
The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics



Abstract
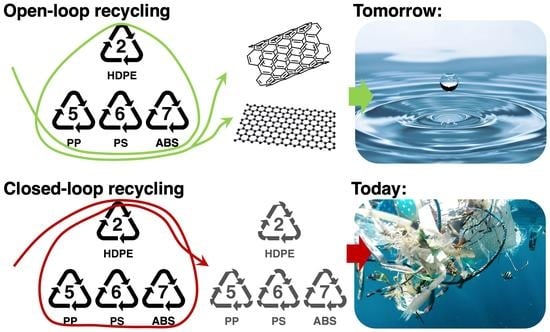
1. Introduction
2. Physical and Chemical Context
2.1. Solubility of Plastics and Basic Polymer Solution Thermodynamics
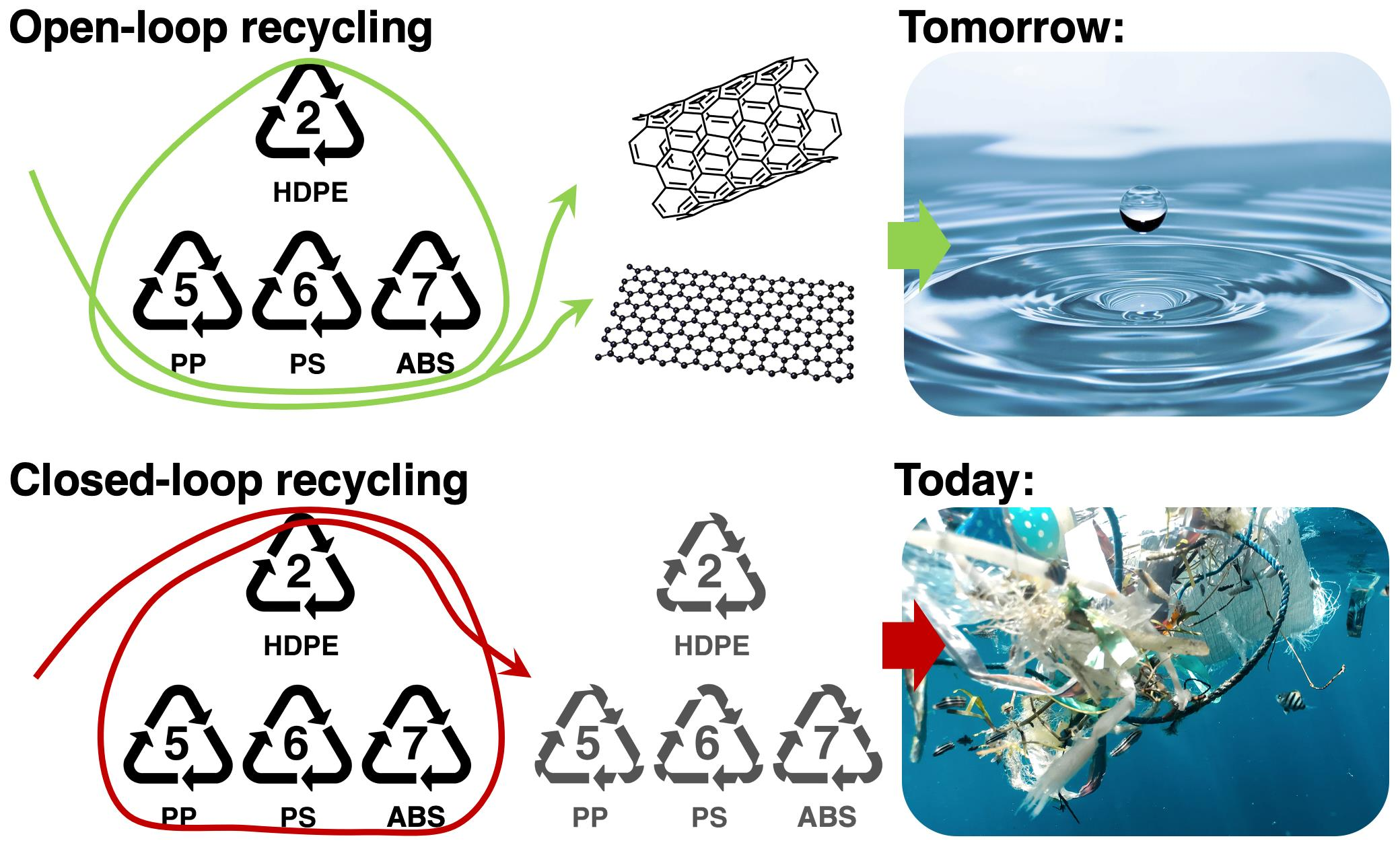
2.1.1. Hildebrand Solubility Parameters and Thermodynamics of Polymer Dissolution
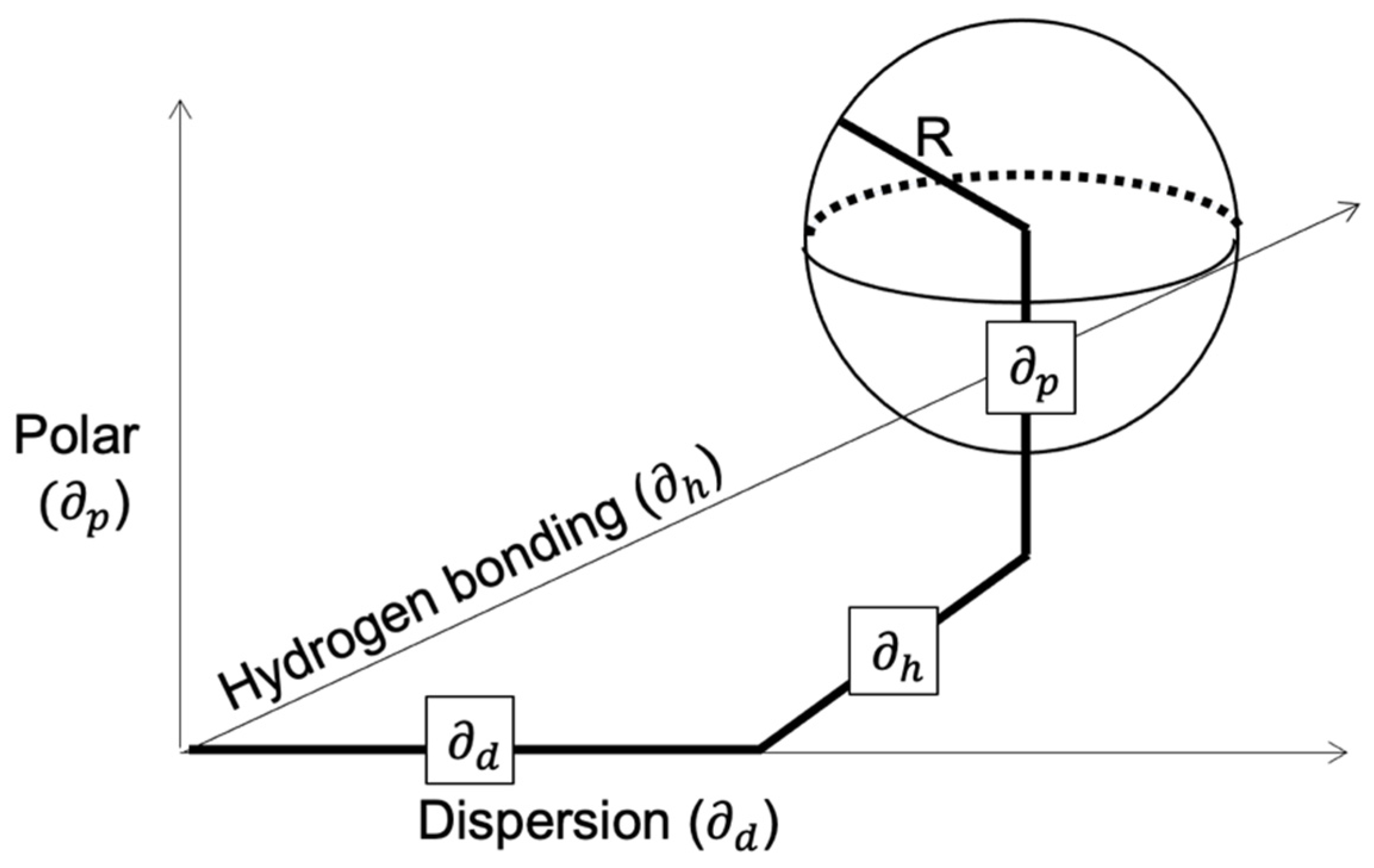
2.1.2. Hansen Solubility Parameters (HSP)
- The cohesive energy from dispersion interactions is described as ED.
- The permanent dipole–dipole interactions. These cause a second type of cohesion energy, the polar cohesive energy, EP. These interaction parameters are also found in most molecules to one extent or another.
- Hydrogen bonding, EH, also known as the electron exchange parameter, resembles the hydrogen polar interactions. In a more simplified approach, this third term is used to collect all the energies from the rest of the interactions not included in polar or dipole cohesive energy components. It has been proven that the Hansen hydrogen bonding has served well and is of practical importance.
2.1.3. Models for the Prediction of Polymer Solubility
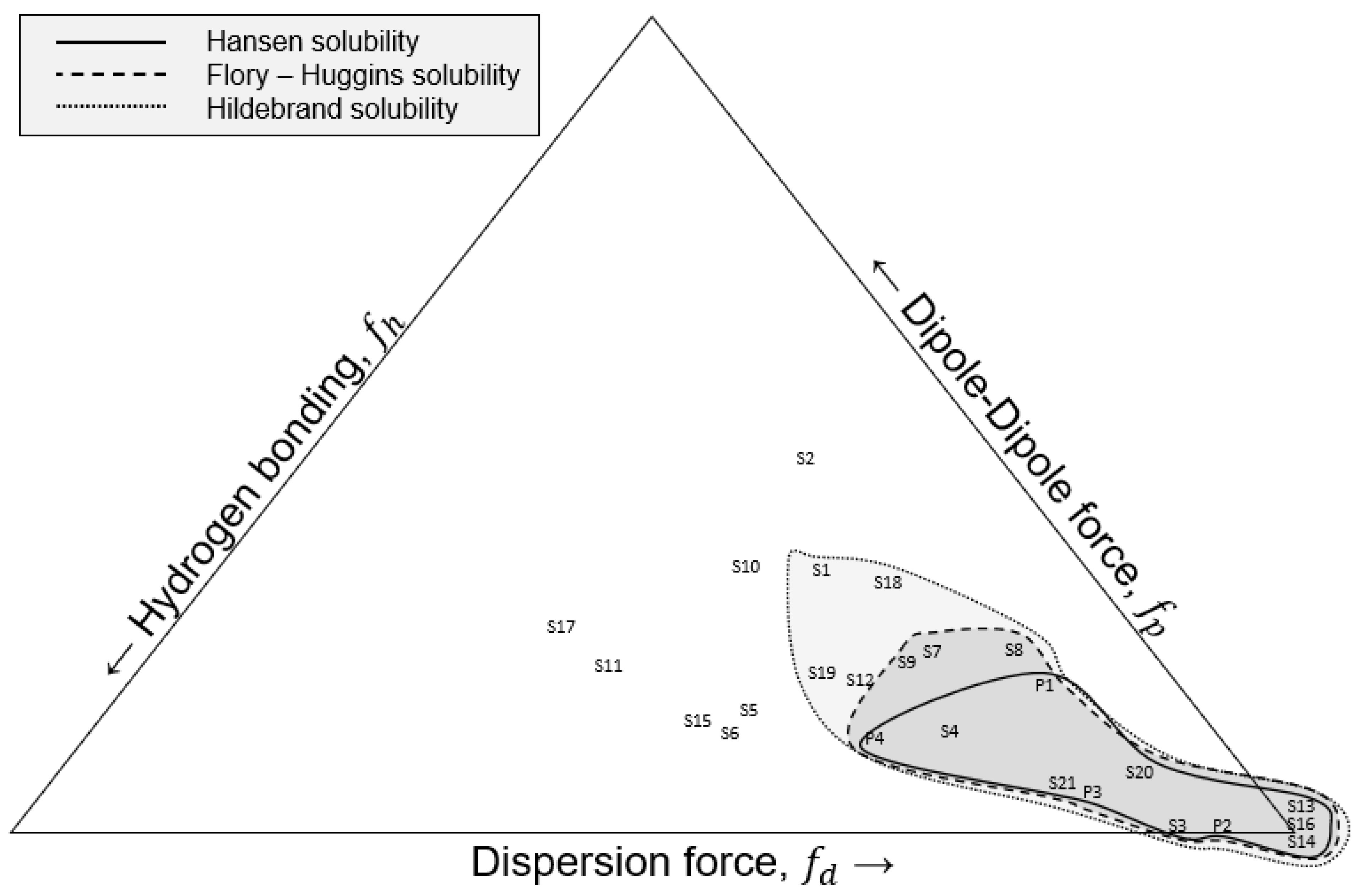
2.1.4. Rules of Thumb and Solvent Selection
- Using the Hildebrand solubility parameters.
- Using Hansen Solubility Parameters (HSP) in the Formula (10) under the condition of R0 ≥ Rα.
- Using the Flory–Huggins parameter, χ12 ≤ 0.5. The lower the parameter, the greater the miscibility. Values above 0.5 indicate insolvency. The ‘chi’ parameter is estimated from the following set of formulas.where V is the reference volume, measured in cm3/mol, R is the gas constant, T is the temperature in Kelvin, and A has units of MPa.
2.1.5. Temperature Dependence on Solubility Parameters
2.2. Polymer Dissolution Models
2.2.1. Phenomenological Models
2.2.2. External Mass Transfer Models, Stress Relaxation, and Molecular Theories
2.2.3. Anomalous Transport Models and Scaling Laws
2.2.4. Molecular Theories in a Continuum Framework
3. Process Selection
3.1. Material Selection
3.2. Operating Procedure
3.3. Material Balance and Dissolution Times
3.3.1. Assumptions
3.3.2. Plastic Dissolution
3.4. Notes on Applicability, Toluene Safety, Energy Requirements, and General Environmental Considerations
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Benefits & Advantages of Plastic. Available online: https://www.bpf.co.uk/industry/benefits_of_plastics.aspx (accessed on 10 November 2021).
- Orbaek White, A. Black Plastic Can’t Be Recycled—But We’ve Just Found a Way to Use the Carbon in Renewable Energy. Available online: http://theconversation.com/black-plastic-cant-be-recycled-but-weve-just-found-a-way-to-use-the-carbon-in-renewable-energy-100037 (accessed on 10 November 2021).
- Orbaek White, A. Plastic Pollution: Why Chemical Recycling Could Provide a Solution. Available online: http://theconversation.com/plastic-pollution-why-chemical-recycling-could-provide-a-solution-129917 (accessed on 10 November 2021).
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef] [PubMed]
- Miller-Chou, B.A.; Koenig, J.L. A review of polymer dissolution. Prog. Polym. Sci. 2003, 28, 1223–1270. [Google Scholar] [CrossRef]
- Hedayati, A.; Barnett, C.; Swan, G.; Orbaek White, A. Chemical recycling of consumer-grade black plastic into electrically conductive carbon nanotubes. C 2019, 5, 32. [Google Scholar] [CrossRef]
- Burke, J. Solubility parameters: Theory and application. Book Pap. Group Annu. 1984, 3, 34. [Google Scholar]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 978-0-8493-7248-3. [Google Scholar]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Stefanis, E.; Panayiotou, C. Prediction of Hansen solubility parameters with a new group-contribution method. Int. J. Thermophys. 2008, 29, 568–585. [Google Scholar] [CrossRef]
- Hansen, C.M.; Abbot, S. Designer Solvent Blends|Hansen Solubility Parameters. Available online: https://www.hansen-solubility.com/HSP-science/solvent-blends.php (accessed on 10 November 2021).
- Teas, J. Graphic analysis of resin solubilities. J. Paint Technol. 1968, 40, 19–25. [Google Scholar]
- Asmussen, F.; Ueberreiter, K. Velocity of dissolution of polymers. Part II. J. Polym. Sci. 1962, 57, 199–208. [Google Scholar] [CrossRef]
- Narasimhan, B.; Peppas, N.A. The physics of polymer dissolution: Modeling approaches and experimental behavior. In Polymer Analysis Polymer Physics; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1997; Volume 128, pp. 157–207. ISBN 978-3-540-61218-6. [Google Scholar]
- Tu, Y.-O.; Ouano, A.C. Model for the kinematics of polymer dissolution. IBM J. Res. Dev. 1977, 21, 131–142. [Google Scholar] [CrossRef]
- Tu, Y. A multi-phase Stefan problem describing the swelling and the dissolution of glassy polymer. Q. Appl. Math. 1977, 35, 269–285. [Google Scholar] [CrossRef][Green Version]
- Devotta, I.; Ambeskar, V.D.; Mandhare, A.B.; Mashelkar, R.A. The life time of a dissolving polymeric particle. Chem. Eng. Sci. 1994, 49, 645–654. [Google Scholar] [CrossRef]
- Peppas, N.A.; Wu, J.C.; von Meerwall, E.D. Mathematical modeling and experimental characterization of polymer dissolution. Macromolecules 1994, 27, 5626–5638. [Google Scholar] [CrossRef]
- Narasimhan, B.; Peppas, N.A. Disentanglement and reptation during dissolution of rubbery polymers. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 947–961. [Google Scholar] [CrossRef]
- Nauman, E.B.; Lynch, J.C. Polymer Recycling by Selective Dissolution. U.S. Patent 5,278,282, 11 January 1994. [Google Scholar]
- Orbaek White, A.; Hedayati, A.; Yick, T.; Gangoli, V.S.; Niu, Y.; Lethbridge, S.; Tsampanakis, I.; Swan, G.; Pointeaux, L.; Crane, A.; et al. On the use of carbon cables from plastic solvent combinations of polystyrene and toluene in carbon nanotube synthesis. Nanomaterials 2021, 12, 9. [Google Scholar] [CrossRef]
- Vieira, O.; Ribeiro, R.S.; Diaz de Tuesta, J.L.; Gomes, H.T.; Silva, A.M.T. A Systematic literature review on the conversion of plastic wastes into valuable 2D graphene-based materials. Chem. Eng. J. 2022, 428, 131399. [Google Scholar] [CrossRef]
- Viswanath, D.S.; Ghosh, T.K.; Prasad, D.H.L.; Dutt, N.V.K.; Rani, K.Y. (Eds.) Viscosities of solutions and mixtures. In Viscosity of Liquids: Theory, Estimation, Experiment, and Data; Springer: Dordrecht, The Netherlands, 2007; pp. 407–442. ISBN 978-1-4020-5482-2. [Google Scholar]
- Tao, S.; Li, B.; Huang, H. Dissolution of expanded polystyrene in cycloalkane solutions. Open J. Appl. Sci. 2018, 8, 465–471. [Google Scholar] [CrossRef][Green Version]
- Flick, E.W. (Ed.) Industrial Solvents Handbook, 5th ed; William Andrew Publishing: Norwich, NY, USA, 1998; p. 832. ISBN 9780815514138. [Google Scholar]
- Toluene Production in the United States from 1990 to 2019 (in 1000 Metric Tons). Available online: https://www.statista.com/statistics/974854/us-toluene-production-volume/ (accessed on 23 November 2021).
- Khalfa, A.L.; Becker, M.L.; Dove, A.P. Stereochemistry-controlled mechanical properties and degradation in 3D-printable photosets. J. Am. Chem. Soc. 2021, 143, 17510–17516. [Google Scholar] [CrossRef] [PubMed]
- Achilias, D.S.; Giannoulis, A.; Papageorgiou, G.Z. Recycling of polymers from plastic packaging materials using the dissolution-reprecipitation technique. Polym. Bull. 2009, 63, 449–465. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, uses, and fate of all plastics ever made. Sci. Adv. 2017, 3, 5. [Google Scholar] [CrossRef]
- Plastic Food Packaging Waste. Available online: https://post.parliament.uk/research-briefings/post-pn-0605/ (accessed on 23 November 2021).
- Energy Recovery from the Combustion of Municipal Solid Waste (MSW). Available online: https://www.epa.gov/smm/energy-recovery-combustion-municipal-solid-waste-msw (accessed on 23 November 2021).
- Schyns, Z.O.G.; Shaver, M.P. Mechanical recycling of packaging plastics: A review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.T. Hydrogen and carbon nanotubes from pyrolysis-catalysis of waste plastics: A review. Waste Biomass Valoriz. 2021, 12, 1–28. [Google Scholar] [CrossRef]
- Jiang, J.; Shi, K.; Zhang, X.; Yu, K.; Zhang, H.; He, J.; Ju, Y.; Liu, J. From plastic waste to wealth using chemical recycling: A review. J. Environ. Chem. Eng. 2022, 10, 106867. [Google Scholar] [CrossRef]
- Jeswani, H.; Krüger, C.; Russ, M.; Horlacher, M.; Antony, F.; Hann, S.; Azapagic, A. Life cycle environmental impacts of chemical recycling via pyrolysis of mixed plastic waste in comparison with mechanical recycling and energy recovery. Sci. Total Environ. 2021, 769, 144483. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
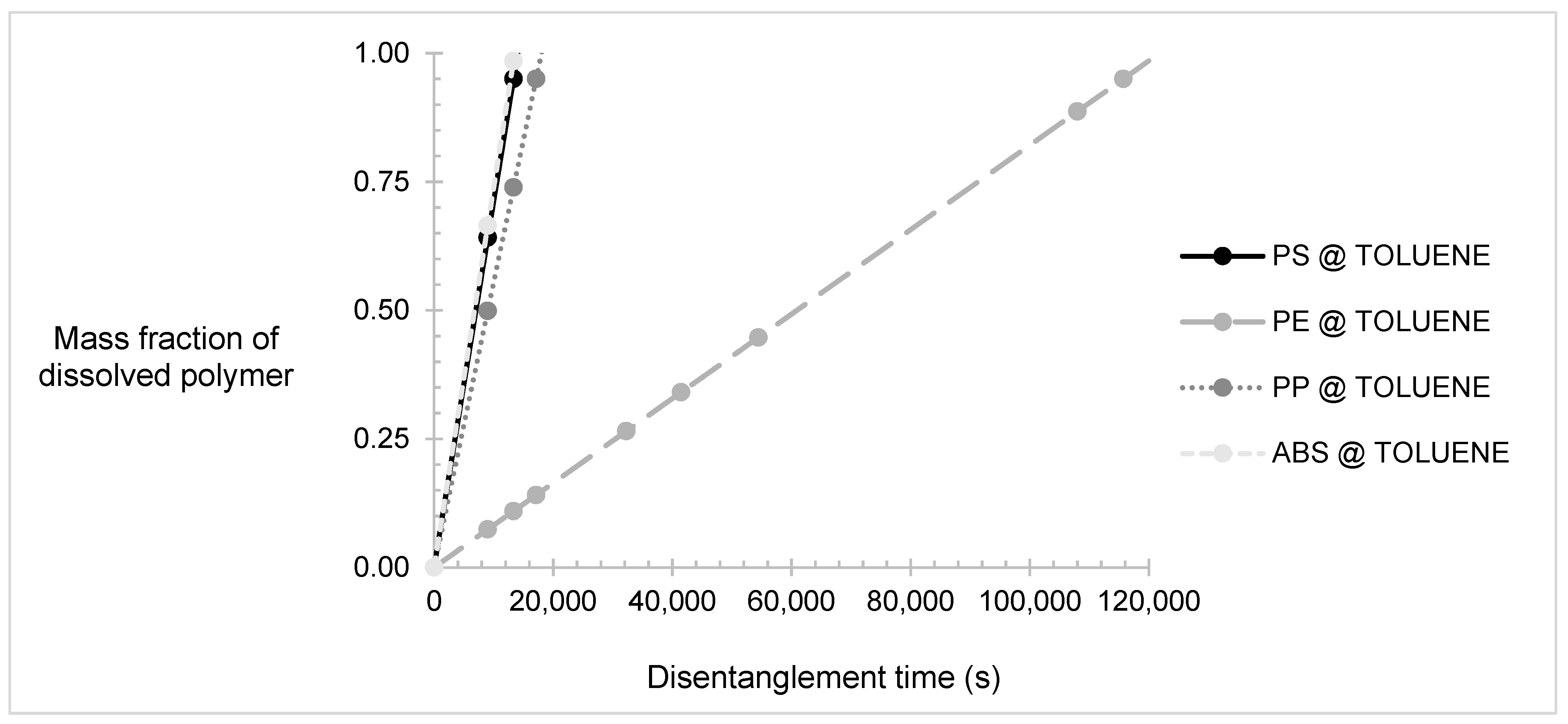{kind=link}
| RED | Average | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent/Polymer | PS | Affinity | PP | Affinity | HDPE | Affinity | ABS | Affinity | RED | Affinity |
| Acetone | 1.8 | Low | 2.2 | Low | 3.3 | Low | 1.0 | Low | 2.1 | Low |
| Acetonitrile | 2.9 | Low | 3.2 | Low | 5.5 | Low | 2.0 | Low | 3.4 | Low |
| Benzene | 0.9 | High | 0.2 | High | 1.5 | Low | 0.9 | High | 0.9 | High |
| Chloroform | 0.6 | High | 0.9 | High | 1.6 | Low | 0.4 | High | 0.9 | High |
| m-Cresol | 1.9 | Low | 2.2 | Low | 3.7 | Low | 0.9 | High | 2.2 | Low |
| Cyclohexanol | 2.0 | Low | 2.2 | Low | 3.6 | Low | 0.9 | High | 2.2 | Low |
| Cyclohexanone | 0.6 | High | 1.3 | Low | 2.2 | Low | 0.7 | High | 1.2 | Low |
| 1,2 Dichlorobenzene | 0.4 | High | 1.2 | Low | 2.6 | Low | 1.0 | Low | 1.3 | Low |
| Dichloromethane | 0.7 | High | 1.4 | Low | 2.4 | Low | 0.7 | High | 1.3 | Low |
| Dimethylformamide | 2.4 | Low | 2.9 | Low | 4.9 | Low | 1.5 | Low | 2.9 | Low |
| Ethanol | 3.4 | Low | 3.5 | Low | 5.8 | Low | 1.8 | Low | 3.6 | Low |
| Ethyl acetate | 1.3 | Low | 1.5 | Low | 2.0 | Low | 0.4 | High | 1.3 | Low |
| Heptane | 1.6 | Low | 0.9 | High | 1.0 | Low | 1.0 | Low | 1.1 | Low |
| Hexadecane | 1.3 | Low | 0.6 | High | 0.9 | High | 1.0 | High | 1.0 | High |
| Hexafluoro-2-propanol | 2.3 | Low | 2.4 | Low | 4.0 | Low | 1.0 | Low | 2.4 | Low |
| Hexane | 1.7 | Low | 1.0 | Low | 1.1 | Low | 1.0 | Low | 1.2 | Low |
| Methanol | 4.1 | Low | 4.2 | Low | 7.1 | Low | 2.3 | Low | 4.4 | Low |
| Methyl Ethyl Ketone | 1.3 | Low | 1.8 | Low | 2.7 | Low | 0.9 | High | 1.7 | Low |
| Tetrahydrofuran | 1.2 | Low | 1.6 | Low | 2.3 | Low | 0.4 | High | 1.4 | Low |
| Toluene | 0.6 | High | 0.3 | High | 1.3 | Low | 0.8 | High | 0.8 | High |
| Xylene | 0.7 | High | 0.4 | High | 1.0 | Low | 0.6 | High | 0.7 | High |
| 15% Acetonitrile 85% Toluene | 0.4 | High | 0.7 | High | 1.4 | Low | 0.7 | High | 0.8 | High |
| 40% Cyclohexanone 60% Toluene | 0.8 | High | 0.2 | High | 1.1 | Low | 0.8 | High | 0.7 | High |
| 50% Cyclohexanone 50% Xylene | 0.4 | High | 1.0 | High | 1.7 | Low | 0.6 | High | 0.9 | High |
| 80% Cyclohexane 20% Cyclohexanol | 0.9 | High | 0.5 | High | 0.6 | High | 0.6 | High | 0.7 | High |
| 87% Cyclohexane 13% Ethanol | 2.2 | Low | 2.4 | Low | 3.8 | Low | 1.0 | High | 2.4 | Low |
| 25% Heptane 75% Xylene | 0.9 | High | 0.4 | High | 0.7 | High | 0.7 | High | 0.7 | High |
| 25% Cyclohexane 75% Toluene | 0.8 | High | 0.2 | High | 1.1 | Low | 0.8 | High | 0.7 | High |
| 40% Cyclohexane 60% Xylene | 0.9 | High | 0.3 | High | 0.8 | High | 0.8 | High | 0.7 | High |
| Solvent | PS | Affinity | PP | Affinity | HDPE | Affinity | ABS | Affinity | Average | Affinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Acetone | 0.7 | High | 1.9 | High | 3.7 | High | 2.0 | High | 2.1 | High |
| Acetonitrile | 5.1 | Low | 6.4 | Low | 8.1 | Low | 6.4 | Low | 6.5 | Low |
| Benzene | 0.8 | High | 0.5 | High | 2.2 | High | 0.5 | High | 1.0 | High |
| Chloroform | 0.3 | High | 0.9 | High | 2.7 | High | 1.0 | High | 1.2 | High |
| m-Cresol | 3.5 | High | 4.7 | Low | 6.5 | Low | 4.7 | Low | 4.8 | Low |
| Cyclohexanol | 3.1 | High | 4.4 | Low | 6.1 | Low | 4.4 | Low | 4.5 | Low |
| Cyclohexanone | 0.3 | High | 1.5 | High | 3.3 | High | 1.6 | High | 1.7 | High |
| 1,2 Dichlorobenzene | 1.2 | High | 2.4 | High | 4.2 | Low | 2.5 | High | 2.6 | High |
| Dichloromethane | 0.9 | High | 2.2 | High | 3.9 | Low | 2.2 | High | 2.3 | High |
| Dimethylformamide | 5.6 | Low | 6.8 | Low | 8.6 | Low | 6.9 | Low | 7.0 | Low |
| Ethanol | 7.3 | Low | 8.5 | Low | 10.3 | Low | 8.5 | Low | 8.6 | Low |
| Ethyl acetate | 1.1 | High | 0.1 | High | 1.9 | High | 0.2 | High | 0.8 | High |
| Heptane | 4.0 | Low | 2.7 | High | 1.0 | High | 2.7 | High | 2.6 | High |
| Hexadecane | 3.0 | High | 1.7 | High | 0.0 | High | 1.7 | High | 1.6 | High |
| Hexafluoro-2-propanol | 3.8 | Low | 5.0 | Low | 6.8 | Low | 5.1 | Low | 5.2 | Low |
| Hexane | 4.4 | Low | 3.1 | High | 1.4 | High | 3.1 | High | 3.0 | High |
| Methanol | 10.3 | Low | 11.6 | Low | 13.3 | Low | 11.6 | Low | 11.7 | Low |
| Methyl ethyl ketone | 0.2 | High | 1.0 | High | 2.8 | High | 1.1 | High | 1.3 | High |
| Tetrahydrofuran | 0.2 | High | 1.4 | High | 3.2 | High | 1.5 | High | 1.6 | High |
| Toluene | 1.1 | High | 0.1 | High | 1.9 | High | 0.2 | High | 0.8 | High |
| Xylene | 1.4 | High | 0.1 | High | 1.6 | High | 0.1 | High | 0.8 | High |
| x1,2 | PS | Affinity | PP | Affinity | HDPE | Affinity | ABS | Affinity | Average | Affinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Acetone | 0.7 | Low | 1.3 | Low | 0.8 | Low | 0.5 | High | 0.9 | Low |
| Acetonitrile | 2.4 | Low | 3.8 | Low | 3.1 | Low | 2.4 | Low | 3.1 | Low |
| Benzene | 0.2 | High | 0.0 | High | 0.2 | High | 0.5 | High | 0.1 | High |
| Chloroform | 0.1 | High | 0.3 | High | 0.2 | High | 0.1 | High | 0.2 | High |
| m-Cresol | 1.0 | Low | 1.7 | Low | 1.4 | Low | 0.5 | Low | 1.4 | Low |
| Cyclohexanol | 1.2 | Low | 1.8 | Low | 1.3 | Low | 0.5 | High | 1.4 | Low |
| Cyclohexanone | 0.1 | High | 0.6 | Low | 0.5 | Low | 0.3 | High | 0.4 | High |
| 1,2 Dichlorobenzene | 0.1 | High | 0.5 | Low | 0.7 | Low | 0.6 | Low | 0.4 | High |
| Dichloromethane | 0.1 | High | 0.7 | Low | 0.6 | Low | 0.3 | High | 0.5 | High |
| Dimethylformamide | 1.6 | Low | 3.0 | Low | 2.5 | Low | 1.4 | Low | 2.4 | Low |
| Ethanol | 3.2 | Low | 4.4 | Low | 3.4 | Low | 1.9 | Low | 3.7 | Low |
| Ethyl acetate | 0.5 | High | 0.9 | Low | 0.4 | High | 0.1 | High | 0.6 | Low |
| Heptane | 0.7 | Low | 0.3 | High | 0.1 | High | 0.6 | Low | 0.4 | High |
| Hexadecane | 0.5 | High | 0.1 | High | 0.1 | High | 0.6 | Low | 0.2 | High |
| Hexafluoro-2-propanol | 1.5 | Low | 2.1 | Low | 1.6 | Low | 0.6 | Low | 1.7 | Low |
| Hexane | 1.1 | Low | 0.5 | Low | 0.2 | High | 0.9 | Low | 0.6 | Low |
| Methanol | 2.0 | Low | 2.6 | Low | 2.1 | Low | 1.4 | Low | 2.2 | Low |
| Methyl ethyl ketone | 0.5 | High | 1.0 | Low | 0.7 | Low | 0.4 | High | 0.7 | Low |
| Tetrahydrofuran | 0.4 | High | 0.9 | Low | 0.5 | Low | 0.1 | High | 0.6 | Low |
| Toluene | 0.1 | High | 0.0 | High | 0.2 | High | 0.4 | High | 0.1 | High |
| Xylene | 0.2 | High | 0.1 | High | 0.1 | High | 0.3 | High | 0.1 | High |
| Polymer Number | Polymer |
|---|---|
| P1 | Polystyrene |
| P2 | Polypropylene |
| P3 | High-density polyethylene |
| P4 | Acrylonitrile–butadiene–styrene |
| Solvent Number | Solvent |
|---|---|
| S1 | Acetone |
| S2 | Acetonitrile |
| S3 | Benzene |
| S4 | Chloroform |
| S5 | m-Cresol |
| S6 | Cyclohexanol |
| S7 | Cyclohexanone |
| S8 | 1,2 Dichlorobenzene |
| S9 | Dichloromethane |
| S10 | Dimethylformamide |
| S11 | Ethanol |
| S12 | Ethyl acetate |
| S13 | Heptane |
| S14 | Hexadecane |
| S15 | Hexafluoro-2-propanol |
| S16 | Hexane |
| S17 | Methanol |
| S18 | Methyl ethyl ketone |
| S19 | Tetrahydrofuran |
| S20 | Toluene |
| S21 | Xylene |
| Solvent | Viscosity | Solvent Volume Fraction | Polymer | Temperature | Polymer Volume Fraction | Reptation Time | Disentanglement Rate | Disentanglement Time |
|---|---|---|---|---|---|---|---|---|
| η | u1 | T | u2 | trep | kd | td | ||
| Pas | °C | s | s−1 | h | ||||
| Toluene | 2.69 × 10−4 | 9.60 × 10−1 | PS | 108 | 1.00 × 10−2 | 8.83 × 10−6 | 7.12 × 10−5 | 4 |
| HDPE | 1.00 × 10−2 | 1.08 × 10−4 | 8.21 × 10−6 | 32 | ||||
| PP | 1.10 × 10−2 | 1.14 × 10−5 | 5.54 × 10−5 | 5 | ||||
| ABS | 0.90 × 10−2 | 8.52 × 10−6 | 7.38 × 10−5 | 4 | ||||
| Xylene | 6.03 × 10−4 | 9.57 × 10−1 | PS | 137 | 1.00 × 10−2 | 2.14 × 10−5 | 2.94 × 10−5 | 9 |
| HDPE | 1.10 × 10−2 | 2.62 × 10−4 | 3.39 × 10−6 | 78 | ||||
| PP | 1.20 × 10−2 | 2.75 × 10−5 | 2.29 × 10−5 | 12 | ||||
| ABS | 1.00 × 10−2 | 2.06 × 10−5 | 3.05 × 10−5 | 9 | ||||
| 80% Cyclohexane 20% Cyclohexanol | 7.58 × 10−4 | 9.59 × 10−1 | PS | 78 | 1.00 × 10−2 | 2.80 × 10−5 | 2.24 × 10−5 | 12 |
| HDPE | 1.10 × 10−2 | 3.44 × 10−4 | 2.59 × 10−6 | 102 | ||||
| PP | 1.10 × 10−2 | 3.60 × 10−5 | 1.75 × 10−5 | 15 | ||||
| ABS | 1.00 × 10−2 | 2.71 × 10−5 | 2.33 × 10−5 | 12 | ||||
| 40% Cyclohexane 60% Xylene | 5.96 × 10−4 | 9.58 × 10−1 | PS | 78 | 1.00 × 10−2 | 2.29 × 10−5 | 2.75 × 10−5 | 9 |
| HDPE | 1.10 × 10−2 | 2.81 × 10−4 | 3.17 × 10−6 | 83 | ||||
| PP | 1.10 × 10−2 | 2.94 × 10−5 | 2.14 × 10−5 | 12 | ||||
| ABS | 1.00 × 10−2 | 2.21 × 10−5 | 2.85 × 10−5 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsampanakis, I.; Orbaek White, A. The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics. Polymers 2022, 14, 112. https://doi.org/10.3390/polym14010112
Tsampanakis I, Orbaek White A. The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics. Polymers. 2022; 14(1):112. https://doi.org/10.3390/polym14010112
Chicago/Turabian StyleTsampanakis, Ioannis, and Alvin Orbaek White. 2022. "The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics" Polymers 14, no. 1: 112. https://doi.org/10.3390/polym14010112
APA StyleTsampanakis, I., & Orbaek White, A. (2022). The Mechanics of Forming Ideal Polymer–Solvent Combinations for Open-Loop Chemical Recycling of Solvents and Plastics. Polymers, 14(1), 112. https://doi.org/10.3390/polym14010112