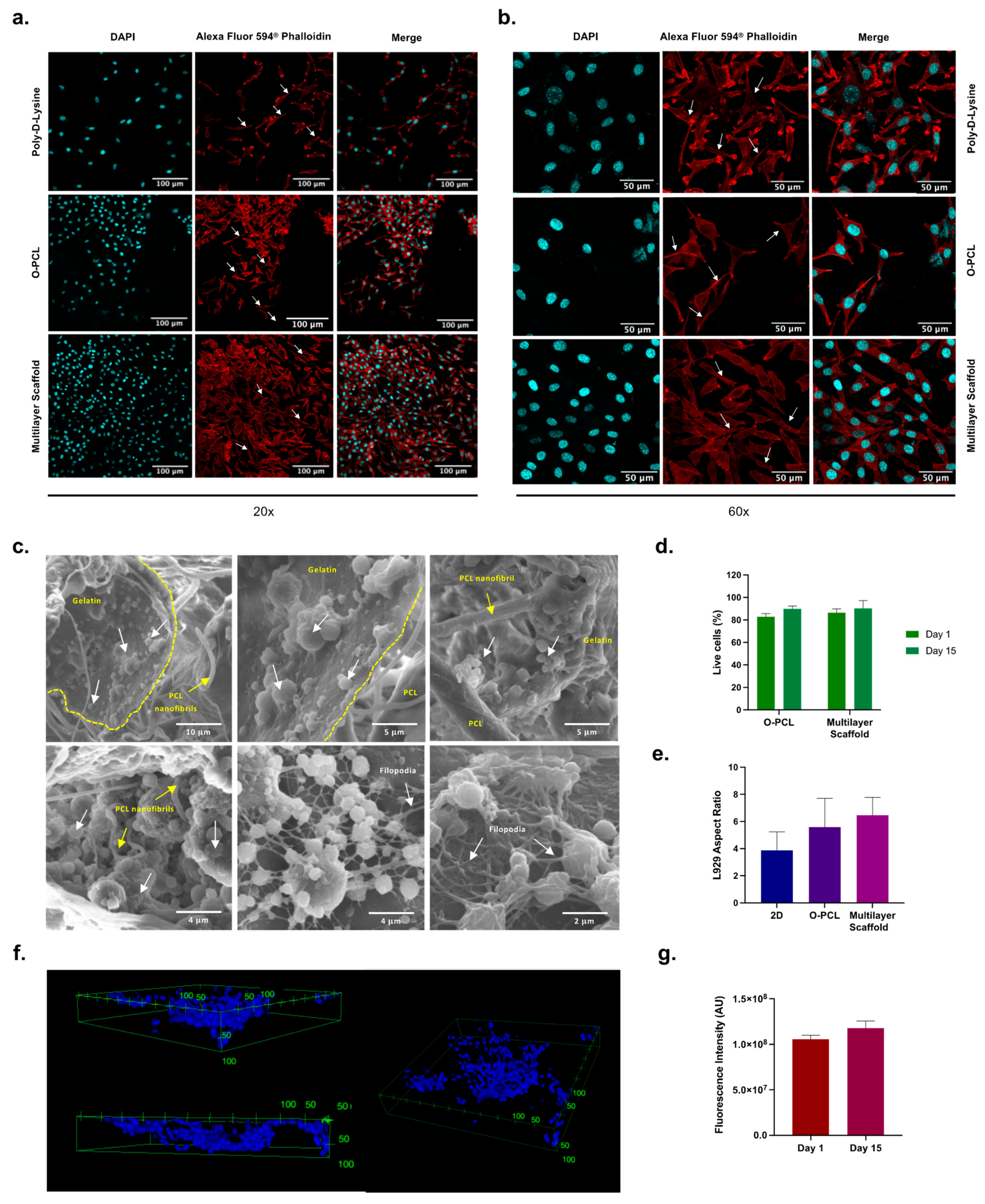
3.4. Cell Attachment Performance In Vitro
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harskamp, R.E.; Lopes, R.D.; Baisden, C.E.; de Winter, R.J.; Alexander, J.H. Saphenous Vein Graft Failure After Coronary Artery Bypass Surgery. Ann. Surg. 2013, 257, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Pashneh-Tala, S.; MacNeil, S.; Claeyssens, F. The Tissue-Engineered Vascular Graft—Past, Present, and Future. Tissue Eng. Part B Rev. 2016, 22, 68–100. [Google Scholar] [CrossRef] [PubMed]
- Tatterton, M.; Wilshaw, S.-P.; Ingham, E.; Homer-Vanniasinkam, S. The Use of Antithrombotic Therapies in Reducing Synthetic Small-Diameter Vascular Graft Thrombosis. Vasc. Endovasc. Surg. 2012, 46, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Soto, M.A.; Vargas, N.S.; Riveros, A.; Camargo, C.M.; Cruz, J.C.; Sandoval, N.; Briceño, J.C. Failure Analysis of TEVG’s I: Overcoming the Initial Stages of Blood Material Interaction and Stabilization of the Immune Response. Cells 2021, 10, 3140. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Joung, Y.K.; Lee, Y.; Bae, J.W.; Park, H.K.; Park, Y.H.; Park, J.-C.; Park, K.D. Enhanced Patency and Endothelialization of Small-Caliber Vascular Grafts Fabricated by Coimmobilization of Heparin and Cell-Adhesive Peptides. ACS Appl. Mater. Interfaces 2016, 8, 4336–4346. [Google Scholar] [CrossRef]
- Qin, L.; Yu, L.; Min, W. Mouse Models for Graft Arteriosclerosis. J. Vis. Exp. 2013, 75, e50290. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.J.; Li, X.; Lv, W.; Yang, C.; Protack, C.D.; Muto, A.; Jadlowiec, C.C.; Shu, C.; Dardik, A. Therapeutic strategies to combat neointimal hyperplasia in vascular grafts. Expert Rev. Cardiovasc. Ther. 2012, 10, 635–647. [Google Scholar] [CrossRef]
- Robert, J.; Weber, B.; Frese, L.; Emmert, M.Y.; Schmidt, D.; Von Eckardstein, A.; Rohrer, L.; Hoerstrup, S.P. A Three-Dimensional Engineered Artery Model for In Vitro Atherosclerosis Research. PLoS ONE 2013, 8, e79821. [Google Scholar] [CrossRef] [Green Version]
- Kabirian, F.; Milan, P.B.; Zamanian, A.; Heying, R.; Mozafari, M. Nitric oxide-releasing vascular grafts: A therapeutic strategy to promote angiogenic activity and endothelium regeneration. Acta Biomater. 2019, 92, 82–91. [Google Scholar] [CrossRef]
- Horst, M.; Madduri, S.; Gobet, R.; Sulser, T.; Hall, H.; Eberli, D. Scaffold Characteristics for Functional Hollow Organ Regeneration. Materials 2010, 3, 241–263. [Google Scholar] [CrossRef]
- Hasan, A.; Memic, A.; Annabi, N.; Hossain, M.; Paul, A.; Dokmeci, M.R.; Dehghani, F.; Khademhosseini, A. Electrospun scaffolds for tissue engineering of vascular grafts. Acta Biomater. 2014, 10, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.; Sankar, M.; Katiyar, V. State of Art on Solvent Casting Particulate Leaching Method for Orthopedic ScaffoldsFabrication. Mater. Today: Proc. 2017, 4, 898–907. [Google Scholar] [CrossRef]
- Adipurnama, I.; Yang, M.-C.; Ciach, T.; Butruk-Raszeja, B. Surface modification and endothelialization of polyurethane for vascular tissue engineering applications: A review. Biomater. Sci. 2016, 5, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, L.; Ren, X.-K.; Guo, J.; Xia, S.; Zhang, W.; Feng, Y. Co-immobilization of ACH11 antithrombotic peptide and CAG cell-adhesive peptide onto vascular grafts for improved hemocompatibility and endothelialization. Acta Biomater. 2019, 97, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, S.; Hou, D.; Gao, J.; Jiang, L.; Shi, J.; Liang, Q.; Kong, D.; Wang, S. Regulation of macrophage polarization and promotion of endothelialization by NO generating and PEG-YIGSR modified vascular graft. Mater. Sci. Eng. C 2018, 84, 1–11. [Google Scholar] [CrossRef]
- Hashi, C.K.; Derugin, N.; Janairo, R.R.R.; Lee, R.; Schultz, D.; Lotz, J.; Li, S. Antithrombogenic Modification of Small-Diameter Microfibrous Vascular Grafts. Arter. Thromb. Vasc. Biol. 2010, 30, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ma, B.; Yin, A.; Zhang, B.; Luo, R.; Pan, J.; Wang, Y. Polycaprolactone vascular graft with epigallocatechin gallate embedded sandwiched layer-by-layer functionalization for enhanced antithrombogenicity and anti-inflammation. J. Control. Release 2020, 320, 226–238. [Google Scholar] [CrossRef]
- Qiu, X.; Lee, B.L.-P.; Ning, X.; Murthy, N.; Dong, N.; Li, S. End-point immobilization of heparin on plasma-treated surface of electrospun polycarbonate-urethane vascular graft. Acta Biomater. 2017, 51, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, N.; Asawa, S.; Birru, B.; Baadhe, R.; Rao, S. PCL-Based Composite Scaffold Matrices for Tissue Engineering Applications. Mol. Biotechnol. 2018, 60, 506–532. [Google Scholar] [CrossRef]
- Salerno, A.; Saurina, J.; Domingo, C. Supercritical CO2 foamed polycaprolactone scaffolds for controlled delivery of 5-fluorouracil, nicotinamide and triflusal. Int. J. Pharm. 2015, 496, 654–663. [Google Scholar] [CrossRef]
- Behtouei, E.; Zandi, M.; Askari, F.; Daemi, H.; Zamanlui, S.; Arabsorkhi-Mishabi, A.; Pezeshki-Modaress, M. Bead-free and tough electrospun PCL/gelatin/PGS ternary nanofibrous scaffolds for tissue engineering application. J. Appl. Polym. Sci. 2021, 139, 51471. [Google Scholar] [CrossRef]
- Liu, T.; Liu, S.; Zhang, K.; Chen, J.; Huang, N. Endothelialization of implanted cardiovascular biomaterial surfaces: The development from in vitro to in vivo. J. Biomed. Mater. Res. Part A 2013, 102, 3754–3772. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, J.; De Lorenzi, F.; Theek, B.; Blaeser, A.; Rommel, D.; Kuehne, A.J.C.; Kießling, F.; Fischer, H. Engineering biofunctional in vitro vessel models using a multilayer bioprinting technique. Sci. Rep. 2018, 8, 10430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Shi, G.-P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Sarkola, T.; Manlhiot, C.; Slorach, C.; Bradley, T.J.; Hui, W.; Mertens, L.; Redington, A.; Jaeggi, E. Evolution of the Arterial Structure and Function From Infancy to Adolescence Is related to Anthropometric and Blood Pressure Changes. Arter. Thromb. Vasc. Biol. 2012, 32, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, D.H.J.; Carter, S.; Green, D.J. Arterial structure and function in vascular ageing: Are you as old as your arteries? J. Physiol. 2016, 594, 2275–2284. [Google Scholar] [CrossRef]
- Vaz, C.; van Tuijl, S.; Bouten, C.; Baaijens, F. Design of scaffolds for blood vessel tissue engineering using a multi-layering electrospinning technique. Acta Biomater. 2005, 1, 575–582. [Google Scholar] [CrossRef]
- Rahmati, M.; Blaker, J.; Lyngstadaas, S.; Mano, J.; Haugen, H. Designing multigradient biomaterials for skin regeneration. Mater. Today Adv. 2020, 5, 100051. [Google Scholar] [CrossRef]
- Kim, T.G.; Chung, H.J.; Park, T.G. Macroporous and nanofibrous hyaluronic acid/collagen hybrid scaffold fabricated by concurrent electrospinning and deposition/leaching of salt particles. Acta Biomater. 2008, 4, 1611–1619. [Google Scholar] [CrossRef]
- Sabino, M.A. Oxidation of polycaprolactone to induce compatibility with other degradable polyesters. Polym. Degrad. Stab. 2007, 92, 986–996. [Google Scholar] [CrossRef]
- Castilla-Cortázar, I.; Vidaurre, A.; Marí, B.; Campillo-Fernández, A.J. Morphology, Crystallinity, and Molecular Weight of Poly(ε-caprolactone)/Graphene Oxide Hybrids. Polymers 2019, 11, 1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Brand, A.J.; Morales, M.A.; Hozman, A.S.; Ramirez, A.C.; Cruz, L.J.; Maranon, A.; Muñoz-Camargo, C.; Cruz, J.C.; Porras, A. Bioactive Poly(lactic acid)–Cocoa Bean Shell Composites for Biomaterial Formulation: Preparation and Preliminary In Vitro Characterization. Polymers 2021, 13, 3707. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Saito, K.; Hearn, M.T.W. Isothermal modelling of protein adsorption to thermo-responsive polymer grafted Sepharose Fast Flow sorbents. J. Sep. Sci. 2021, 44, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi-Mobarakeh, L.; Prabhakaran, M.P.; Morshed, M.; Nasr-Esfahani, M.-H.; Ramakrishna, S. Bio-functionalized PCL nanofibrous scaffolds for nerve tissue engineering. Mater. Sci. Eng. C 2010, 30, 1129–1136. [Google Scholar] [CrossRef]
- Rueda-Gensini, L.; A Serna, J.; Cifuentes, J.; Cruz, J.C.; Muñoz-Camargo, C. Graphene Oxide-Embedded Extracellular Matrix- Derived Hydrogel as a Multiresponsive Platform for 3D Bioprinting Applications. Int. J. Bioprinting 2021, 7, 353. [Google Scholar] [CrossRef] [PubMed]
- Seredych, M.; Mikhalovska, L.; Mikhalovsky, S.; Gogotsi, Y. Adsorption of Bovine Serum Albumin on Carbon-Based Materials. C 2018, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Simonin, J.P. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics. Chem. Eng. J. 2016, 300, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Trakoolwannachai, V.; Kheolamai, P.; Ummartyotin, S. Characterization of hydroxyapatite from eggshell waste and polycaprolactone (PCL) composite for scaffold material. Compos. Part B Eng. 2019, 173, 106974. [Google Scholar] [CrossRef]
- Honma, T.; Senda, T.; Inoue, Y. Thermal properties and crystallization behaviour of blends of poly(-caprolactone) with chitin and chitosan. Polym. Int. 2003, 52, 1839–1846. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Li, B.; Wang, J.; Han, S.; Liu, L.; Wei, H. A bio-inspired nacre-like layered hybrid structure of calcium carbonate under the control of carboxyl graphene. CrystEngComm 2014, 17, 520–525. [Google Scholar] [CrossRef]
- Mohammed, Z.; Jeelani, S.; Rangari, V. Effect of Low-Temperature Plasma Treatment on Surface Modification of Polycaprolactone Pellets and Thermal Properties of Extruded Filaments. JOM 2020, 72, 1523–1532. [Google Scholar] [CrossRef]
- Sánchez-González, S.; Diban, N.; Urtiaga, A. Hydrolytic Degradation and Mechanical Stability of Poly(ε-Caprolactone)/Reduced Graphene Oxide Membranes as Scaffolds for In Vitro Neural Tissue Regeneration. Membranes 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Abdallah, A.; Kallel, A.; Gamaoun, F.; Tcharkhtchi, A. Enzymatic Hydrolysis of Poly (Caprolactone) and its Blend with Styrene–Butadiene–Styrene (40% PCL/60% SBS). J. Polym. Environ. 2019, 27, 2341–2351. [Google Scholar] [CrossRef]
- Patil, V.; Dennis, R.V.; Rout, T.K.; Banerjee, S.; Yadav, G.D. Graphene oxide and functionalized multi walled carbon nanotubes as epoxy curing agents: A novel synthetic approach to nanocomposites containing active nanostructured fillers. RSC Adv. 2014, 4, 49264–49272. [Google Scholar] [CrossRef]
- Buchanan, L.A.; El-Ghannam, A. Effect of bioactive glass crystallization on the conformation and bioactivity of adsorbed proteins. J. Biomed. Mater. Res. Part A 2009, 93, 537–546. [Google Scholar] [CrossRef]
- Hu, X.N.; Yang, B.C. Conformation change of bovine serum albumin induced by bioactive titanium metals and its effects on cell behaviors. J. Biomed. Mater. Res. Part A 2013, 102, 1053–1062. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Wang, M.; Zhang, D.; Yang, Q.; Liu, T.; Lei, R.; Zhu, S.; Zhao, Y.; Chen, C. Probing Adsorption Behaviors of BSA onto Chiral Surfaces of Nanoparticles. Small 2018, 14, e1703982. [Google Scholar] [CrossRef]
- Koblinski, J.E.; Wu, M.C.; Demeler, B.; Jacob, K.; Kleinman, H.K. Matrix cell adhesion activation by non-adhesion proteins. J. Cell Sci. 2005, 118, 2965–2974. [Google Scholar] [CrossRef] [Green Version]
- Thamma, U.; Kowal, T.J.; Falk, M.M.; Jain, H. Nanostructure of bioactive glass affects bone cell attachment via protein restructuring upon adsorption. Sci. Rep. 2021, 11, 5763. [Google Scholar] [CrossRef]
- Abstiens, K.; Figueroa, S.M.; Gregoritza, M.; Goepferich, A.M. Interaction of functionalized nanoparticles with serum proteins and its impact on colloidal stability and cargo leaching. Soft Matter 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Campaña, A.; Guillén, A.; Rivas, R.; Akle, V.; Cruz, J.; Osma, J. Functionalization and Evaluation of Inorganic Adsorbents for the Removal of Cadmium in Wastewater. Molecules 2021, 26, 4150. [Google Scholar] [CrossRef] [PubMed]
- Toledano, M.; Carrasco-Carmona, Á.; Medina-Castillo, A.L.; Toledano-Osorio, M.; Osorio, R. Protein adsorption and bioactivity of functionalized electrospun membranes for bone regeneration. J. Dent. 2020, 102, 103473. [Google Scholar] [CrossRef] [PubMed]
- Raslan, A.; Ciriza, J.; de Retana, A.M.O.; Sanjuán, M.L.; Toprak, M.S.; Galvez-Martin, P.; Saenz-Del-Burgo, L.; Pedraz, J.L. Modulation of Conductivity of Alginate Hydrogels Containing Reduced Graphene Oxide through the Addition of Proteins. Pharmaceutics 2021, 13, 1473. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Fan, J.; Chu, C.-C.; Wu, J. Electrospinning of small diameter 3-D nanofibrous tubular scaffolds with controllable nanofiber orientations for vascular grafts. J. Mater. Sci. Mater. Med. 2010, 21, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, J.; Lee, M.-K.; Park, C.; Jung, H.-D.; Kim, H.-E.; Jang, T.-S. Fabrication of strong, bioactive vascular grafts with PCL/collagen and PCL/silica bilayers for small-diameter vascular applications. Mater. Des. 2019, 181, 108079. [Google Scholar] [CrossRef]
- Han, D.G.; Ahn, C.B.; Lee, J.-H.; Hwang, Y.; Kim, J.H.; Park, K.Y.; Lee, J.W.; Son, K.H. Optimization of Electrospun Poly(caprolactone) Fiber Diameter for Vascular Scaffolds to Maximize Smooth Muscle Cell Infiltration and Phenotype Modulation. Polymers 2019, 11, 643. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, D.; Goedecke, N.; Snedeker, J.; Ferguson, S. Mechanical evaluation of electrospun poly(ε-caprolactone) single fibers. Mater. Today Commun. 2020, 24, 101211. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Geever, L.M.; Higginbotham, C.L.; Devine, D.M. Analysis of the Mechanical Properties of Solvent Cast Blends of PLA/PCL. Appl. Mech. Mater. 2014, 679, 50–56. [Google Scholar] [CrossRef]
- Al-Baadani, M.A.; Yie, K.H.R.; Al-Bishari, A.M.; Alshobi, B.A.; Zhou, Z.; Fang, K.; Dai, B.; Shen, Y.; Ma, J.; Liu, J.; et al. Co-electrospinning polycaprolactone/gelatin membrane as a tunable drug delivery system for bone tissue regeneration. Mater. Des. 2021, 209, 109962. [Google Scholar] [CrossRef]
- Yao, R.; He, J.; Meng, G.; Jiang, B.; Wu, F. Electrospun PCL/Gelatin composite fibrous scaffolds: Mechanical properties and cellular responses. J. Biomater. Sci. Polym. Ed. 2016, 27, 824–838. [Google Scholar] [CrossRef]
- Saharudin, M.S.; Atif, R.; Shyha, I.; Inam, F. The degradation of mechanical properties in polymer nano-composites exposed to liquid media—A review. RSC Adv. 2016, 6, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Wang, Q.; Wang, T. The effects of crystallinity on the mechanical properties and the limiting PV (pressure×velocity) value of PTFE. Tribol. Int. 2016, 93, 1–10. [Google Scholar] [CrossRef]
- Dwivedi, R.; Kumar, S.; Pandey, R.; Mahajan, A.; Nandana, D.; Katti, D.S.; Mehrotra, D. Polycaprolactone as biomaterial for bone scaffolds: Review of literature. J. Oral Biol. Craniofacial Res. 2019, 10, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.R.; Serra, T.; Oliveira, M.I.; Planell, J.A.; Barbosa, M.A.; Navarro, M. Impact of 3-D printed PLA- and chitosan-based scaffolds on human monocyte/macrophage responses: Unraveling the effect of 3-D structures on inflammation. Acta Biomater. 2014, 10, 613–622. [Google Scholar] [CrossRef]
- Provin, C.; Takano, K.; Sakai, Y.; Fujii, T.; Shirakashi, R. A method for the design of 3D scaffolds for high-density cell attachment and determination of optimum perfusion culture conditions. J. Biomech. 2008, 41, 1436–1449. [Google Scholar] [CrossRef]
- Xiang, L.; Cui, W. Biomedical application of photo-crosslinked gelatin hydrogels. J. Leather Sci. Eng. 2021, 3, 770049. [Google Scholar] [CrossRef]
- Wattamwar, P.P.; Mo, Y.; Wan, R.; Palli, R.; Zhang, Q.; Dziubla, T.D. Antioxidant Activity of Degradable Polymer Poly(trolox ester) to Suppress Oxidative Stress Injury in the Cells. Adv. Funct. Mater. 2010, 20, 147–154. [Google Scholar] [CrossRef]
- Lu, X.; Guo, X.; Wassall, C.D.; Kemple, M.D.; Unthank, J.L.; Kassab, G.S. Reactive oxygen species cause endothelial dysfunction in chronic flow overload. J. Appl. Physiol. 2011, 110, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.F.; Ma, M.; Bratlie, K.M.; Dang, T.; Langer, R.; Anderson, D.G. Real-time in vivo detection of biomaterial-induced reactive oxygen species. Biomaterials 2011, 32, 1796–1801. [Google Scholar] [CrossRef] [Green Version]
- Blanchette, J.; Jabbarzadeh, E. Modulation of Inflammatory Response to Implanted Biomaterials Using Natural Compounds. Curr. Pharm. Des. 2018, 23, 6347–6357. [Google Scholar] [CrossRef] [Green Version]
- Bracaglia, L.G.; Fisher, J.P. Extracellular Matrix-Based Biohybrid Materials for Engineering Compliant, Matrix-Dense Tissues. Adv. Heal. Mater. 2015, 4, 2475–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barro, R.; Regueiro, J.; Llompart, M.; Garcia-Jares, C. Analysis of industrial contaminants in indoor air: Part 1. Volatile organic compounds, carbonyl compounds, polycyclic aromatic hydrocarbons and polychlorinated biphenyls. J. Chromatogr. A 2009, 1216, 540–566. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Lee, B.H.; Irvine, S.A.; An, J.; Bhuthalingam, R.; Singh, V.; Low, K.Y.; Chua, C.K.; Venkatraman, S. Smooth Muscle Cell Alignment and Phenotype Control by Melt Spun Polycaprolactone Fibers for Seeding of Tissue Engineered Blood Vessels. Int. J. Biomater. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, G.; Li, M.; Wang, L.; Elson, E.L.; Lu, T.J.; Genin, G.M.; Xu, F. An approach to quantifying 3D responses of cells to extreme strain. Sci. Rep. 2016, 6, 19550. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Badylak, S.F. Phenotypic changes in cultured smooth muscle cells: Limitation or opportunity for tissue engineering of hollow organs? J. Tissue Eng. Regen. Med. 2012, 6, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Stradins, P.; Lācis, R.; Ozolanta, I.; Purina, B.; Ose, V.; Feldmane, L.; Kasyanov, V. Comparison of biomechanical and structural properties between human aortic and pulmonary valve. Eur. J. Cardio-Thoracic Surg. 2004, 26, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.; Lee, H.; Kim, G. A Surface-Modified Poly(ε-caprolactone) Scaffold Comprising Variable Nanosized Surface-Roughness Using a Plasma Treatment. Tissue Eng. Part C Methods 2014, 20, 951–963. [Google Scholar] [CrossRef] [Green Version]
- Sownthari, K.; Suthanthiraraj, S.A. Synthesis and characterization of an electrolyte system based on a biodegradable polymer. Express Polym. Lett. 2013, 7, 495–504. [Google Scholar] [CrossRef]
- Borjigin, M.; Eskridge, C.; Niamat, R.; Strouse, B.; Bialk, P.; Kmiec, E.B. Electrospun fiber membranes enable proliferation of genetically modified cells. Int. J. Nanomed. 2013, 8, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.E.; Henry, L.; McGennisken, E.; Onofrillo, C.; Bella, C.D.; Duchi, S.; O’Connell, C.D.; Pirogova, E. Characterization of Polycaprolactone Nanohydroxyapatite Composites with Tunable Degradability Suitable for Indirect Printing. Polymers 2021, 13, 295. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Maximum Absorbance (cm−1) | PCL | O-PCL |
|---|---|---|---|
| Amorphous | 1735 | 14.18% | 33.73% |
| Crystaline | 1719 | 78.15% | 48.18% |
| H-bonding | 1698 | 7.67% | 18.10% |
| Sample | Cooling Scan | Second Heating Scan | Xc (%) | ||
|---|---|---|---|---|---|
| Tc (°C) | ∆Hc (J/g) | Tm (°C) | ∆Hm (J/g) | ||
| PCL | 32.41 | 62.60 | 56.85 | 61.63 | 38.40 |
| O-PCL | 28.53 | 55.50 | 55.70 | 53.56 | 34.05 |
| Samples | β-Structures (%) | α-Helix | Intermolecular β-Sheet (%) | Random Coil (%) |
|---|---|---|---|---|
| BSA | 37.4 | 51.27 | 11.86 | - |
| O-PCL | 47.36 | 44.51 | 7.60 | - |
| PCL | 44.87 | - | 1.27 | 52.93 |
| Multilayer Scaffold | 75.57 | 20.24 | 3.90 | - |
| Langmuir Model | Freundlich Model | ||||||
|---|---|---|---|---|---|---|---|
| Isotherm Parameters | PCL | O-PCL | Multilayer Scaffold | Isotherm Parameters | PCL | O-PCL | Multilayer Scaffold |
| qm (μg/cm3) | 11.12 | 23.98 | 32.15 | N | 2.68 | 5.52 | 1.79 |
| KL * | 0.15 | 0.44 | 0.12 | Kf * | 2.61 | 12.2 | 4.56 |
| RL | 0.93 | 0.81 | 0.94 | - | - | - | - |
| R2 | 0.98 | 0.87 | 0.95 | R2 | 0.85 | 0.97 | 0.97 |
| Parameters | PFO | PSO | ||||
|---|---|---|---|---|---|---|
| PCL | O-PCL | Multilayer Scaffold | PCL | O-PCL | Multilayer Scaffold | |
| qe (μg/cm3) | 3.21 | 18.05 | 31.84 | 5.03 | 25.83 | 31.84 |
| K1 (min−1) | 0.20 | 0.03 | 0.03 | - | - | - |
| K2 (min−1) | - | - | - | 0.11 | 0.006 | 0.031 |
| R2 | 0.93 | 0.98 | 0.94 | 0.99 | 0.99 | 0.99 |
| MAE | 0.47 | 0.065 | 0.53 | 0.13 | 0.024 | 0.009 |
| RMSE | 0.69 | 0.26 | 0.73 | 0.35 | 0.16 | 0.092 |
| Mechanical Properties | Before FBS Protein Adsorption | After FBS Protein Adsorption | ||||
|---|---|---|---|---|---|---|
| PCL a | PCL-O a | Multilayered Scaffold b | PCL a | PCL-O a | Multilayered Scaffold b | |
| Tensile strength (MPa) | 8.59 ± 0.24 | 7.413 ± 1.34 | 4.60 ± 0.21 | 27.97 ± 0.43 | 25.17 ± 1.01 | 8.45 ± 1.12 |
| Tensile elongation (%) | 250.32 ± 1.7 | 7.5 ± 0.36 | 19.42 ± 0.11 | 250.32 ± 1.7 | 248.76 ± 2.34 | 10.89 ± 0.43 |
| Young’s modulus (GPa) | 1.43 ± 0.26 | 1.74 ± 0.97 | 1.57 ± 0.35 | 1.56 ± 0.12 | 2.06 ± 0.32 | 1.73 ± 0.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Soto, M.A.; Garcia-Brand, A.J.; Riveros, A.; Suarez, N.A.; Serrano, F.; Osma, J.F.; Muñoz Camargo, C.; Cruz, J.C.; Sandoval, N.; Briceño, J.C. Blood-Vessel-Inspired Hierarchical Trilayer Scaffolds: PCL/Gelatin-Driven Protein Adsorption and Cellular Interaction. Polymers 2022, 14, 2135. https://doi.org/10.3390/polym14112135
Rodriguez-Soto MA, Garcia-Brand AJ, Riveros A, Suarez NA, Serrano F, Osma JF, Muñoz Camargo C, Cruz JC, Sandoval N, Briceño JC. Blood-Vessel-Inspired Hierarchical Trilayer Scaffolds: PCL/Gelatin-Driven Protein Adsorption and Cellular Interaction. Polymers. 2022; 14(11):2135. https://doi.org/10.3390/polym14112135
Chicago/Turabian StyleRodriguez-Soto, Maria A., Andres J. Garcia-Brand, Alejandra Riveros, Natalia A. Suarez, Fidel Serrano, Johann F. Osma, Carolina Muñoz Camargo, Juan C. Cruz, Nestor Sandoval, and Juan C. Briceño. 2022. "Blood-Vessel-Inspired Hierarchical Trilayer Scaffolds: PCL/Gelatin-Driven Protein Adsorption and Cellular Interaction" Polymers 14, no. 11: 2135. https://doi.org/10.3390/polym14112135
APA StyleRodriguez-Soto, M. A., Garcia-Brand, A. J., Riveros, A., Suarez, N. A., Serrano, F., Osma, J. F., Muñoz Camargo, C., Cruz, J. C., Sandoval, N., & Briceño, J. C. (2022). Blood-Vessel-Inspired Hierarchical Trilayer Scaffolds: PCL/Gelatin-Driven Protein Adsorption and Cellular Interaction. Polymers, 14(11), 2135. https://doi.org/10.3390/polym14112135