
Computational Study of Catalytic Urethane Formation

 , , , and
, , , and 
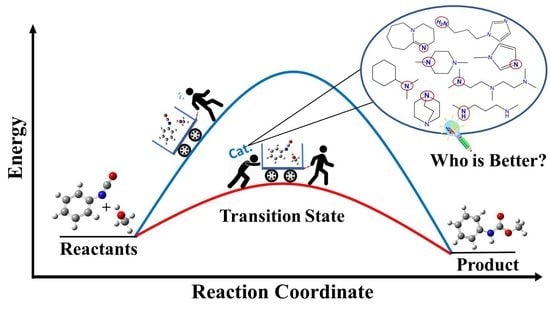
Abstract
:
1. Introduction
2. Methods
3. Results and Discussion
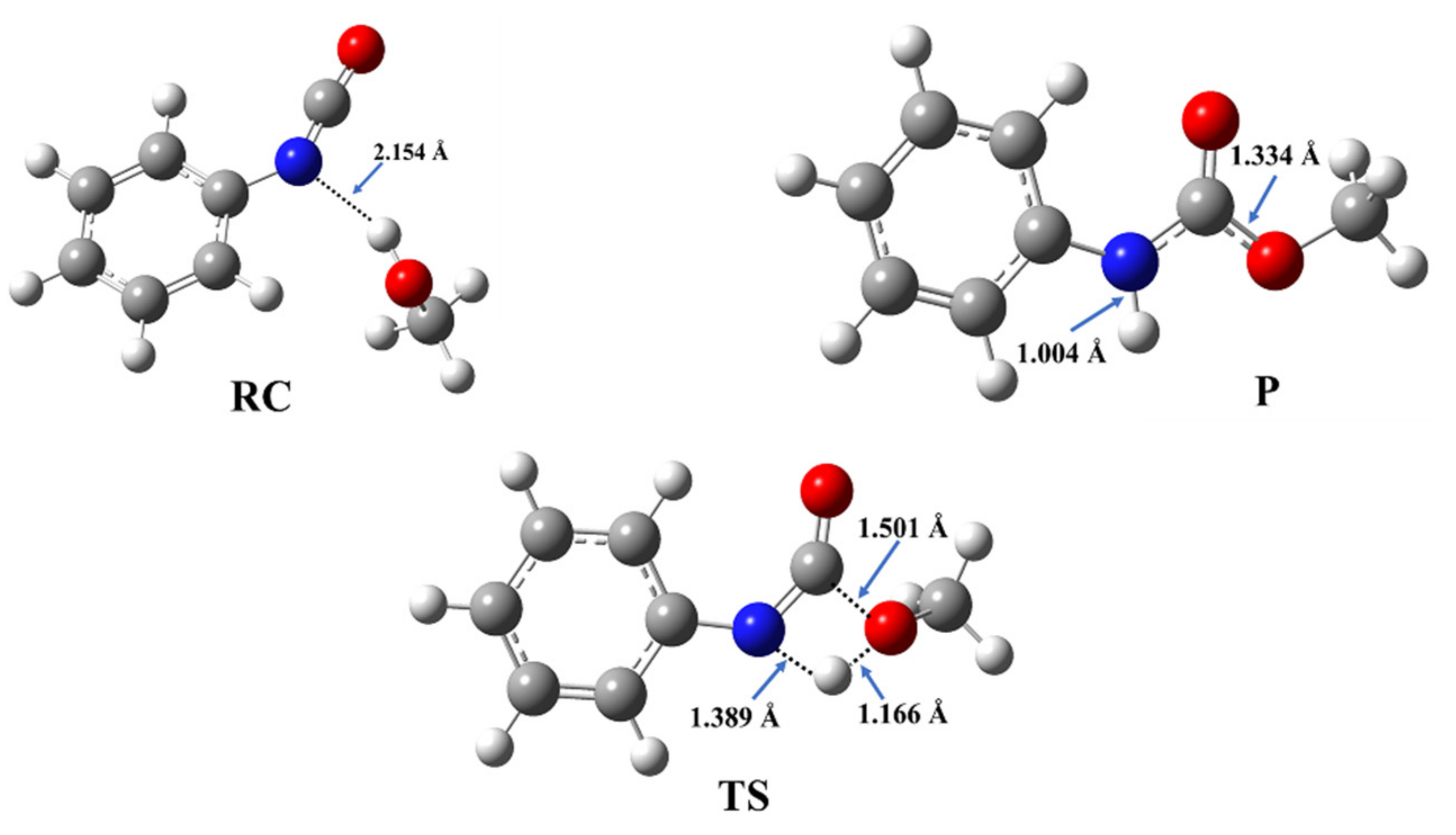
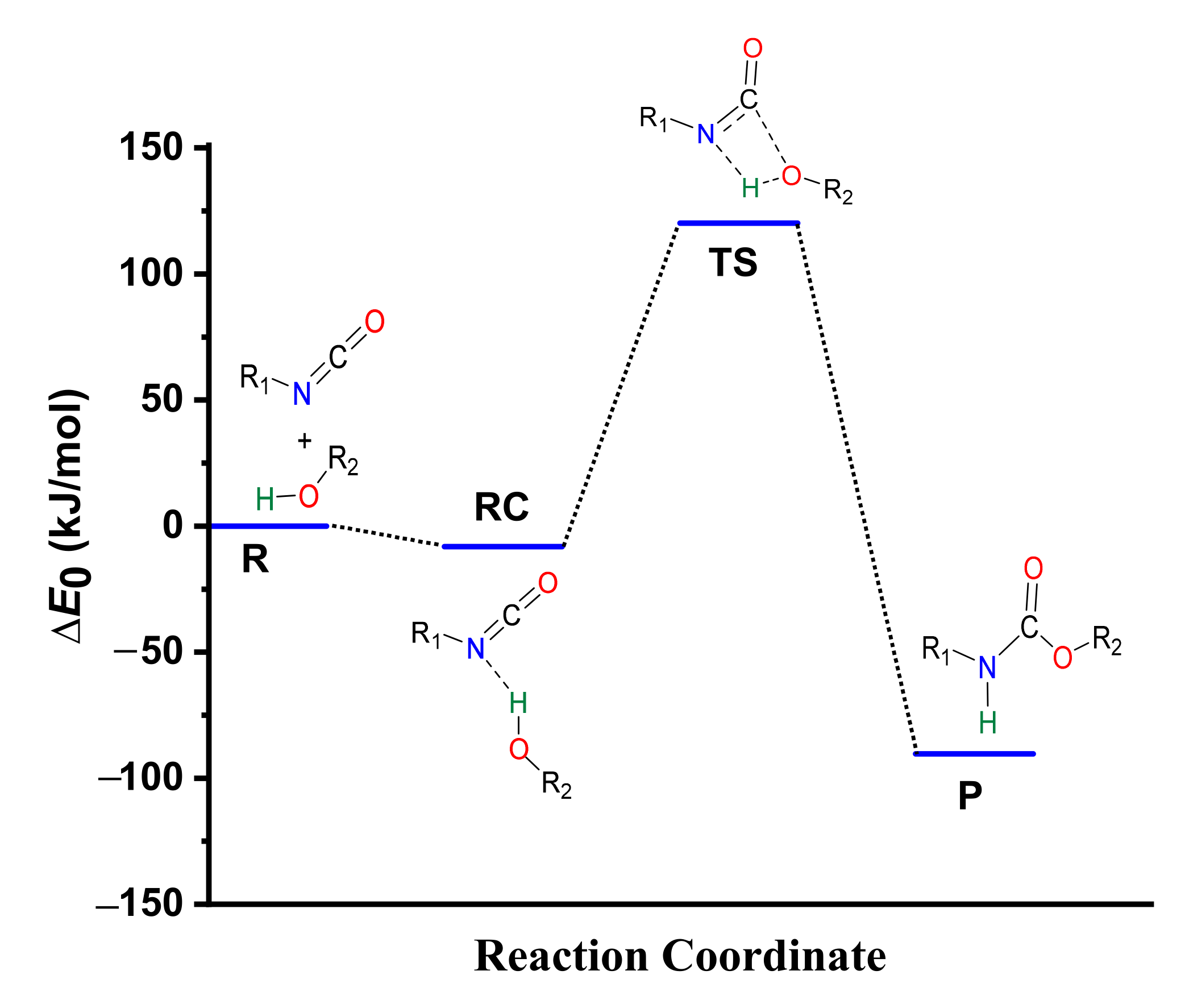
3.1. Catalyst-Free Reaction Mechanism of Urethane Formation
3.2. Structural and Energetic Features of a Catalyst-Free Model Reaction
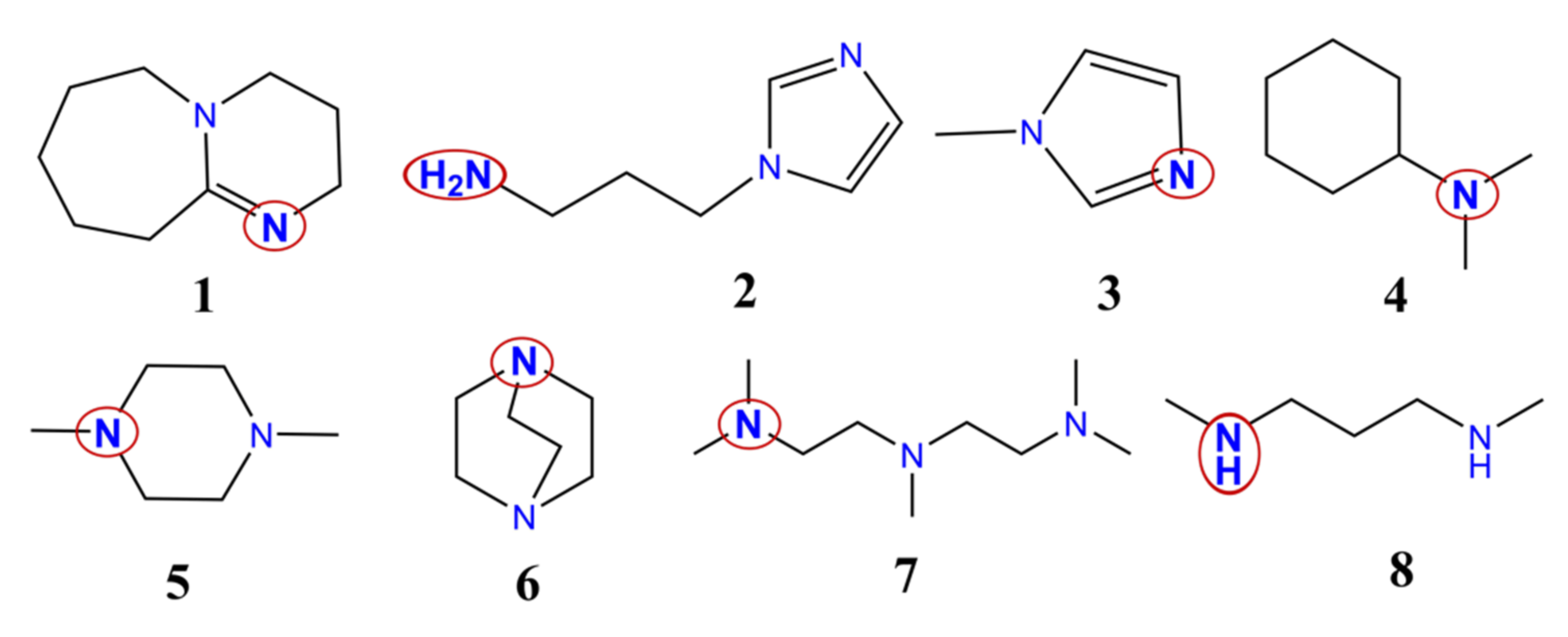
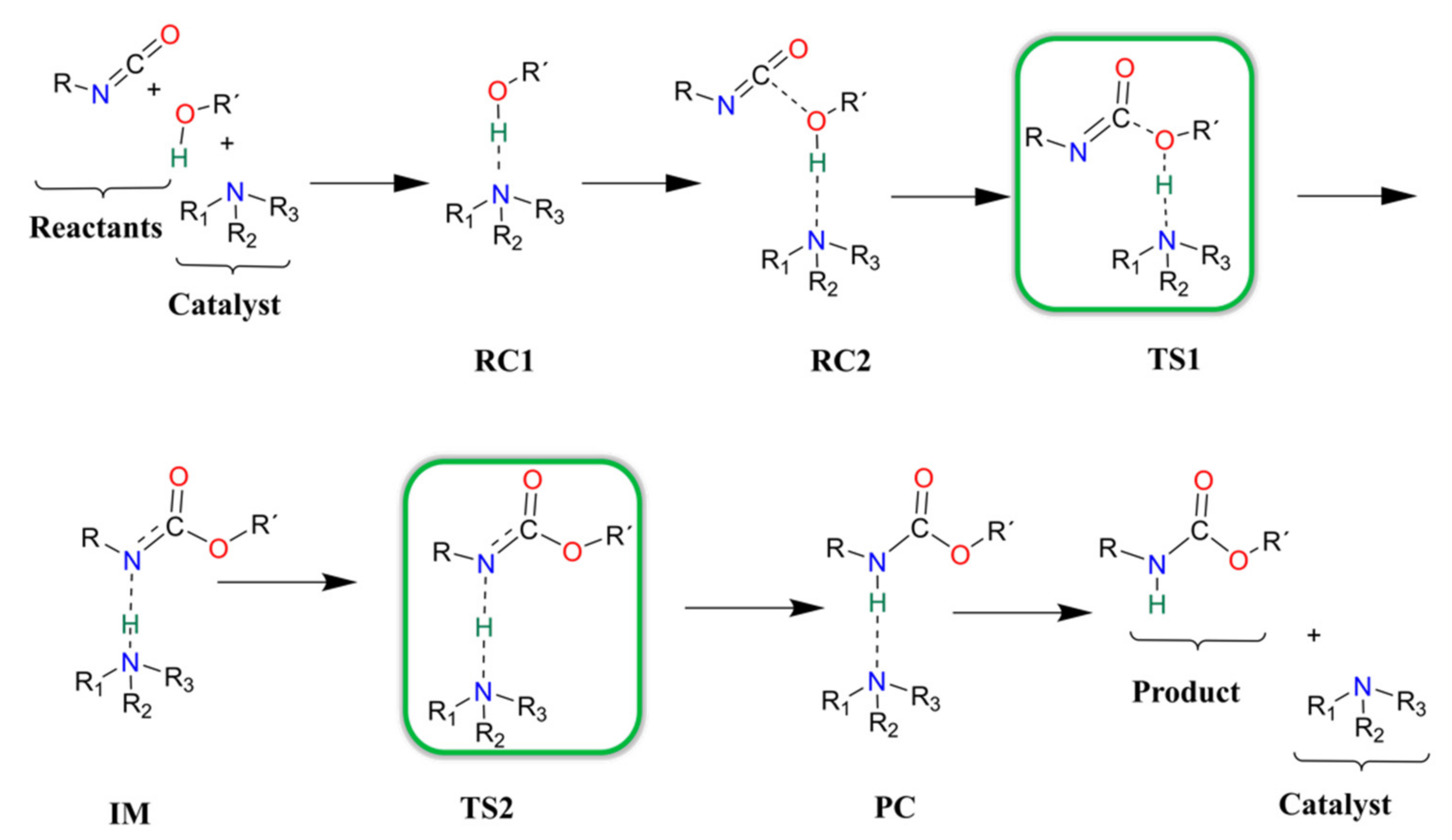
3.3. Urethane Formation Reaction in the Presence of Nitrogen-Containing Catalysts
3.4. Urethane Formation Mechanism in the Presence of Catalysts
3.5. Proton Affinity (PA) of the Studied Catalysts
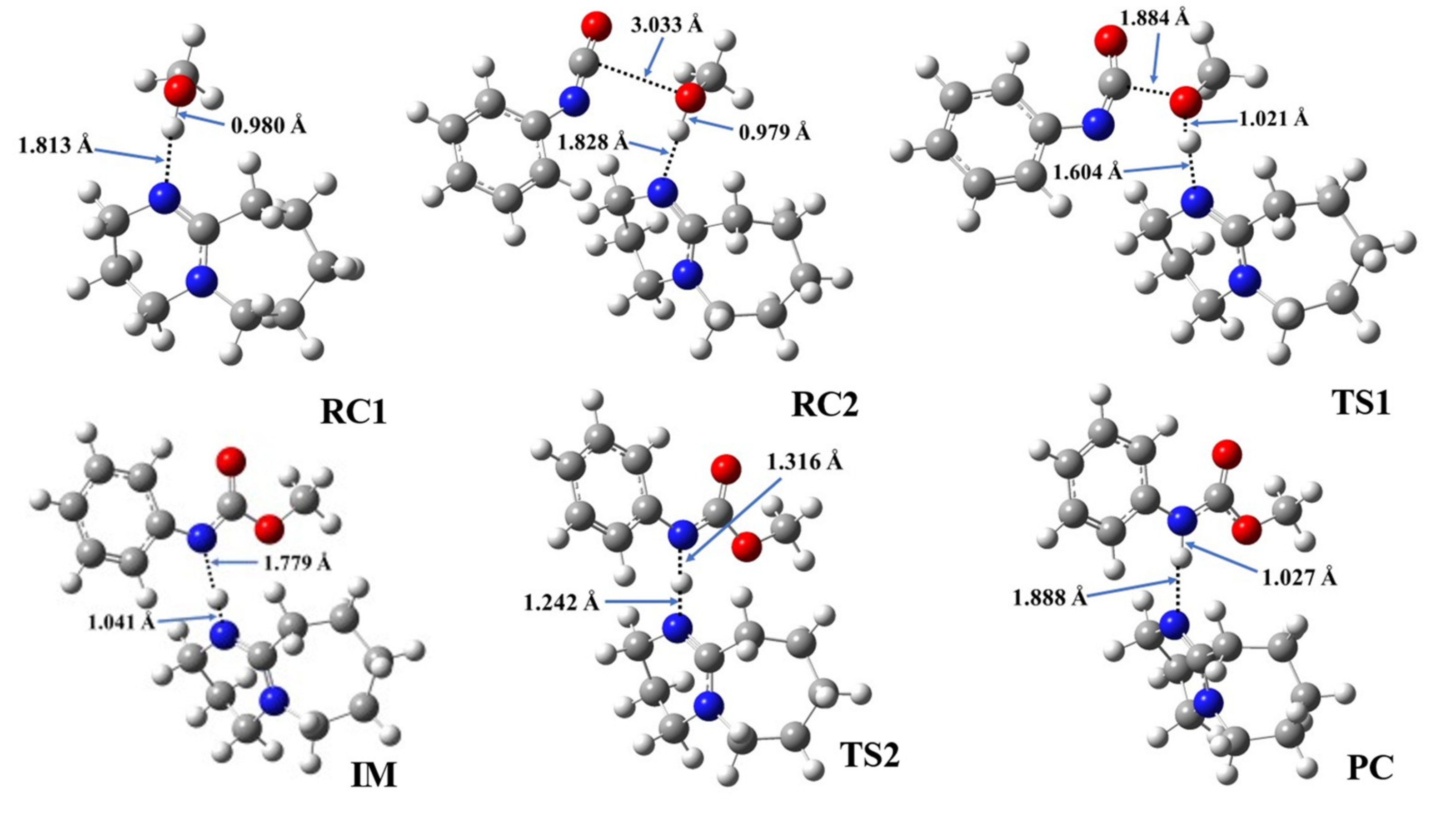
3.6. Structural Features of Urethane Formation in the Presence of the Studied Catalysts
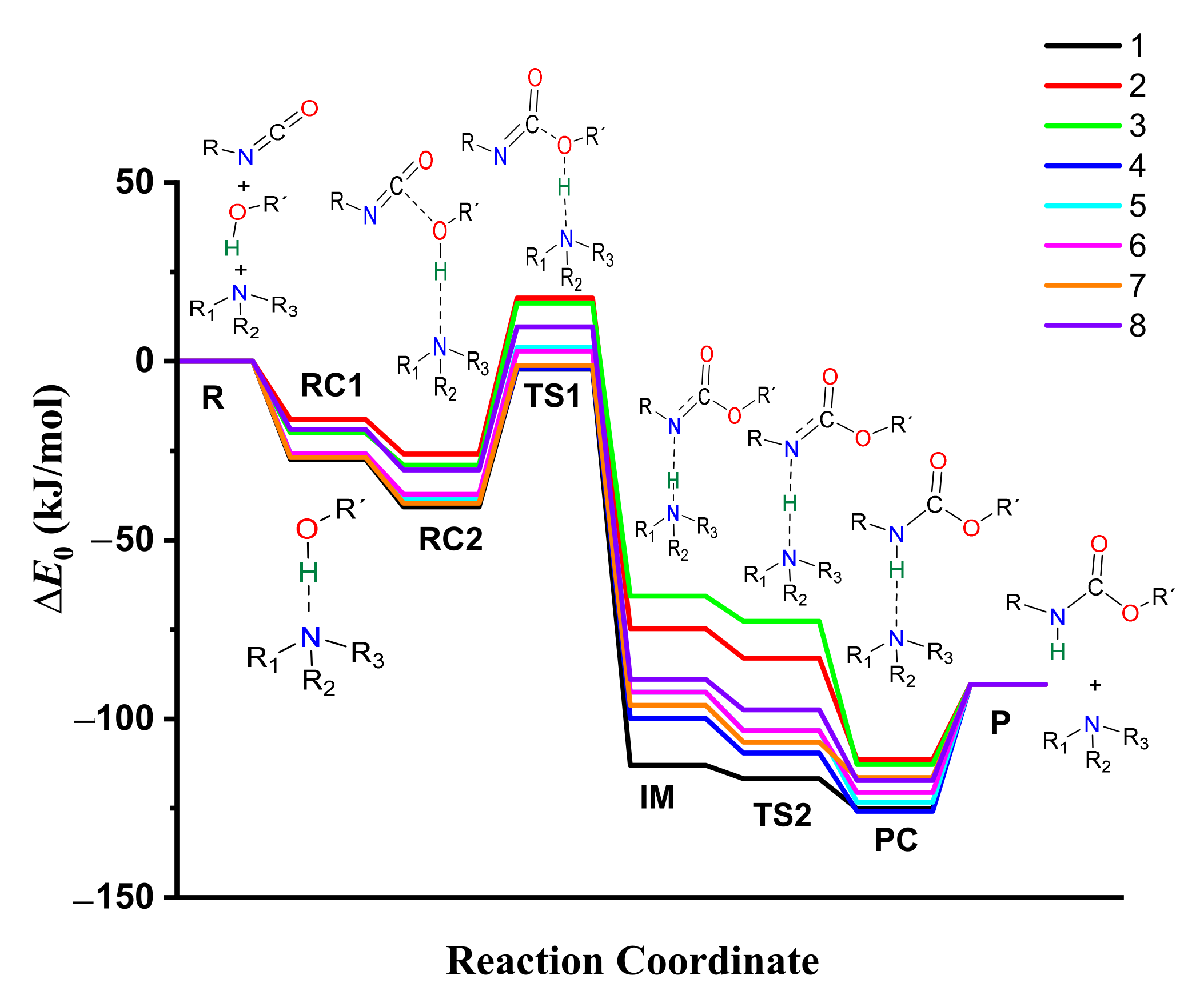
3.7. Energetics of Urethane Formation in the Presence of Different Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szycher, M. Structure–Property Relations in Polyurethanes. In Szycher’s Handbook of Polyurethanes; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Szycher, M. Handbook of Polyurethanes; CRC Press: Boca Raton, FL, USA, 1938; p. 142. [Google Scholar]
- Cavaco, L.I. Polyurethanes Properies, Structure and Applicatiovs; Melo, J.A., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2012. [Google Scholar]
- Sabrina, S.S.A.; Denilson, A.S.; Danielle, M.A. Physico-Chemical Analysis of Flexible Polyurethane Foams Containing Commercial Calcium Carbonate. Mater. Res. 2008, 11, 433–438. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane Types, Synthesis and Applications—A Review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, C.; Yang, W.; Tang, Q. Thermoplastic Polyurethanes Stemming from Castor Oil: Green Synthesis and Their Application in Wood Bonding. Coatings 2017, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Marrs, K.D. Bio-Based Polyurethanes-A Computational and Experimental Study. In Proceedings of the 8th Visegrad Symposium on Structural Systems Biology, Lucenec, Slovakia, 20–23 June 2018; pp. 1–30. [Google Scholar]
- Gama, N.V.; Ferreira, A.; Barros-Timmons, A. Polyurethane Foams: Past, Present, and Future. Materials 2018, 11, 1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.L.; Bordado, J.C. Recent Developments in Polyurethane Catalysis: Catalytic Mechanisms Review. Catal. Rev. Sci. Eng. 2004, 46, 31–51. [Google Scholar] [CrossRef]
- Li, R.; Liu, L.; Liu, Y.; Wang, B.; Yang, J.J.; Zhang, J. Research Progress of Amine Catalysts for Polyurethane. Top. Chem. Mater. Eng. 2018, 1, 54–57. [Google Scholar] [CrossRef]
- Samuilov, A.Y.; Balabanova, F.B.; Kamalov, T.A.; Samuilov, Y.D.; Konovalov, A.I. Quantum-Chemical Study on Reactions of Isocyanates with Linear Methanol Associates: III.* Reaction of Merhyl Isocyanate with Linear Methanol Associates. Russ. J. Org. Chem. 2010, 46, 1450–1457. [Google Scholar] [CrossRef]
- Hatanaka, M. DFT Analysis of Catalytic Urethanation. Bull. Chem. Soc. Jpn. 2011, 84, 933–935. [Google Scholar] [CrossRef]
- Wen, Z. DFT Study of the Catalytic Mechanism for Urethane Formation in the Presence of Basic Catalyst 1,4-Diazabicyclo[2.2.2]Octane. Commun. Comput. Chem. 2014, 2, 22–35. [Google Scholar] [CrossRef]
- Cheikh, W.; Rózsa, Z.B.; López, C.O.C.; Mizsey, P.; Viskolcz, B.; Szori, M.; Fejes, Z. Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study. Polymers 2019, 11, 1543. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Szori, M.; Abou-Abdo, T.; Fittschen, C.; Csizmadia, I.G.; Viskolcz, B. Allylic Hydrogen Abstraction II. H-Abstraction from 1,4 Type Polyalkenes as a Model for Free Radical Trapping by Polyunsaturated Fatty Acids (PUFAs). Phys. Chem. Chem. Phys. 2007, 9, 1931–1940. [Google Scholar] [CrossRef]
- Szori, M.; Fittschen, C.; Csizmadia, I.G.; Viskolcz, B. Allylic H-Abstraction Mechanism: The Potential Energy Surface of the Reaction of Propene with OH Radical. J. Chem. Theory Comput. 2006, 2, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Izsák, R.; Szori, M.; Knowles, P.J.; Viskolcz, B. High Accuracy Ab Initio Calculations on Reactions of OH with 1-Alkenes. The Case of Propene. J. Chem. Theory Comput. 2009, 5, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Janoschek, R.; Rossi, M.J. Thermochemical Properties of Free Radicals from G3MP2B3 Calculations. Int. J. Chem. Kinet. 2002, 34, 550–560. [Google Scholar] [CrossRef]
- Hadjadj, R.; Csizmadia, I.G.; Mizsey, P.; Jensen, S.; Viskolcz, B.; Fiser, B. Water Enhanced Mechanism for CO2—Methanol Conversion. Chem. Phys. Lett. 2020, 746, 137298. [Google Scholar] [CrossRef]
- Wang, X.; HU, W.; Gui, D.; Chi, X.; Wang, M.; Tian, D.; Liu, J.; Ma, X.; Pang, A. DFT Study of the Proton Transfer in the Urethane Formation between 2,4-Diisocyanatotoluene and Methanol. Bull. Chem. Soc. Jpn. Vol. 2013, 86, 255–265. [Google Scholar] [CrossRef]
- Kolboe, S. Proton Affinity Calculations with High Level Methods. J. Chem. Theory Comput. 2014, 10, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Beran, R.; Zarybnicka, L.; Machova, D. Recycling of Rigid Polyurethane Foam: Micro-Milled Powder Used as Active Filler in Polyurethane Adhesives. J. Appl. Polym. Sci. 2020, 137, 49095. [Google Scholar] [CrossRef]
- Chuayjuljit, S.; Maungchareon, A.; Saravari, O. Preparation and Properties of Palm Oil-Based Rigid Polyurethane Nanocomposite Foams. J. Reinf. Plast. Compos. 2010, 29, 218–225. [Google Scholar] [CrossRef]
- Samarappuli, I.P.; Liyanage, N.M.V.K. Evaluation of the Suitability of 1,4-Dimethylpiperazine as a Substitute Catalyst in Polyurethane Foam Production. In Proceedings of the MERCon 2018—4th International Multidisciplinary Moratuwa Engineering Research Conference, Moratuwa, Sri Lanka, 30 May–1 June 2018; pp. 282–287. [Google Scholar] [CrossRef]
- Kennedy, W.A.; Austin, T. Catalyst Combination for Polyurethanes. U.S. Patent 3,661,808, 9 May 1972. [Google Scholar]
- El Ghobary, H.; Muller, L. Process for Preparing Polyurethane Foam. EP Patent 1 018 525 A1, 12 July 2000. [Google Scholar]
- Shokuhi, A.; Ardjmand, M. Studying on the Mechanism and Raw Materials Used to Manufacturing Polyurethane. Chem. Technol. Indian J. 2008, 3, 60–71. [Google Scholar]
- NIST Chemistry WebBook. Available online: https://webbook.nist.gov/chemistry/ (accessed on 13 April 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆E0 (kJ/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|
| R | RC1 | RC2 | TS1 | IM | TS2 | PC | P | |
| Cat.-free | 0.00 | - | −8.22 * | 120.18 | - | - | - | −90.33 |
| 1 | 0.00 | −27.43 | −40.71 | −2.20 | −112.98 | −116.72 | −125.14 | −90.33 |
| 2 | 0.00 | −16.27 | −25.95 | 17.72 | −74.79 | −82.97 | −111.38 | −90.33 |
| 3 | 0.00 | −19.99 | −29.04 | 16.33 | −65.63 | −72.68 | −112.74 | −90.33 |
| 4 | 0.00 | −26.81 | −38.76 | −1.96 | −99.90 | −109.55 | −125.82 | −90.33 |
| 5 | 0.00 | −25.77 | −38.73 | 3.84 | −92.55 | −103.27 | −123.31 | −90.33 |
| 6 | 0.00 | −25.85 | −37.16 | 2.85 | −92.53 | −103.34 | −120.54 | −90.33 |
| 7 | 0.00 | −26.88 | −39.61 | −1.08 | −96.15 | −106.49 | −116.42 | −90.33 |
| 8 | 0.00 | −19.03 | −30.45 | 9.63 | −88.89 | −97.52 | −117.25 | −90.33 |
| Cat. | Resulted Polyurethane Type | Structural Property of PU | Functional Groups | References | |||
|---|---|---|---|---|---|---|---|
| RNH2 | R2NH | R3N | R2C=N-R | ||||
| 1 | Flexible foam | Cyclic | • | • | [9] | ||
| 2 | Rigid foam | Aromatic, Linear | • | • | • | [9] | |
| 3 | Rigid foam | Aromatic | • | • | [26] | ||
| 4 | Rigid foam | Linear | • | [27] | |||
| 5 | Semi-rigid | Cyclic | • • | [28,29] | |||
| 6 | Semi-rigid | Cyclic | • • | [30] | |||
| 7 | Semi-rigid | Linear | • • • | [9] | |||
| 8 | Other | Linear | • • | [31] | |||
| Catalysts | PAcalc (kJ/mol) | PAexp (kJ/mol) [32] | |||
|---|---|---|---|---|---|
| RNH2 | R2NH | R3N | R2C=N-R | ||
| 1 | - | - | 937.50 | 1070.34 | - |
| 2 | 905.75 | 994.19 | 800.93 | - | - |
| 3 | - | 983.68 | 785.51 | - | 959.6 |
| 4 | - | - | 998.68 | - | 983.6 |
| 5 | - | - | 984.84 | - | - |
| 6 | - | - | 983.91 | - | 963.4 |
| 7 | - | - | 984.71 • 982.62 •• | - | - |
| 8 | - | 966.54 | - | - | 1035.2 |
| Cat. | RC1 | RC2 | TS1 | IM | TS2 | PC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-H* | O-H | N-H* | O-H | C-O | N-H* | O-H | C-O | N-H* | N-H** | N-H* | N-H** | N-H* | N-H** | |
| 1 | 1.813 | 0.980 | 1.828 | 0.979 | 3.033 | 1.604 | 1.021 | 1.884 | 1.041 | 1.779 | 1.242 | 1.316 | 1.888 | 1.027 |
| 2 | 1.848 | 0.977 | 1.853 | 0.977 | 3.026 | 1.653 | 1.011 | 1.869 | 1.092 | 1.605 | 1.168 | 1.437 | 1.920 | 1.026 |
| 3 | 1.882 | 0.972 | 1.855 | 0.974 | 3.064 | 1.690 | 1.002 | 1.828 | 1.088 | 1.597 | 1.134 | 1.481 | 1.967 | 1.019 |
| 4 | 1.864 | 0.977 | 1.868 | 0.977 | 3.036 | 1.670 | 1.014 | 1.845 | 1.065 | 1.725 | 1.223 | 1.370 | 2.012 | 1.023 |
| 5 | 1.879 | 0.975 | 1.883 | 0.975 | 3.062 | 1.695 | 1.009 | 1.836 | 1.068 | 1.718 | 1.212 | 1.384 | 2.022 | 1.021 |
| 6 | 1.851 | 0.976 | 1.827 | 0.980 | 3.078 | 1.657 | 1.013 | 1.857 | 1.073 | 1.670 | 1.211 | 1.372 | 1.996 | 1.023 |
| 7 | 1.870 | 0.976 | 1.872 | 0.976 | 3.053 | 1.669 | 1.012 | 1.845 | 1.070 | 1.703 | 1.213 | 1.381 | 1.987 | 1.023 |
| 8 | 1.849 | 0.978 | 1.850 | 0.978 | 3.052 | 1.653 | 1.013 | 1.866 | 1.077 | 1.660 | 1.203 | 1.387 | 1.927 | 1.026 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waleed, H.Q.; Csécsi, M.; Hadjadj, R.; Thangaraj, R.; Pecsmány, D.; Owen, M.; Szőri, M.; Fejes, Z.; Viskolcz, B.; Fiser, B. Computational Study of Catalytic Urethane Formation. Polymers 2022, 14, 8. https://doi.org/10.3390/polym14010008
Waleed HQ, Csécsi M, Hadjadj R, Thangaraj R, Pecsmány D, Owen M, Szőri M, Fejes Z, Viskolcz B, Fiser B. Computational Study of Catalytic Urethane Formation. Polymers. 2022; 14(1):8. https://doi.org/10.3390/polym14010008
Chicago/Turabian StyleWaleed, Hadeer Q., Marcell Csécsi, Rachid Hadjadj, Ravikumar Thangaraj, Dániel Pecsmány, Michael Owen, Milán Szőri, Zsolt Fejes, Béla Viskolcz, and Béla Fiser. 2022. "Computational Study of Catalytic Urethane Formation" Polymers 14, no. 1: 8. https://doi.org/10.3390/polym14010008
APA StyleWaleed, H. Q., Csécsi, M., Hadjadj, R., Thangaraj, R., Pecsmány, D., Owen, M., Szőri, M., Fejes, Z., Viskolcz, B., & Fiser, B. (2022). Computational Study of Catalytic Urethane Formation. Polymers, 14(1), 8. https://doi.org/10.3390/polym14010008