
Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods
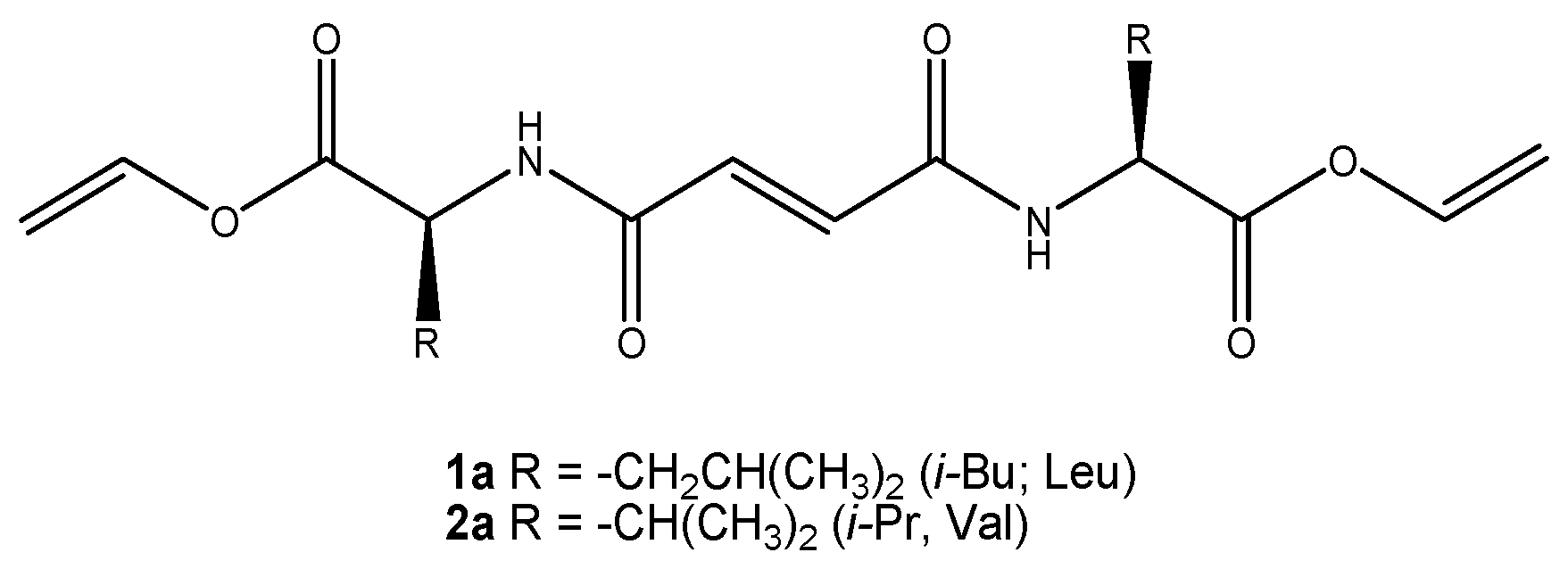
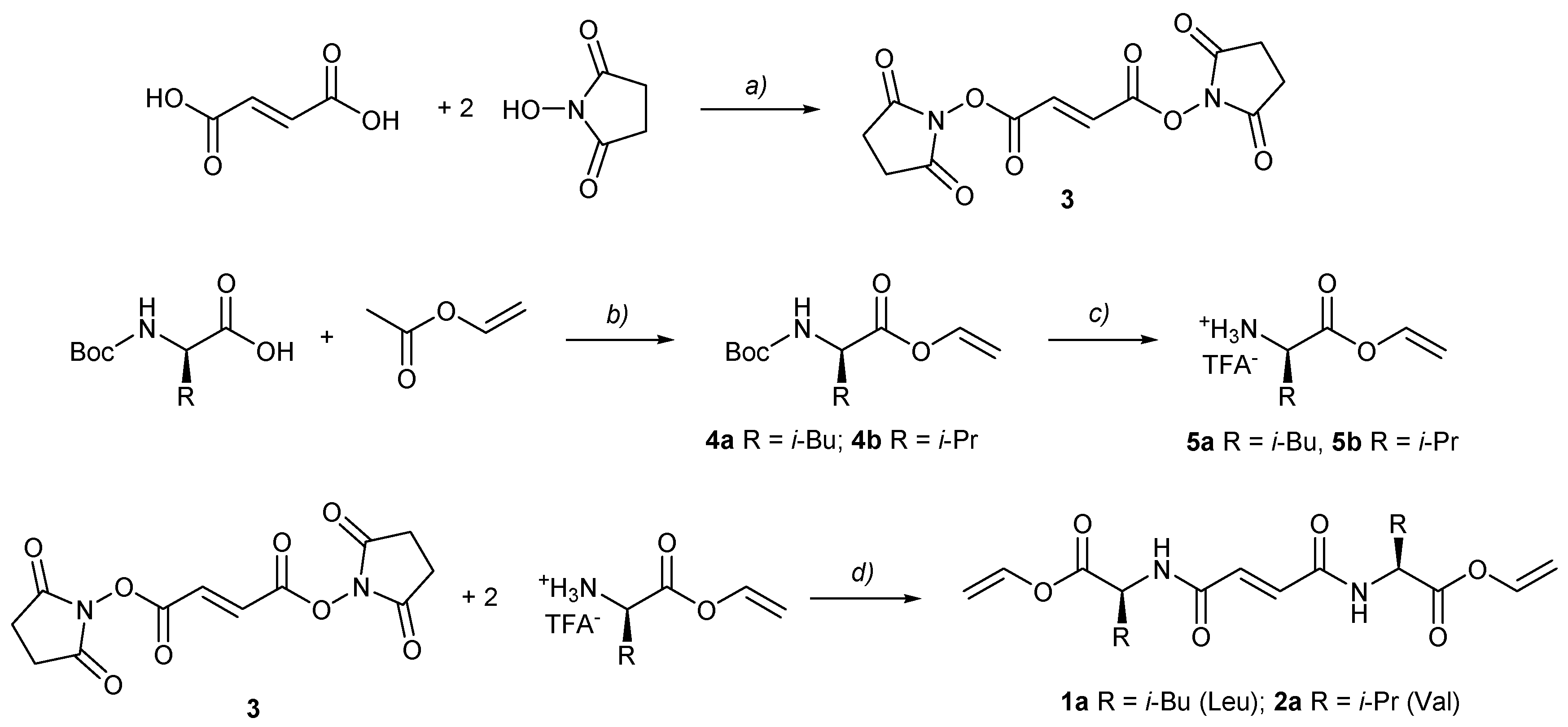
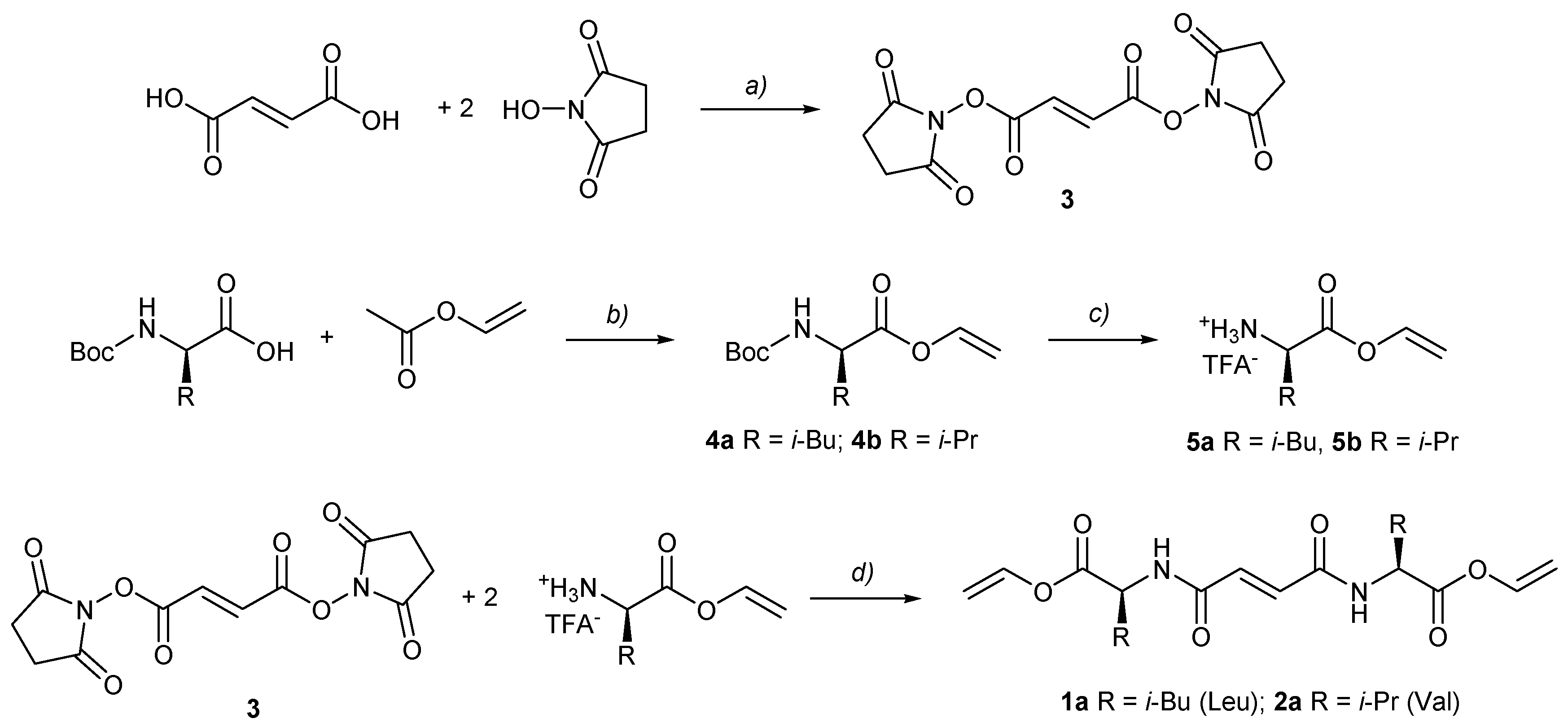
2.2. Synthesis, General Procedures for the Preparation of Fumaryl Diesters 1a and 2a
2.3. General Procedure for Synthesis Vinyl Ester Bis(leu and val)fumaramides (1a and 2a)
2.4. General Procedure for Synthesis of Methyl Ester Bis(leu and val)fumaramides (1b and 2b)
3. Results and Discussion
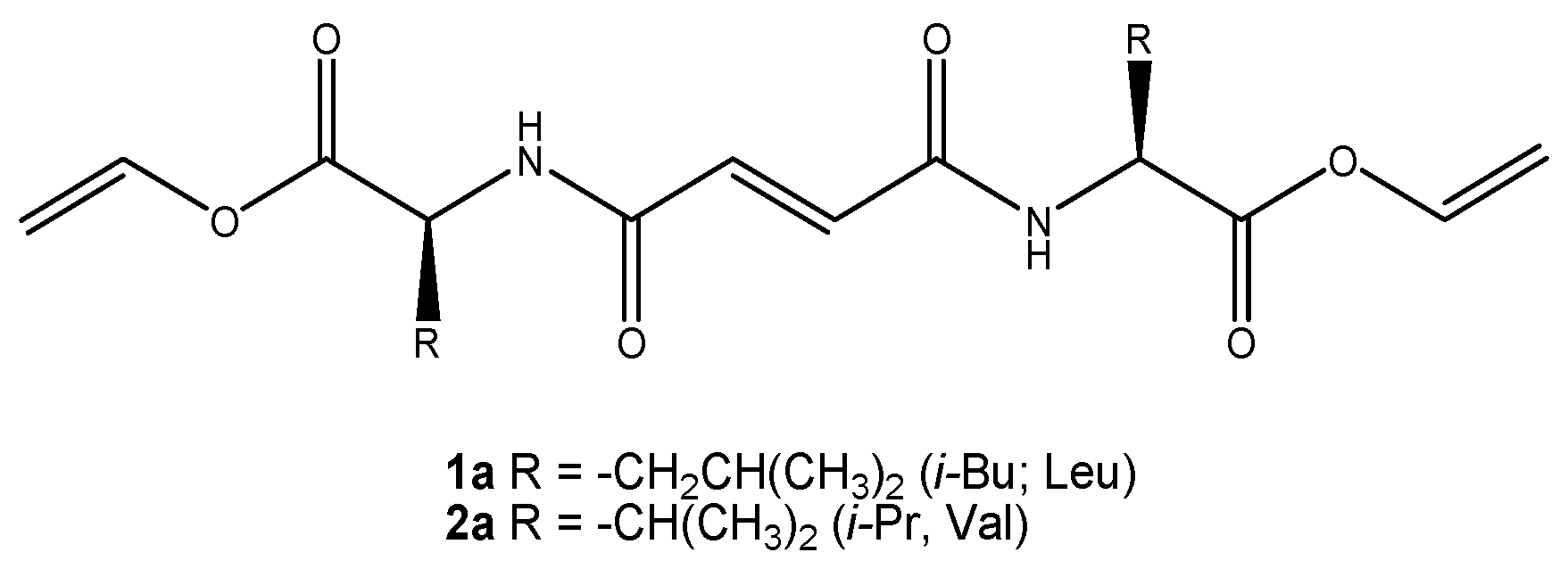
3.1. Molecular Design and Synthesis of Fumaramide Gelators
3.2. Gelation Properties
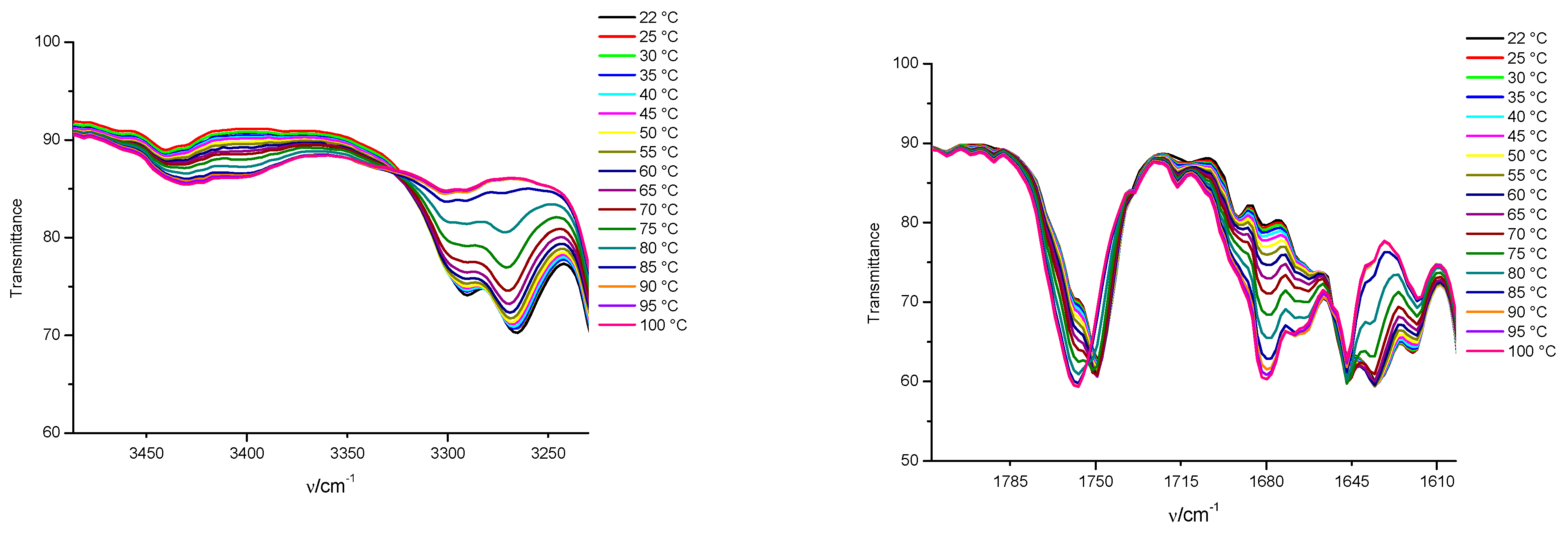
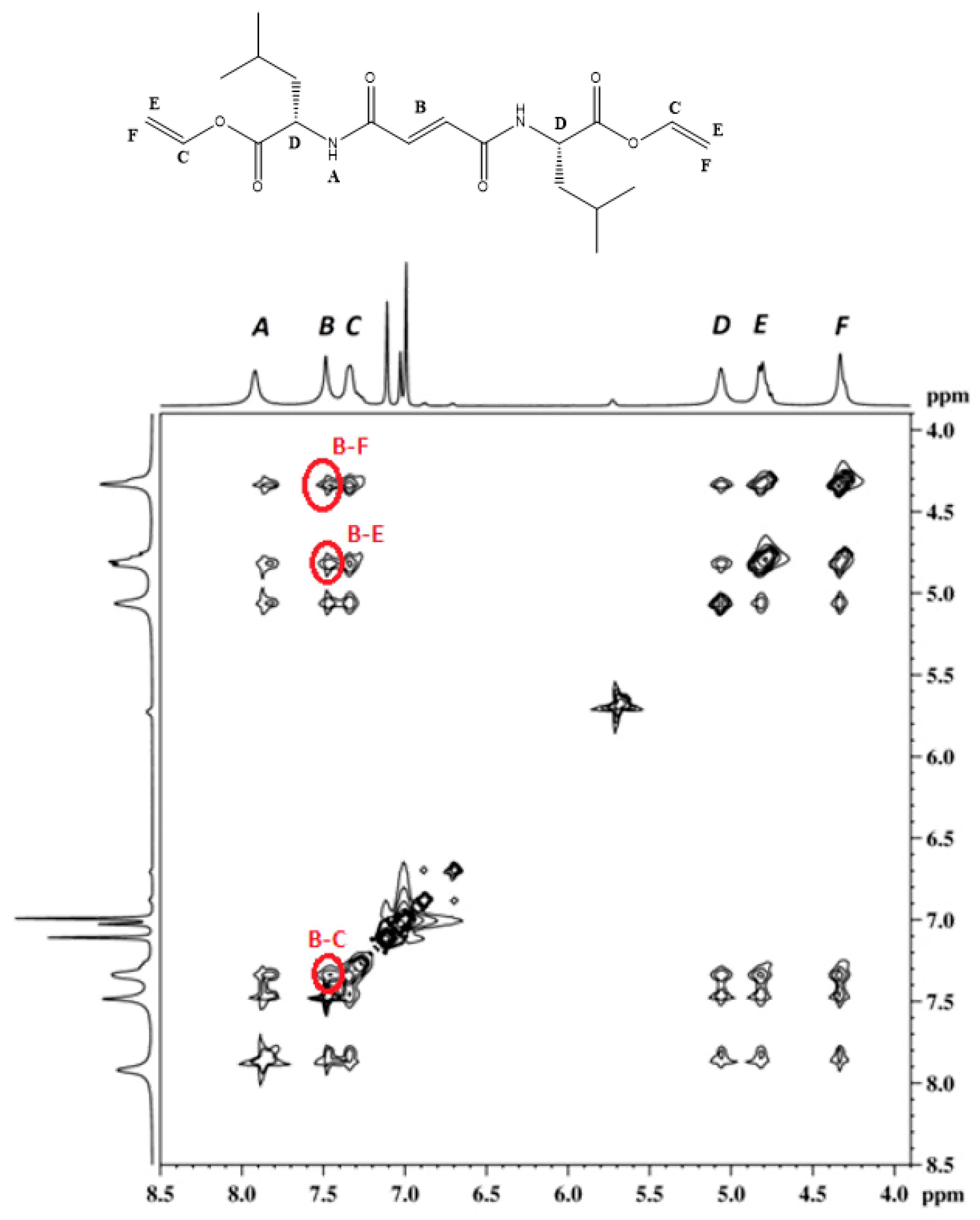
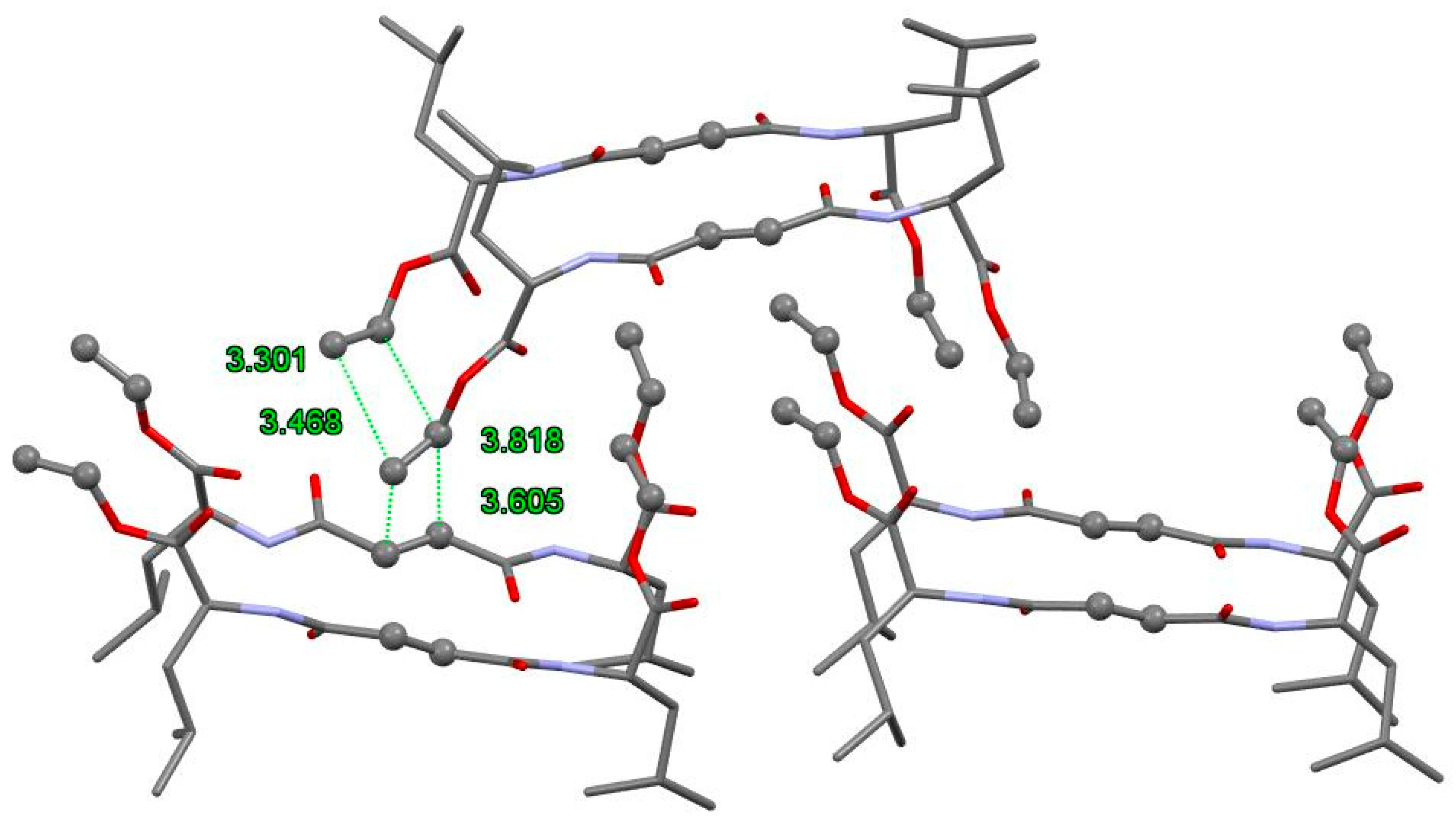
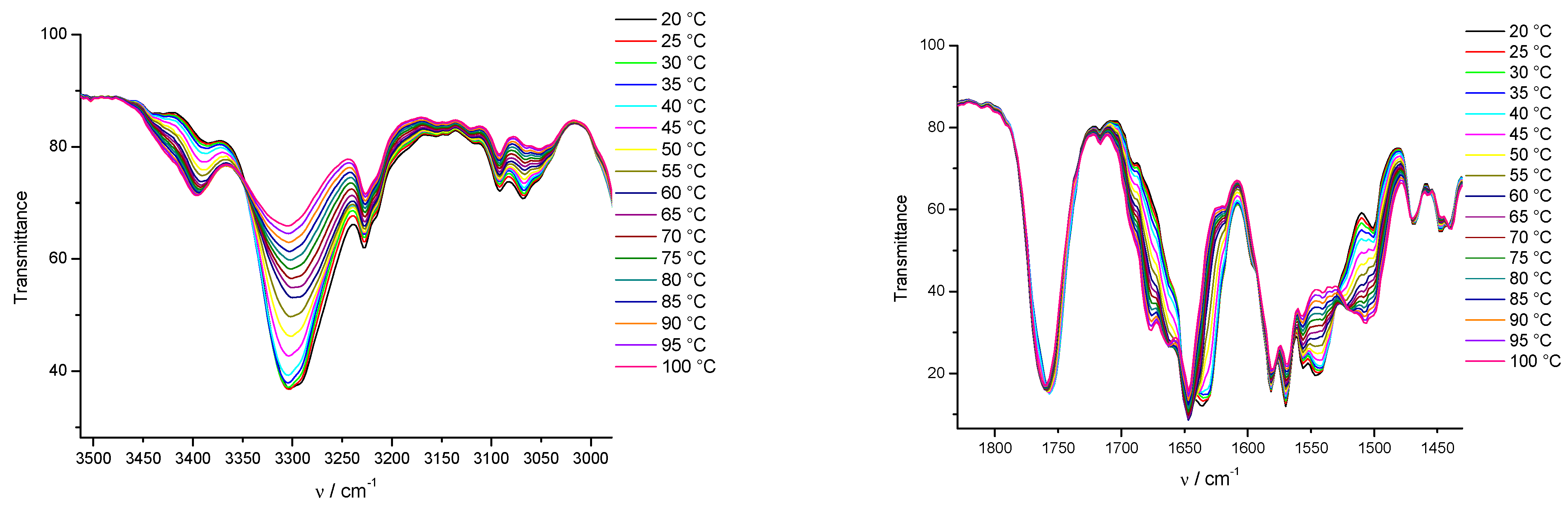
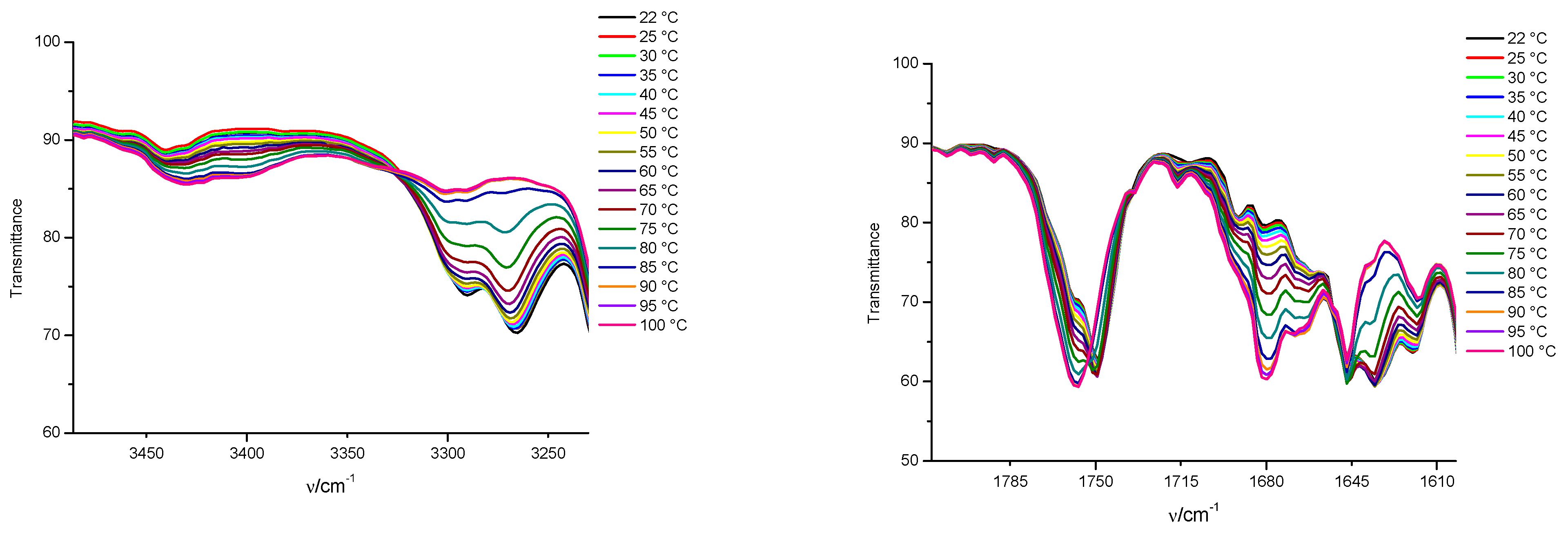
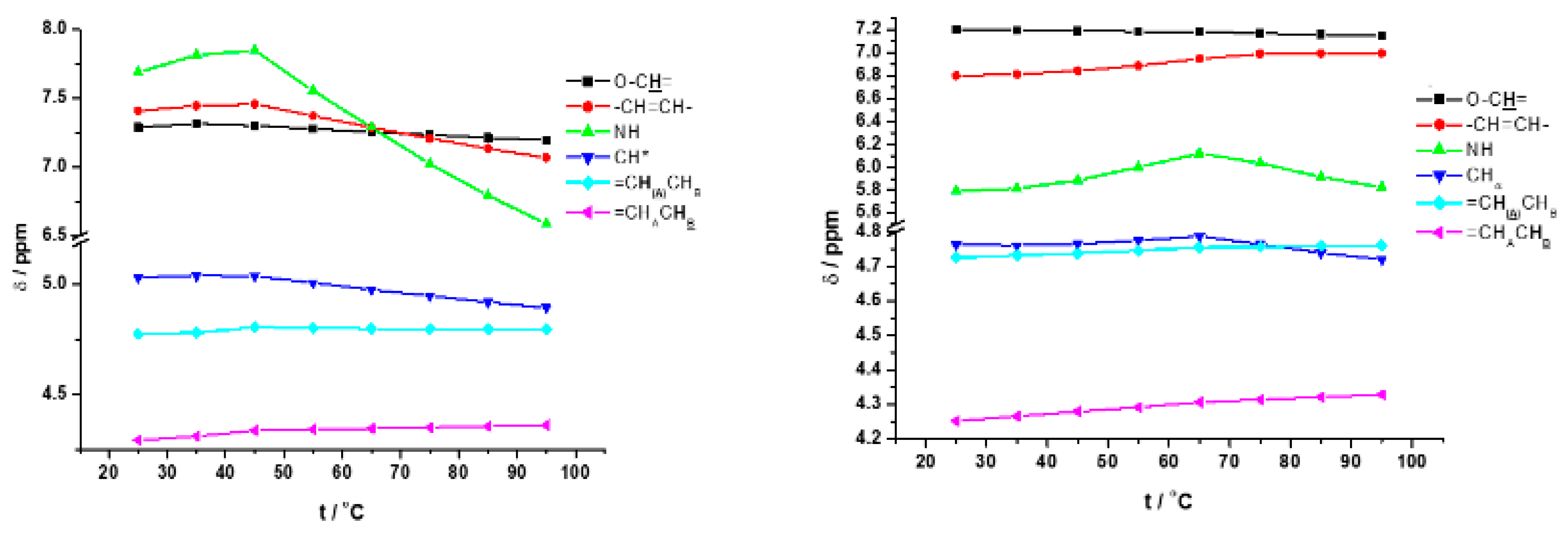
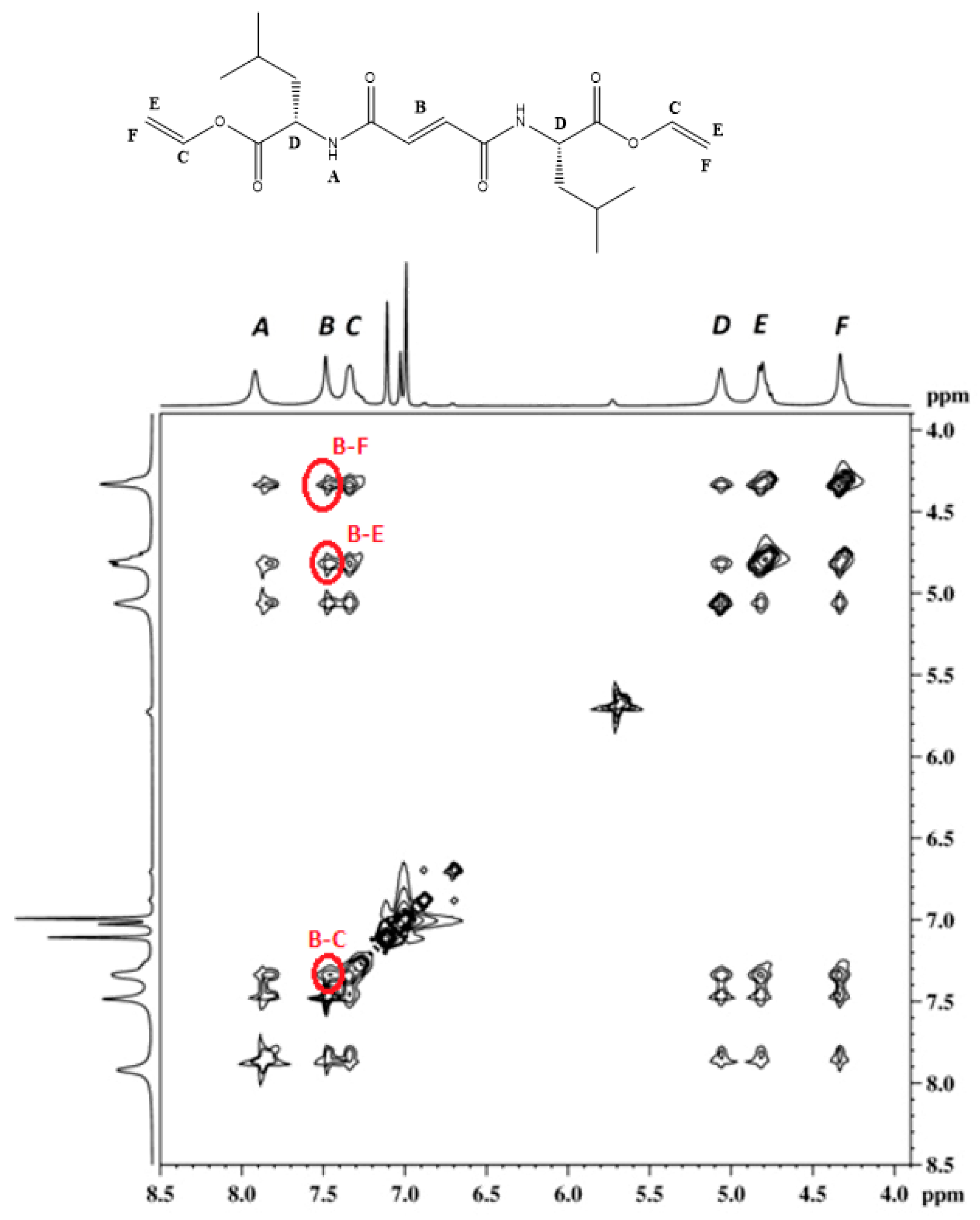
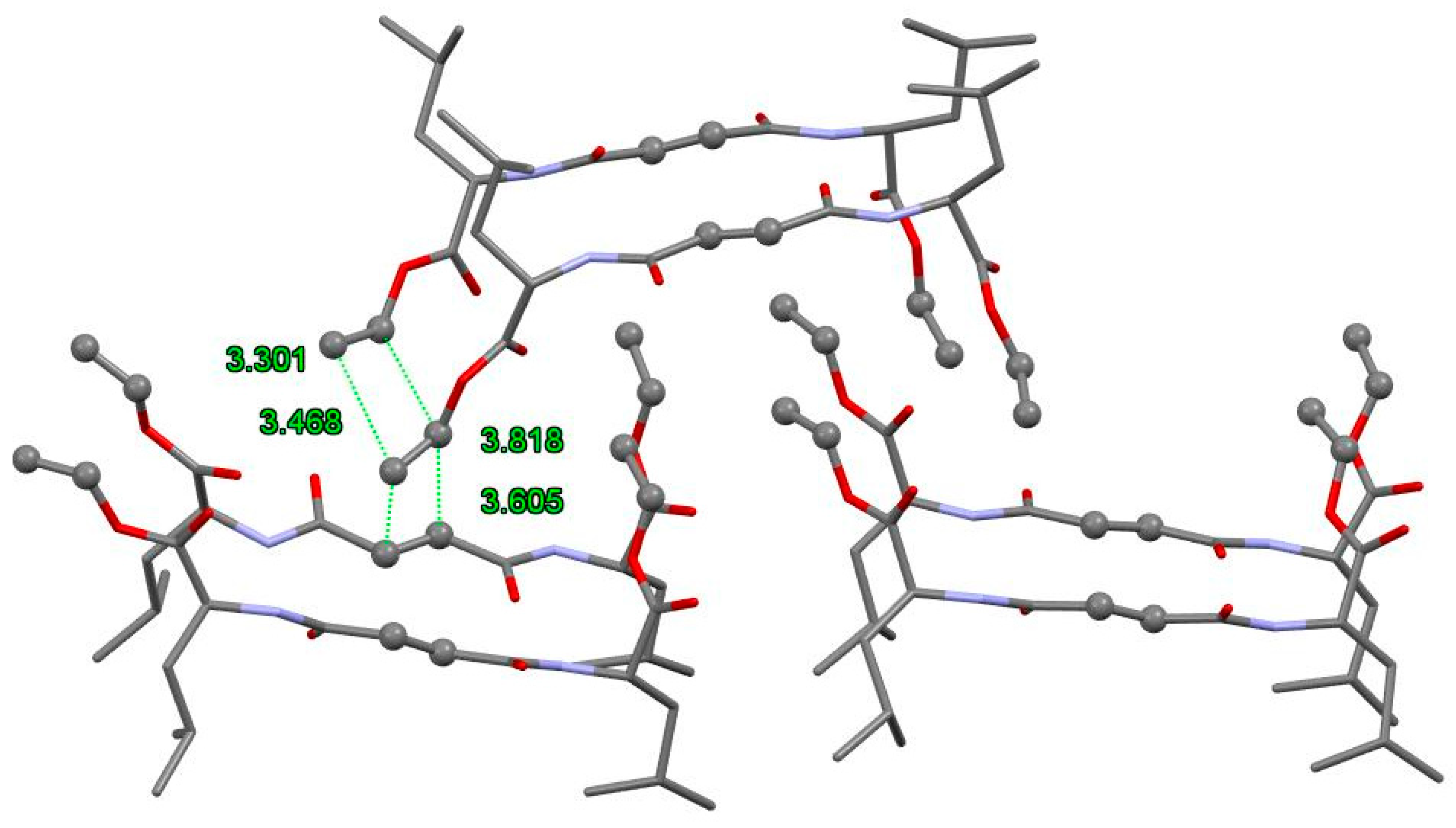
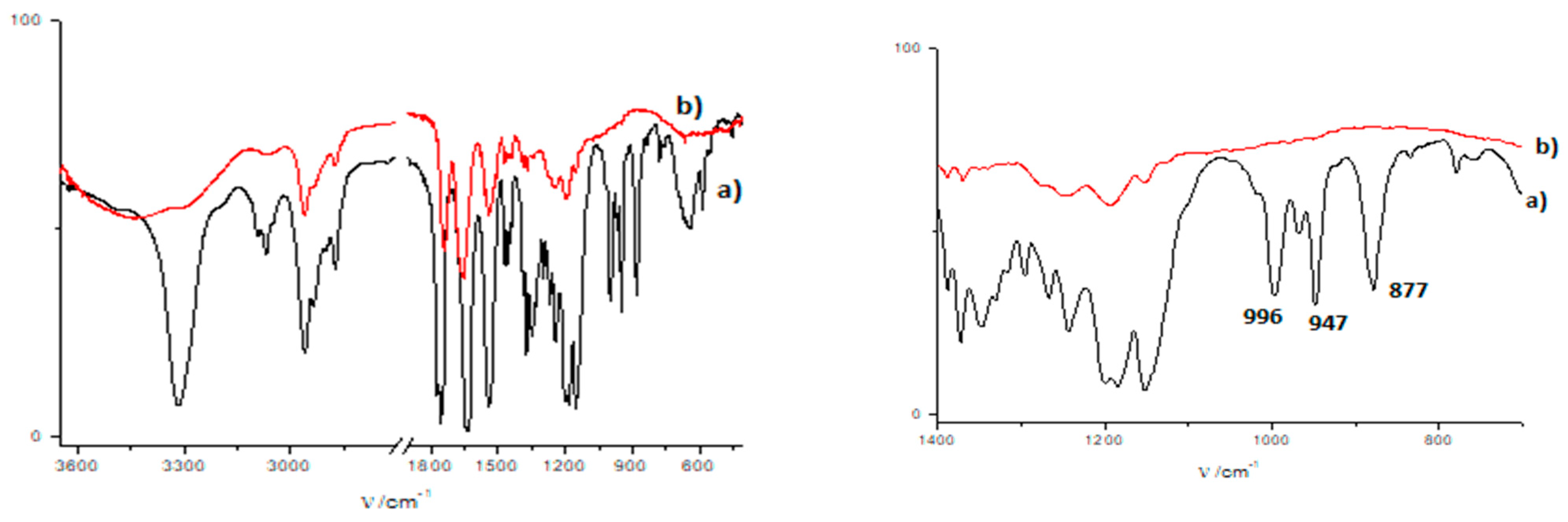
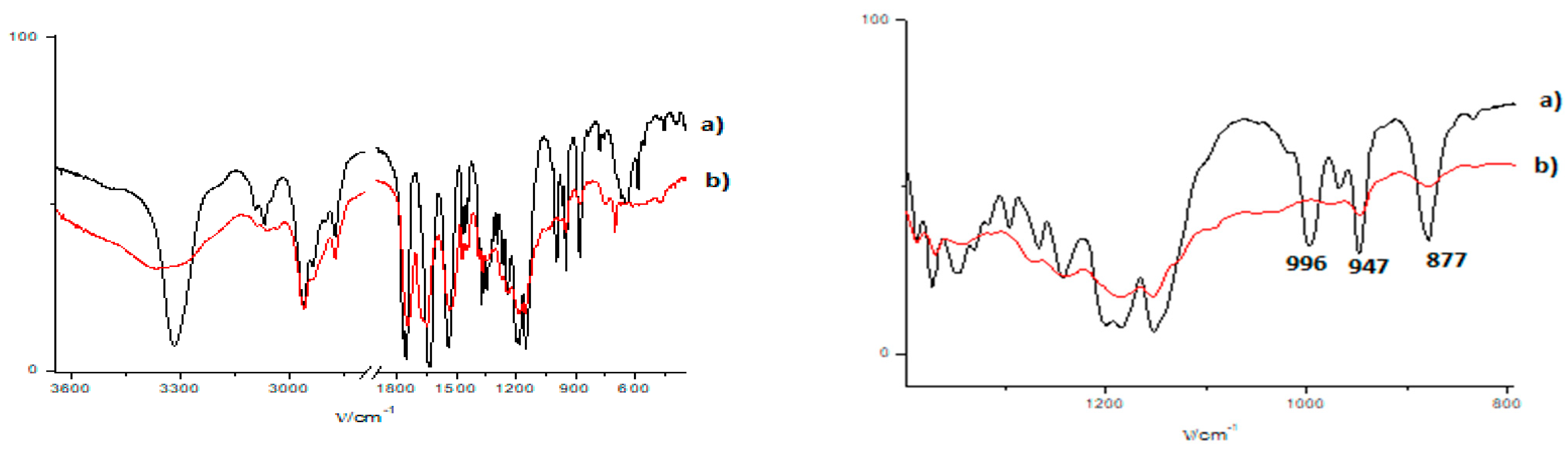
3.3. FTIR and 1H NMR Investigations of 1a and 2a Self-Assemblies, and Molecular Modeling
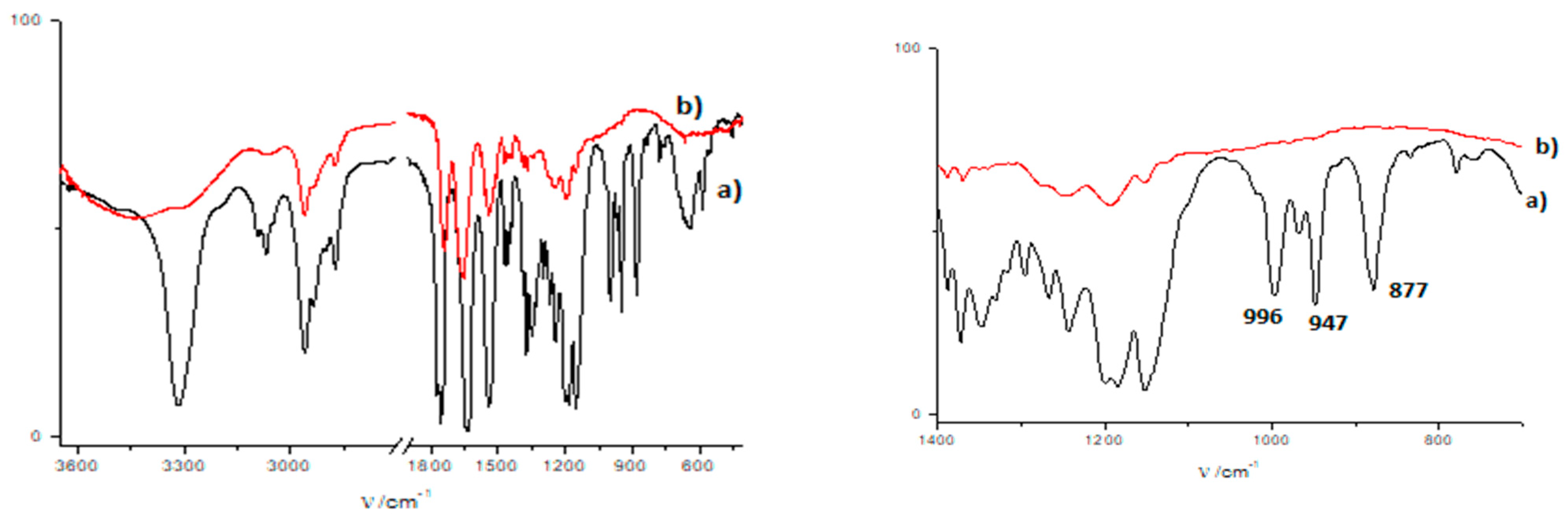
3.4. Gamma-Ray- and Ultraviolet-Induced Polymerization of Gels 1a and 2a
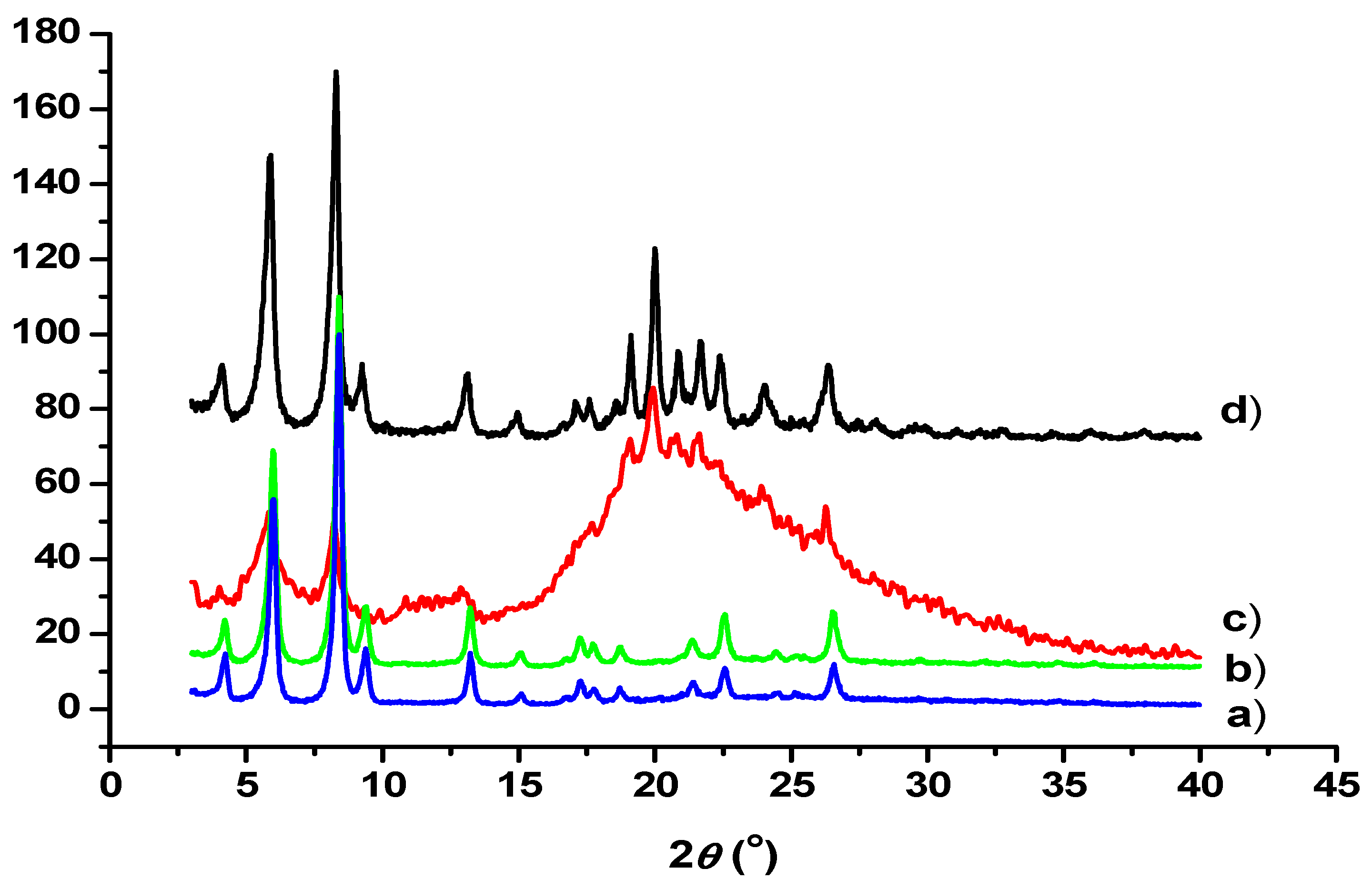
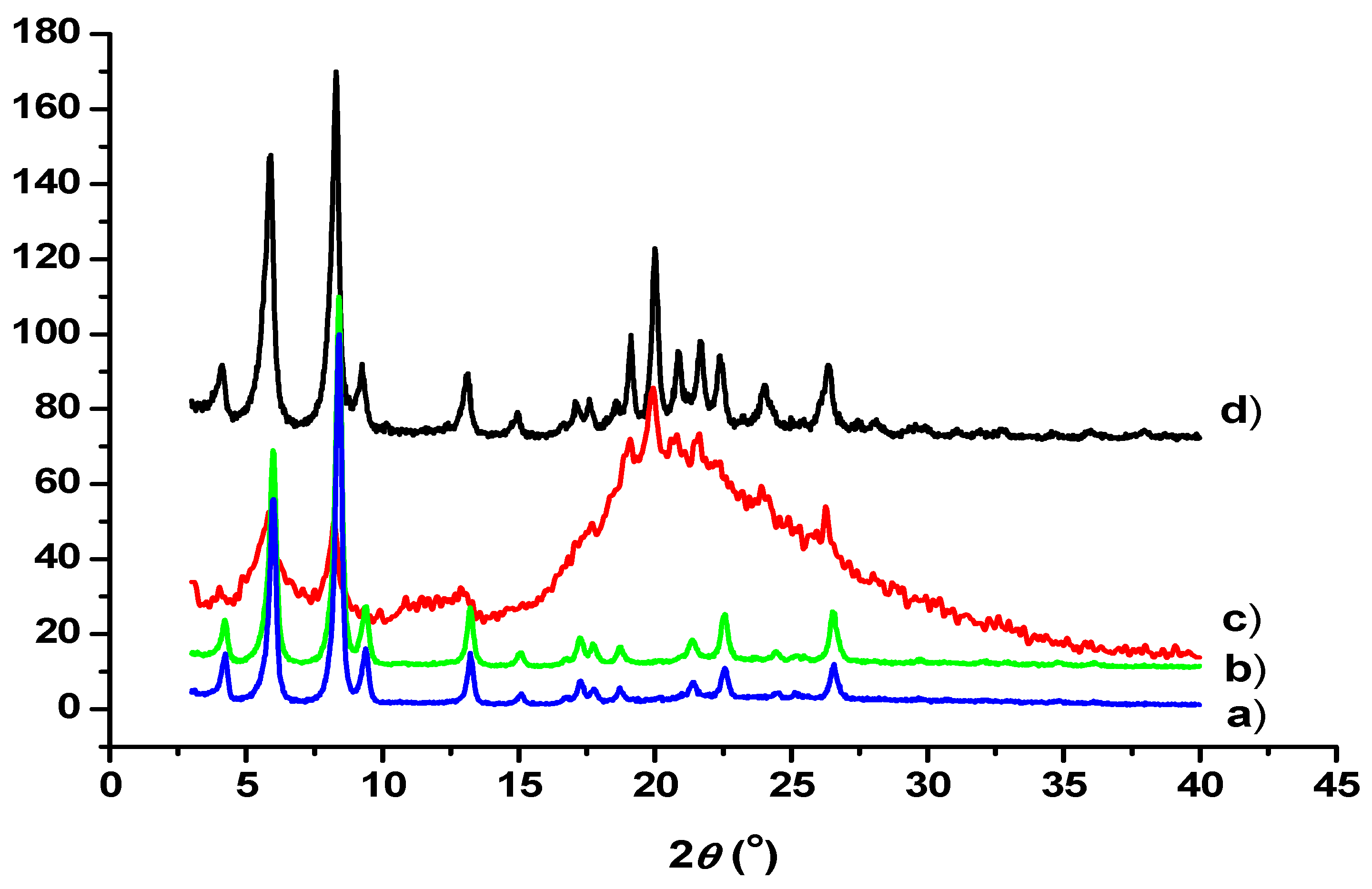
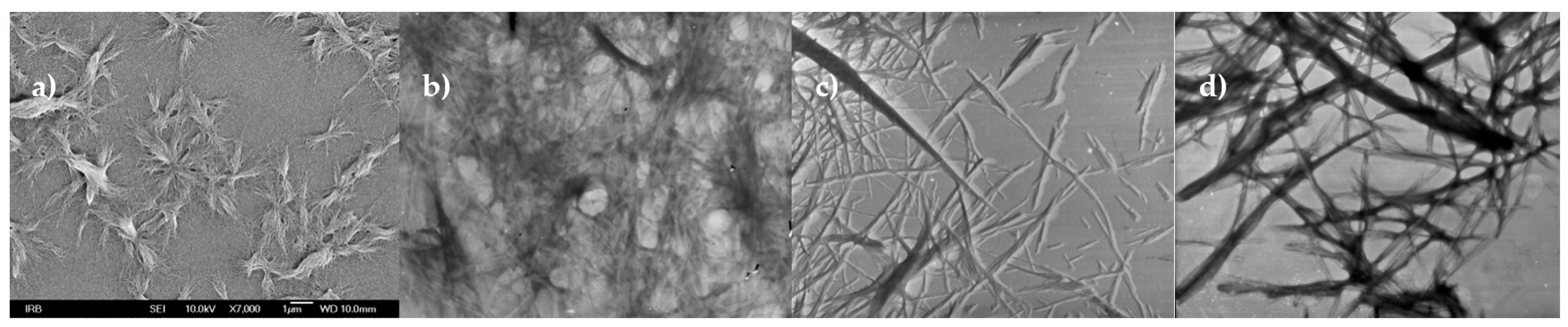
3.5. TEM and SEM Investigations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weiss, R.G.; Terech, P. Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Weiss, R.G., Terech, P., Eds.; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Dastidar, P. Supramolecular gelling agents: Can they be designed? Chem. Soc. Rev. 2008, 37, 2699–2715. [Google Scholar] [CrossRef]
- Terech, P.; Weiss, R.G. Low molecular mass gelators of organic liquids and the properties of their gels. Chem. Rev. 1997, 97, 3133–3159. [Google Scholar] [CrossRef]
- Frkanec, L.; Žinić, M. Chiral bis(amino acid)- and bis(amino alcohol)-oxalamidegelators. Gelation properties, self-assembly motifs and chirality effects. Chem. Commun. 2010, 46, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Corradini, M.G.; Weiss, R.G.; Raghavan, S.R.; Rogers, M.A. To gel or not to gel: Correlating molecular gelation with solvent parameters. Chem. Soc. Rev. 2015, 44, 6035–6058. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y. Gelation with small molecules: From formation mechanism to nanostructure architecture. Top. Curr. Chem. 2005, 256, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Dipeptide and tripeptide conjugates as low-molecular-weight hydrogelators. Macromol. Biosci. 2011, 11, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, G.O.; Steed, J.W. Anion-tuning of supramolecular gel properties. Nat. Chem. 2009, 1, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Sangeetha, N.M.; Maitra, U. Supramolecular gels: Functions and uses. Chem. Soc. Rev. 2005, 34, 821–836. [Google Scholar] [CrossRef] [Green Version]
- Duan, P.; Cao, H.; Zhang, L.; Liu, M. Gelation induced supramolecular chirality: Chirality transfer, amplification and application. Soft Matter 2014, 10, 5428–5448. [Google Scholar] [CrossRef]
- de Loos, M.; Feringa, B.L.; van Esch, J.H. Design and application of self-assembled low molecular weight hydrogels. Eur. J. Org. Chem. 2005, 2005, 3615–3631. [Google Scholar] [CrossRef]
- Babu, S.S.; Praveen, V.K.; Ajayaghosh, A. Functional π-gelators and their applications. Chem. Rev. 2014, 114, 1973–2129. [Google Scholar] [CrossRef]
- Sahoo, S.; Kumar, N.; Bhattacharya, C.; Sagiri, S.S.; Jain, K.; Pal, K.; Ray, S.S.; Nayak, B. Organogels: Properties and applications in drug deliver. Des. Monomers Polym. 2011, 14, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Cornwell, D.J.; Smith, D.K. Expanding the scope of gels—Combining polymers with low-molecular-weight gelators to yield modified self-assembling smart materials with high-tech applications. Mater. Horiz. 2015, 2, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Escuder, B.; Rodríguez-Llansola, F.; Miravet, J.F. Supramolecular gels as active media for organic reactions and catalysis. New J. Chem. 2010, 34, 1044–1054. [Google Scholar] [CrossRef]
- Sagiri, S.S.; Behera, B.; Rafanan, R.R.; Bhattacharya, C.; Pal, K.; Banerjee, I.; Rousseau, D. Organogels as matrices for controlled drug delivery: A review on the current state. Soft Mater. 2014, 12, 47–72. [Google Scholar] [CrossRef]
- Foster, J.A.; Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Howard, J.A.K.; Steed, J.W. Anion-switchable supramolecular gels for controlling pharmaceutical crystal growth. Nat. Chem. 2010, 2, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Makarević, J.; Jokić, M.; Frkanec, L.; Katalenić, D.; Žinić, M.M. Gels with exceptional thermal stability formed by bis(amino acid) oxalamide gelators and solvents of low polarity. Chem. Commun. 2002, 19, 2238–2239. [Google Scholar] [CrossRef]
- Čaplar, V.; Frkanec, L.; Vujičić, N.Š.; Žinić, M. Positionally isomeric organic gelators: Structure–gelation study, racemic versus enantiomeric gelators, and solvation effects. Chem. Eur. J. 2010, 16, 3066–3082. [Google Scholar] [CrossRef]
- Makarević, J.; Jokić, M.; Frkanec, L.; Čaplar, V.; Vujičić, N.Š.; Žinić, M. Oxalyl retro-peptide gelators. Synthesis, gelation properties and stereochemical effects. Beilstein J. Org. Chem. 2010, 6, 945–959. [Google Scholar] [CrossRef]
- Jung, J.H.; Shinkai, S. Gels as templates for nanotubes. Top. Curr. Chem. 2004, 248, 223–260. [Google Scholar] [CrossRef]
- Anilkumar, P.; Jayakannan, M. A novel supramolecular organogel nanotubular template approach for conducting nanomaterials. J. Phys. Chem. B 2010, 114, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qiao, Y.; Gao, C.; Tang, P.; Liu, Y.; Li, Z.; Yan, Y.; Huang, J. Tunable one-dimensional helical nanostructures: From supramolecular self-assemblies to silica nanomaterials. J. Chem. Mater. 2010, 22, 6711–6717. [Google Scholar] [CrossRef]
- Jung, J.H.; Park, M.; Shinkai, S. Fabrication of silica nanotubes by using self-assembled gels and their applications in environmental and biological fields. Chem. Soc. Rev. 2010, 39, 4286–4302. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Chen, H.; Lin, Y.; Yang, Z.; Cheng, X.; Huang, J. Photoluminescent lanthanide-doped silica nanotubes: Sol−gel transcription from functional template. J. Phys. Chem. C 2011, 115, 7323–7330. [Google Scholar] [CrossRef]
- van Esch, J.H.; Feringa, B.L. New functional materials based on self-assembling organogels: From serendipity towards design. Agew. Chem. Int. Ed. 2000, 39, 2263–2266. [Google Scholar] [CrossRef]
- Christoff-Tempesta, T.; Lew, A.J.; Ortony, J.H. Beyond covalent crosslinks: Applications of supramolecular gels. Gels 2018, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Busseron, E.; Ruff, Y.; Moulin, E.; Giuseppone, N. Supramolecular self-assemblies as functional nanomaterials. Nanoscale 2013, 5, 7098–7140. [Google Scholar] [CrossRef] [Green Version]
- Miljanić, S.; Frkanec, L.; Biljan, T.; Meić, Z.; Žinić, M. Surface-enhanced raman scattering on molecular self-assembly in nanoparticle-hydrogel composite. Langmuir 2006, 22, 9079–9081. [Google Scholar] [CrossRef]
- Miljanić, S.; Frkanec, L.; Biljan, T.; Meić, Z.; Žinić, M. Surface-enhanced raman scattering on colloid gels originated from low molecular weight gelator. J. Raman Spectrosc. 2008, 39, 1799–1804. [Google Scholar] [CrossRef]
- Steed, J.W. Supramolecular gel chemistry: Developments over the last decade. Chem. Commun. 2011, 47, 1379–1383. [Google Scholar] [CrossRef]
- Hanabusa, K.; Suzuki, M. Development of low-molecular-weight gelators and polymer-based gelators. Polym. J. 2014, 46, 776–782. [Google Scholar] [CrossRef]
- Paramonov, S.E.; Jun, H.-W.; Hartgerink, J.D. Self-assembly of peptide—Amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc. 2006, 128, 7291–7298. [Google Scholar] [CrossRef]
- Palmer, L.C.; Stupp, S. Molecular self-assembly into one-dimensional nanostructures. Acc. Chem. Res. 2008, 41, 1674–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashfaq, A.; Clochard, M.-C.; Coqueret, X.; Dispenza, C.; Driscoll, M.S.; Ulański, P.; Al-Sheikhly, M. Polymerization reactions and modifications of polymers by ionizing radiation. Polymers 2020, 12, 2877. [Google Scholar] [CrossRef]
- Kim, W.J.; Jung, B.M.; Kang, S.H.; Chang, J.Y. Molecular imprinting into organogel nanofibers. Soft Matter 2011, 7, 4160–4162. [Google Scholar] [CrossRef]
- Mueller, A.; O’Brien, D.F. Supramolecular materials via polymerization of mesophases of hydrated. Amphiphileschem. Rev. 2002, 102, 727–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.H.; Jung, B.M.; Chang, J.Y. Polymerization of an organogel formed by a hetero-bifunctional gelator in a monomeric solvent: Preparation of nanofibers embedded in a polymer matrix. Adv. Mater. 2007, 19, 2780–2784. [Google Scholar] [CrossRef]
- Wang, G.; Hamilton, A.D. Synthesis and self-assembling properties of polymerizable organogelators. Chem. Eur. J. 2002, 8, 1954–1961. [Google Scholar] [CrossRef]
- de Loos, M.; van Esch, J.; Stokroos, I.; Kellogg, R.M.; Feringa, B.L. Remarkable stabilization of self-assembled organogels by polymerization. J. Am. Chem. Soc. 1997, 119, 12675–12676. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Wang, G. Synthesis and self-assembling properties of diacetylene-containing glycolipids. J. Org. Chem. 2006, 71, 4734–4741. [Google Scholar] [CrossRef]
- Tamaoki, N.; Shimada, S.; Okada, Y.; Belaissaoui, A.; Kruk, G.; Yase, K.; Matsuda, H. Polymerization of a diacetylene dicholesteryl ester having two urethanes in organic gel states. Langmuir 2000, 16, 7545–7547. [Google Scholar] [CrossRef]
- George, M.; Weiss, R.G. Low molecular-mass gelators with diyne functional groups and their unpolymerized and polymerized gel assemblies. Chem. Mater. 2003, 15, 2879–2888. [Google Scholar] [CrossRef]
- Wang, G.; Yang, H.; Cheuk, S.; Coleman, S. Synthesis and self-assembly of 1-deoxyglucose derivatives as low molecular weight organogelators. Beilstein J. Org. Chem. 2011, 7, 234–242. [Google Scholar] [CrossRef]
- Dautel, O.J.; Robitzer, M.; Lere-Porte, J.-P.; Serein-Spirau, F.; Moreau, J.J.E. Self-organized ureido substituted diacetylenic organogel. Photopolymerization of one-dimensional supramolecular assemblies to give conjugated nanofibers. J. Am. Chem. Soc. 2006, 128, 16213–16223. [Google Scholar] [CrossRef] [PubMed]
- Altoè, P.; Haraszkiewicz, N.; Gatti, F.G.; Wiering, P.G.; Frochot, C.; Brouwer, A.M.; Balkowski, G.; Shaw, D.; Woutersen, S.; Buma, W.J.; et al. Multistate photo-induced relaxation and photoisomerization ability of fumaramide threads: A computational and experimental study. J. Am. Chem. Soc. 2009, 131, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Miljanić, S.; Frkanec, L.; Meić, Z.; Žinić, M. Photoinduced gelation by stilbene oxalyl amide compounds. Langmuir 2005, 21, 2754–2760. [Google Scholar] [CrossRef] [PubMed]
- Miljanić, S.; Frkanec, L.; Meić, Z.; Žinić, M. Gelation ability of novel oxamide-based derivatives bearing a stilbene as a photo-responsive unit. Eur. J. Org. Chem. 2006, 2005, 1323–1334. [Google Scholar] [CrossRef]
- Frkanec, L.; Jokić, M.; Makarević, J.; Wolsperger, K.; Žinić, M. Bis(PheOH) maleic acid amide-fumaric acid amide photoizomerization induces microsphere-to-gel fiber morphological transition: The photoinduced gelation system. J. Am. Chem. Soc. 2002, 124, 9716–9717. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yamaguchi, S.; Ueno, S.; Komatsu, H.; Ikeda, M.; Ishizuka, K.; Iko, Y.; Tabata, K.V.; Aoki, H.; Ito, S.; et al. Photo gel–sol/sol–gel transition and its patterning of a supramolecular hydrogel as stimuli-responsive biomaterials. Chem. Eur. J. 2008, 14, 3977–3986. [Google Scholar] [CrossRef]
- van Dongen, S.F.M.; Cantekin, S.; Elemans, J.A.A.W.; Rowan, A.E.; Nolte, R.J.M. Functional interlocked systems. Chem. Soc. Rev. 2014, 43, 99–122. [Google Scholar] [CrossRef] [Green Version]
- Gatti, F.G.; León, S.; Wong, J.K.Y.; Bottari, G.; Altieri, A.; Morales, M.A.F.; Teat, S.J.; Frochot, C.; Leigh, D.A.; Brouwer, A.M.; et al. Photoisomerization of a rotaxane hydrogen bonding template: Light-induced acceleration of a large amplitude rotational motion. Proc. Natl. Acad. Sci. USA 2003, 100, 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, J.V.; Kay, E.R.; Leigh, D.A. A reversible synthetic rotary molecular motor. Science 2004, 306, 1532–1537. [Google Scholar] [CrossRef] [Green Version]
- Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D.A.; Wong, J.K.Y.; Zerbetto, F. Remarkable positional discrimination in bistable light- and heat-switchable hydrogen-bonded molecular shuttles. Angew. Chem. Int. Ed. 2003, 42, 2296–2300. [Google Scholar] [CrossRef]
- Fisher, J.P.; Dean, D.; Mikos, A.G. Photocrosslinking characteristics and mechanical properties of diethyl fumarate/poly(propylene fumarate) biomaterials. Biomaterials 2002, 23, 4333–4343. [Google Scholar] [CrossRef]
- Azuma, C.; Ogata, N. Radical polymerizability of fumaramide derivatives. J. Polym. Sci. 1974, 12, 759–768. [Google Scholar] [CrossRef]
- Wei, H.; Lee, T.Y.; Miao, W.; Fortenberry, R.; Magers, D.H.; Hait, S.; Guymon, A.C.; Jonsson, S.E.; Hoyle, C.E. Characterization and photopolymerization of divinyl fumarate. Macromolecules 2007, 40, 6172–6180. [Google Scholar] [CrossRef]
- Nguyen, K.T.; West, J.L. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials 2002, 23, 4307–4314. [Google Scholar] [CrossRef]
- Cortizoa, M.S.; Laurellaa, S.; Alessandrini, J.L. Microwave-assisted radical polymerization of dialkyl fumarates. Radiat. Phys. Chem. 2007, 76, 1140–1146. [Google Scholar] [CrossRef]
- Alkassiri, H. Radiation polymerization of diethyl fumarate. Radiat. Phys. Chem. 2005, 73, 61–63. [Google Scholar] [CrossRef]
- Zayzafoon, G.; Alkassiri, H. Diethyl fumarate dimethyl formamide solution for high dose dosimetry. Radiat. Meas. 2008, 43, 1550–1553. [Google Scholar] [CrossRef]
- Michinobu, T.; Nakada, K.; Shigehara, K. Radical polymerization of fumaramide and fumaramate derivatives for homogeneous Langmuir monolayers. Polym. Bull. 2008, 60, 49–55. [Google Scholar] [CrossRef]
- Diaz, D.D.; Rajagopal, K.; Strable, E.; Schneider, J.; Finn, M.G. “Click” chemistry in a supramolecular environment: Stabilization of organogels by copper(I)-catalyzed azide-alkyne [3 + 2] cycloadd. J. Am. Chem. Soc. 2006, 128, 6056–6057. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann Elsevier Inc.: Oxford, UK, 2009. [Google Scholar]
- Menger, F.M.; Caran, K.L. Anatomy of a gel. Amino acid derivatives that rigidify water at submillimolar concentrations. J. Am. Chem. Soc. 2000, 122, 11679. [Google Scholar] [CrossRef]
- Vemula, P.K.; John, G. Smart amphiphiles: Hydro/organogelators for in situreduction of gold. Chem. Commun. 2006, 21, 2218. [Google Scholar] [CrossRef] [PubMed]
- Leonard, N.M.; Brunckova, J. In situ formation of N-trifluoroacetoxy succinimide (TFA-NHS): One-pot formation of succinimidyl esters, N-trifluoroacetyl amino acid succinimidyl esters, and N-maleoyl amino acid succinimidyl esters. J. Org. Chem. 2011, 76, 9169–9174. [Google Scholar] [CrossRef]
- Thomas, G.B.; Lipscomb, C.E.; Mahanthappa, M.K. Amino acid vinyl esters: A new monomer palette for degradable polycationic materials. Polym. Chem. 2012, 3, 741–750. [Google Scholar] [CrossRef]
- Baures, P.W.; Beatty, A.M.; Dhanasekaran, M.; Helfrich, B.A.; Pérez-Segarra, W.; Desper, J. Solution and solid-state models of peptide CH···O hydrogen bonds. J. Am. Chem. Soc. 2002, 124, 11315–11323. [Google Scholar] [CrossRef]
- Streuff, J.; Nieger, M.; Muniz, K. Synthesis of small tripeptide molecules through a catalysis sequence comprising metathesis and aminohydroxylation. Chem. Eur. J. 2006, 12, 4362–4371. [Google Scholar] [CrossRef]
- Popp, B.V.; Thorman, J.L.; Stahl, S.S. Similarities between the reactions of dioxygen and alkenes with palladium(0): Relevance to the use of benzoquinone and molecular oxygen as stoichiometric oxidants in palladium-catalyzed oxidation reactions. J. Mol. Catal. A Chem. 2006, 251, 2–7. [Google Scholar] [CrossRef]
- Sybyl-X Molecular Modeling Software Packages, Version 2.0; Certara, Tripos Inc.: St. Louis, MO, USA, 2012.
- Christensen, I.T.; Jørgensen, F.S. Molecular mechanics calculations of proteins. Comparison of different energy minimization strategies. J. Biomol. Struct. Dyn. 1997, 15, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Hema, K.; Ravi, A.; Raju, C.; Pathan, J.R.; Rai, R.; Sureshan, K.M. Topochemical polymerizations for the solid-state synthesis of organic polymers. Chem. Soc. Rev. 2021, 50, 4062–4099. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | 1a | 2a |
|---|---|---|
| H2O | ns | ns. |
| H2O/DMSO | ng | 6.2 + s5.3 * |
| H2O/DMF | 0.44 + 0.48 * | cryst. |
| EtOH | sol. | cryst. |
| ±2-octanol | sol. | cryst. |
| THF | 0.26 + 2.8 ** | sol. |
| dioxane | sol. | sol. |
| acetone | 0.1 + 1.3 **US | cryst. |
| EtOAc | 0.2 + 1.2 **US | cryst. |
| CH2Cl2 | ng | sol. |
| CH3CN | cryst. | cryst. |
| toluene | 0.2 US | 1.6 US |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorić, T.; Makarević, J.; Štefanić, Z.; Žinić, M.; Frkanec, L. Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies. Polymers 2022, 14, 214. https://doi.org/10.3390/polym14010214
Gregorić T, Makarević J, Štefanić Z, Žinić M, Frkanec L. Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies. Polymers. 2022; 14(1):214. https://doi.org/10.3390/polym14010214
Chicago/Turabian StyleGregorić, Tomislav, Janja Makarević, Zoran Štefanić, Mladen Žinić, and Leo Frkanec. 2022. "Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies" Polymers 14, no. 1: 214. https://doi.org/10.3390/polym14010214
APA StyleGregorić, T., Makarević, J., Štefanić, Z., Žinić, M., & Frkanec, L. (2022). Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies. Polymers, 14(1), 214. https://doi.org/10.3390/polym14010214