
Revised Atomic Charges for OPLS Force Field Model of Poly(Ethylene Oxide): Benchmarks and Applications in Polymer Electrolyte



Abstract
1. Introduction
2. Methods
2.1. Force Field and Atomic Charge Reparameterization
2.2. Molecular Dynamics Simulation Details
3. Results and Discussion
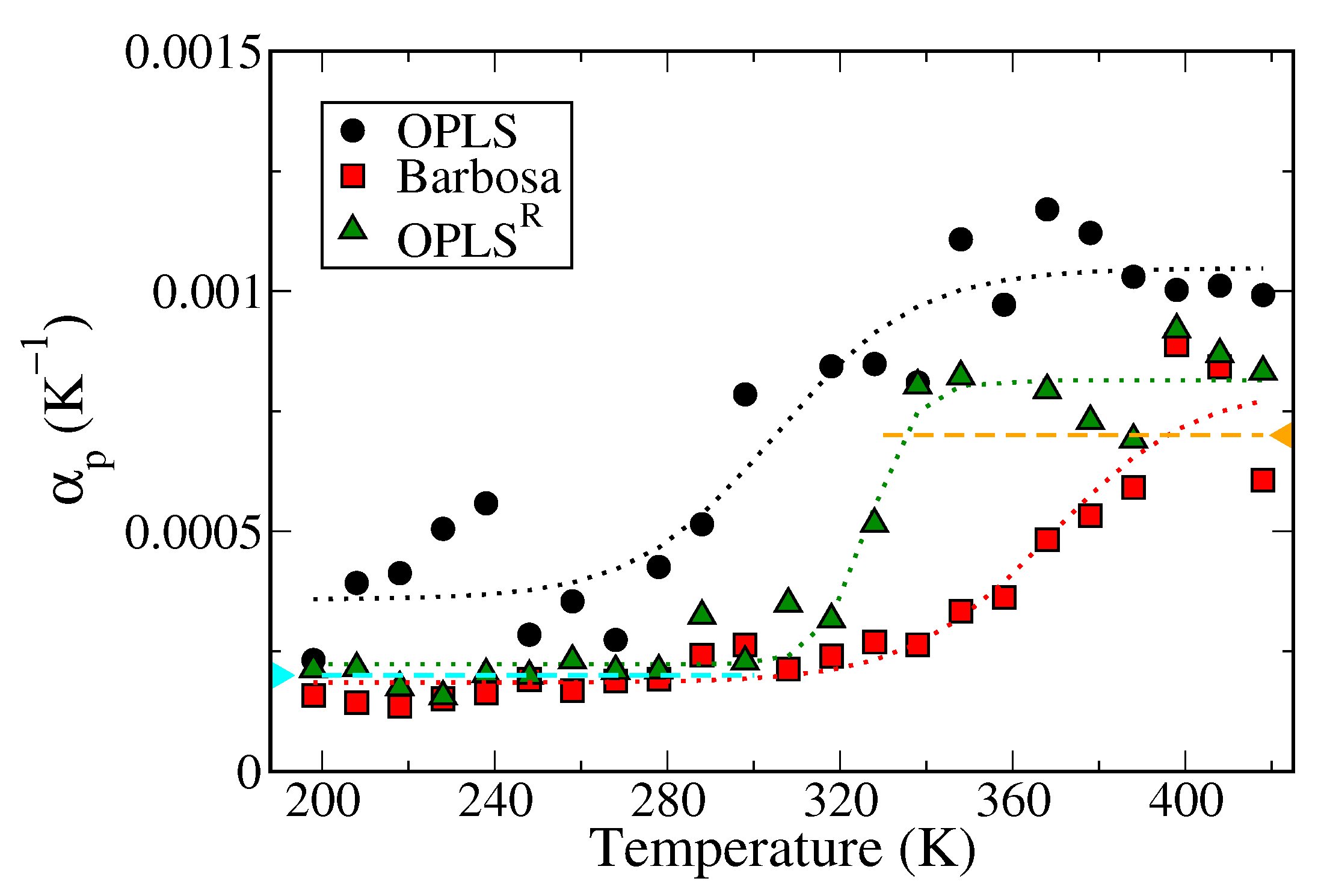
3.1. Polymer Melts System
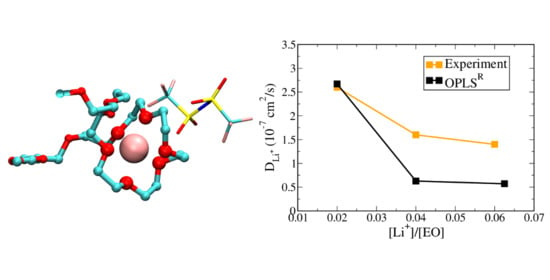
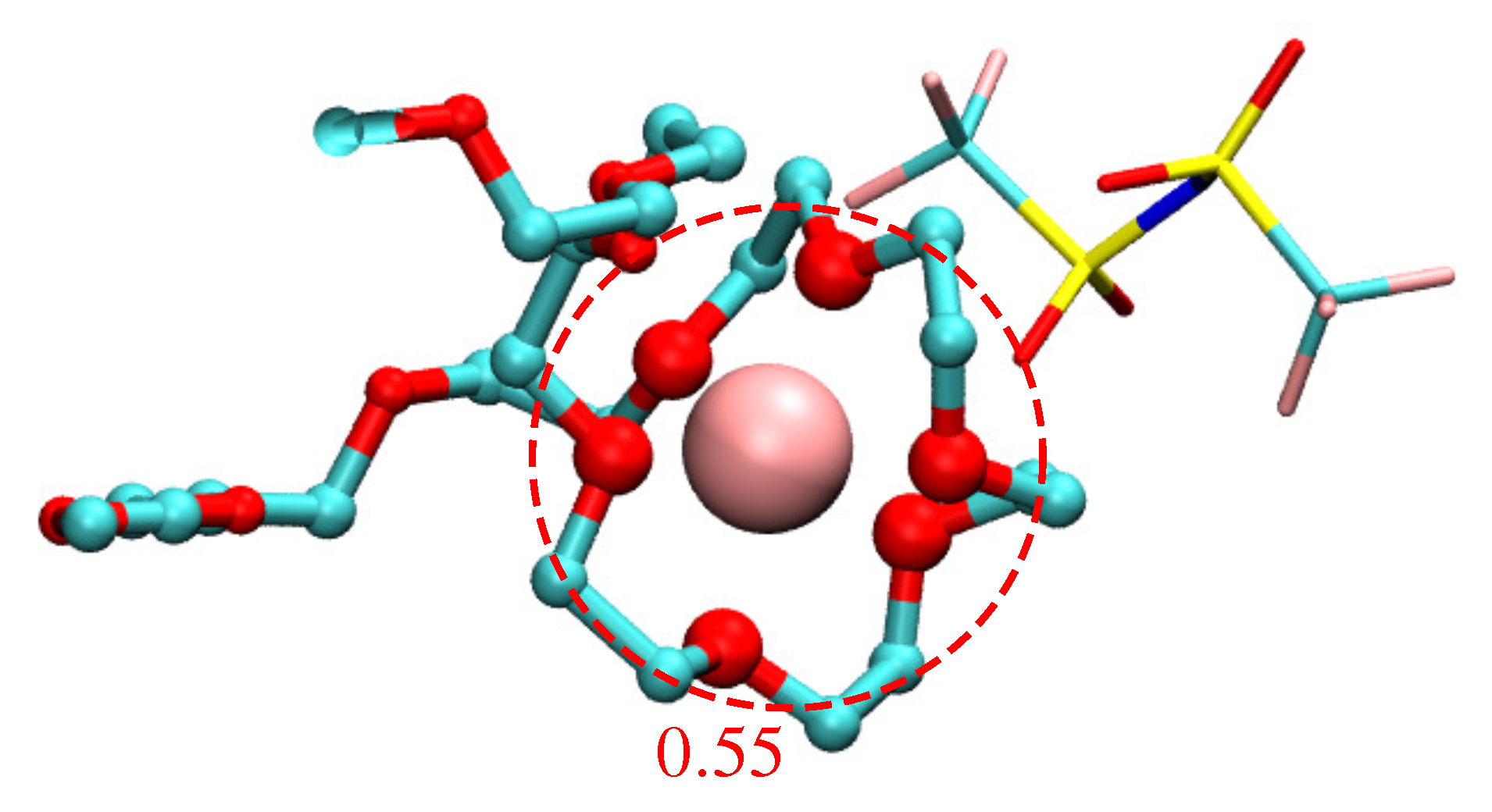
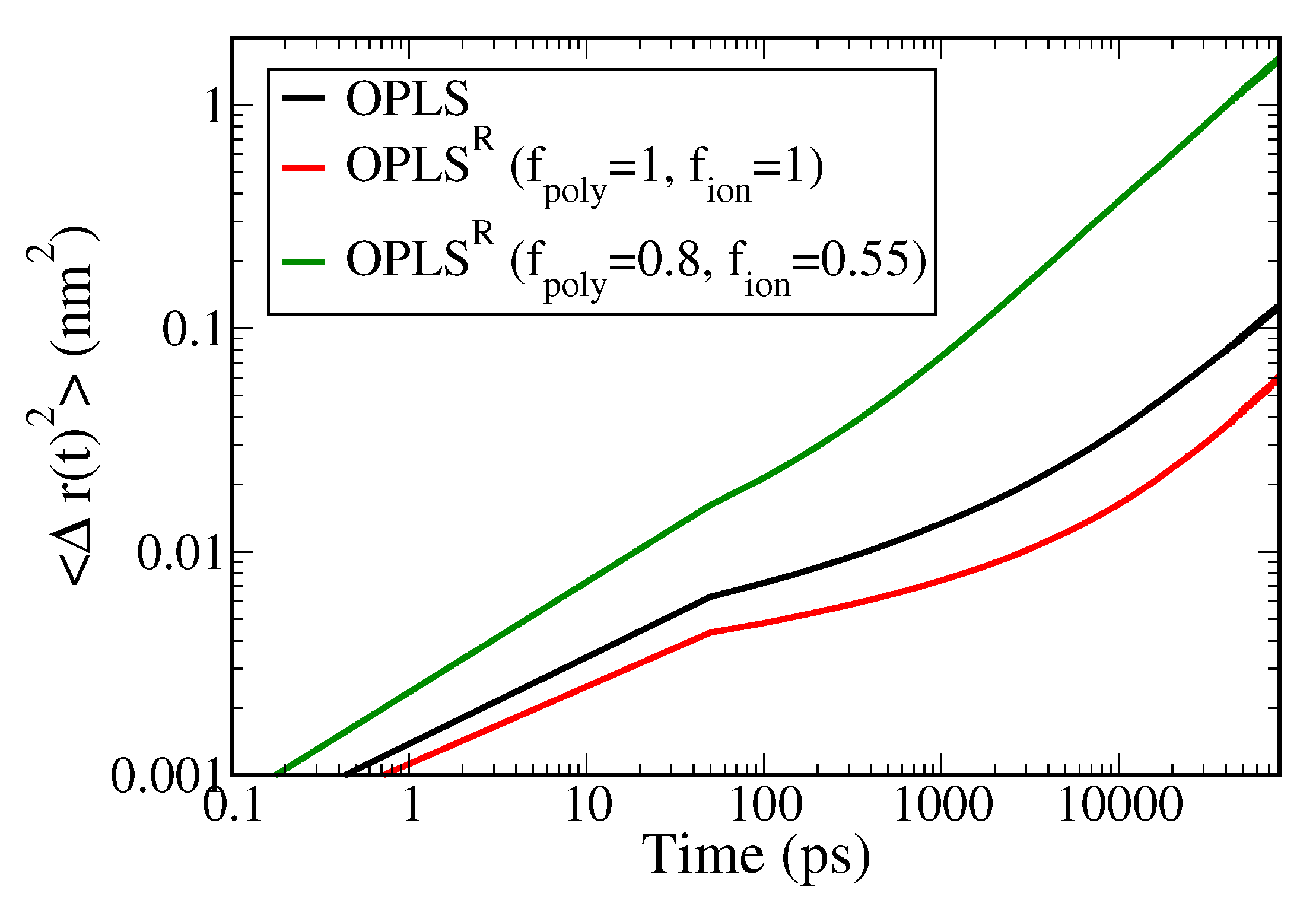
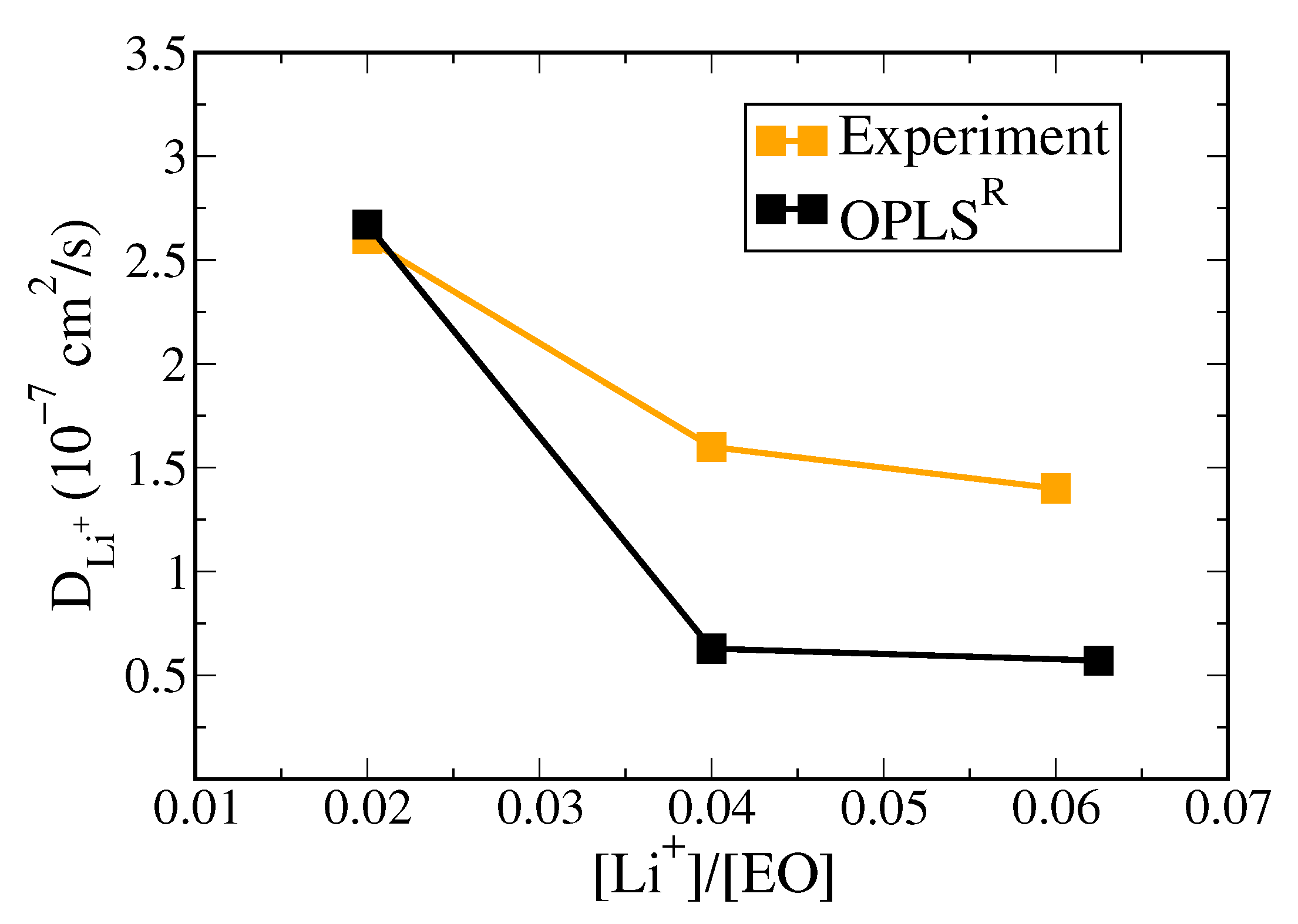
3.2. Polymer Electrolyte
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| OPLS | optimized potentials for liquid simulations |
| RESP | the restrained electrostatic potential |
| OPLSR | OPLS model with RESP charges |
| PEO | polyethylene oxide |
| PEODME | poly(ethylene oxide) dimethyl ether |
| DP | degree of polymerization |
| SPE | solid polymer electrolyte |
References
- Mauger, A.; Armand, M.; Julien, C.M.; Zaghib, K. Challenges and issues facing lithium metal for solid-state rechargeable batteries. J. Power Sources 2017, 353, 333–342. [Google Scholar] [CrossRef]
- Mindemark, J.; Lacey, M.J.; Bowden, T.; Brandell, D. Beyond PEO—Alternative host materials for Li+-conducting solid polymer electrolytes. Prog. Polym. Sci. 2018, 81, 114–143. [Google Scholar] [CrossRef]
- Lopez, J.; Mackanic, D.G.; Cui, Y.; Bao, Z. Designing polymers for advanced battery chemistries. Nat. Rev. Mater. 2019, 4, 312–330. [Google Scholar] [CrossRef]
- Su, Y.H.; Lin, Y.H.; Tseng, Y.H.; Lee, Y.L.; Jan, J.S.; Chiu, C.C.; Hou, S.S.; Teng, H. Postinjection gelation of an electrolyte with high storage permittivity and low loss permittivity for electrochemical capacitors. J. Power Sources 2021, 481, 228869. [Google Scholar] [CrossRef]
- Li, W.; Pang, Y.; Liu, J.; Liu, G.; Wang, Y.; Xia, Y. A PEO-based gel polymer electrolyte for lithium ion batteries. RSC Adv. 2017, 7, 23494–23501. [Google Scholar] [CrossRef]
- Xue, Z.; He, D.; Xie, X. Poly(ethylene oxide)-based electrolytes for lithium-ion batteries. J. Mater. Chem. A 2015, 3, 19218–19253. [Google Scholar] [CrossRef]
- Mogurampelly, S.; Borodin, O.; Ganesan, V. Computer Simulations of Ion Transport in Polymer Electrolyte Membranes. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 349–371. [Google Scholar] [CrossRef]
- Lin, K.J.; Maranas, J.K. Cation Coordination and Motion in a Poly(ethylene oxide)-Based Single Ion Conductor. Macromolecules 2012, 45, 6230–6240. [Google Scholar] [CrossRef]
- Salomon, M.; Xu, M.; Eyring, E.M.; Petrucci, S. Molecular structure and dynamics of LiClO4-polyethylene oxide-400 (dimethyl ether and diglycol systems) at 25 C. J. Phys. Chem. 1994, 98, 8234–8244. [Google Scholar] [CrossRef]
- Frech, R.; Huang, W. Conformational Changes in Diethylene Glycol Dimethyl Ether and Poly(ethylene oxide) Induced by Lithium Ion Complexation. Macromolecules 1995, 28, 1246–1251. [Google Scholar] [CrossRef]
- Wang, H.; Im, D.; Lee, D.J.; Matsui, M.; Takeda, Y.; Yamamoto, O.; Imanishi, N. A Composite Polymer Electrolyte Protect Layer between Lithium and Water Stable Ceramics for Aqueous Lithium-Air Batteries. J. Electrochem. Soc. 2013, 160, A728. [Google Scholar] [CrossRef]
- Saito, S.; Watanabe, H.; Ueno, K.; Mandai, T.; Seki, S.; Tsuzuki, S.; Kameda, Y.; Dokko, K.; Watanabe, M.; Umebayashi, Y. Li+ Local Structure in Hydrofluoroether Diluted Li-Glyme Solvate Ionic Liquid. J. Phys. Chem. D 2016, 120, 3378–3387. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Shinoda, W.; Matsugami, M.; Umebayashi, Y.; Ueno, K.; Mandai, T.; Seki, S.; Dokko, K.; Watanabe, M. Structures of [Li(glyme)]+ complexes and their interactions with anions in equimolar mixtures of glymes and Li[TFSA]: Analysis by molecular dynamics simulations. Phys. Chem. Chem. Phys. 2015, 17, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Didwal, P.N.; Kim, J.M.; Chang, D.R.; Park, C.J. Poly(ethylene oxide)-based composite solid polymer electrolyte containing Li7La3Zr2O12 and poly(ethylene glycol) dimethyl ether. J. Membr. Sci. 2020, 595, 117538. [Google Scholar] [CrossRef]
- Costa, L.T.; Ribeiro, M.C.C. Molecular dynamics simulation of polymer electrolytes based on poly(ethylene oxide) and ionic liquids. I. Structural properties. J. Chem. Phys. 2006, 124, 184902. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.T.; Ribeiro, M.C.C. Molecular dynamics simulation of polymer electrolytes based on poly(ethylene oxide) and ionic liquids. II. Dynamical properties. J. Chem. Phys. 2007, 127, 164901. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.T.; Lavall, R.L.; Borges, R.S.; Rieumont, J.; Silva, G.G.; Ribeiro, M.C.C. Polymer electrolytes based on poly(ethylene glycol) dimethyl ether and the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate: Preparation, physico-chemical characterization, and theoretical study. Electrochim. Acta 2007, 53, 1568–1574. [Google Scholar] [CrossRef]
- Maitra, A.; Heuer, A. Cation Transport in Polymer Electrolytes: A Microscopic Approach. Phys. Rev. Lett. 2007, 98, 227802. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. Mechanism of Ion Transport in Amorphous Poly(ethylene oxide)/LiTFSI from Molecular Dynamics Simulations. Macromolecules 2006, 39, 1620–1629. [Google Scholar] [CrossRef]
- Sambasivarao, S.V.; Acevedo, O. Development of OPLS-AA Force Field Parameters for 68 Unique Ionic Liquids. J. Chem. Theory Comput. 2009, 5, 1038–1050. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids Composed of Triflate or Bistriflylimide Anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory Comput. 2017, 13, 6131–6145. [Google Scholar] [CrossRef]
- Costa, L.T.; Sun, B.; Jeschull, F.; Brandell, D. Polymer-ionic liquid ternary systems for Li-battery electrolytes: Molecular dynamics studies of LiTFSI in a EMIm-TFSI and PEO blend. J. Chem. Phys. 2015, 143, 024904. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.G.; Hariharan, K.S.; Park, D.H.; Kang, H.R.; Kolake, S.M. Effects of variation in chain length on ternary polymer electrolyte—Ionic liquid mixture—A molecular dynamics simulation study. J. Power Sources 2015, 293, 983–992. [Google Scholar] [CrossRef]
- Barbosa, N.S.V.; Zhang, Y.; Lima, E.R.A.; Tavares, F.W.; Maginn, E.J. Development of an AMBER-compatible transferable force field for poly(ethylene glycol) ethers (glymes). J. Mol. Model 2017, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Douglas, R.; Smith, G.D.; Trouw, F.; Petrucci, S. MD simulations and experimental study of structure, dynamics, and thermodynamics of poly(ethylene oxide) and its oligomers. J. Phys. Chem. B 2003, 107, 6813–6823. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D.; Jaffe, R.L. Ab initio quantum chemistry and molecular dynamics simulations studies of LiPF6/poly(ethylene oxide) interactions. J. Comput. Chem. 2001, 22, 641–654. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. Development of many-body polarizable force fields for Li-battery components: 1. Ether, alkane, and carbonate-based solvents. J. Phys. Chem. B 2006, 110, 6279–6292. [Google Scholar] [CrossRef]
- Diddens, D.; Heuer, A.; Borodin, O. Understanding the Lithium Transport within a Rouse-Based Model for a PEO/LiTFSI Polymer Electrolyte. Macromolecules 2010, 43, 2028–2036. [Google Scholar] [CrossRef]
- Siqueira, L.J.A.; Ribeiro, M.C.C. Molecular dynamics simulation of the polymer electrolyte poly(ethyleneoxide)/LiClO4. I. Structural properties. J. Chem. Phys. 2005, 122, 194911. [Google Scholar] [CrossRef]
- Siqueira, L.J.A.; Ribeiro, M.C.C. Molecular dynamics simulation of the polymer electrolyte poly(ethylene oxide)/LiClO[sub 4]. II. Dynamical properties. J. Chem. Phys. 2006, 125, 214903. [Google Scholar] [CrossRef]
- van Zon, A.; Mos, B.; Verkerk, P.; de Leeuw, S.W. On the dynamics of PEO-NaI polymer electrolytes. Electrochim. Acta 2001, 46, 1717–1721. [Google Scholar] [CrossRef]
- Müller-Plathe, F.; van Gunsteren, W.F. Computer simulation of a polymer electrolyte: Lithium iodide in amorphous poly(ethylene oxide). J. Chem. Phys. 1995, 103, 4745–4756. [Google Scholar] [CrossRef]
- Halley, J.W.; Duan, Y.; Curtiss, L.A.; Baboul, A.G. Lithium perchlorate ion pairing in a model of amorphous polyethylene oxide. J. Chem. Phys. 1999, 111, 3302. [Google Scholar] [CrossRef]
- Mos, B.; Verkerk, P.; Pouget, S.; van Zon, A.; Bel, G.J.; de Leeuw, S.W.; Eisenbach, C.D. The dynamics in polyethyleneoxide–alkali iodide complexes investigated by neutron spin-echo spectroscopy and molecular dynamics simulations. J. Chem. Phys. 2000, 113, 4. [Google Scholar] [CrossRef][Green Version]
- Liu, Z.; Timmermann, J.; Reuter, K.; Scheurer, C. Benchmarks and Dielectric Constants for Reparametrized OPLS and Polarizable Force Field Models of Chlorinated Hydrocarbons. J. Phys. Chem. B 2018, 122, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Caleman, C.; van Maaren, P.J.; Hong, M.; Hub, J.S.; Costa, L.T.; van der Spoel, D. Force field benchmark of organic liquids: Density, enthalpy of vaporization, heat capacities, surface tension, isothermal compressibility, volumetric expansion coefficient, and dielectric constant. J. Chem. Theory Comput. 2012, 8, 61–74. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP Charges To Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, H.L.T.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8596. [Google Scholar] [CrossRef]
- Nose, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Polyethylene glycol [MAK Value Documentation, 1998]. In The MAK-Collection for Occupational Health and Safety; American Cancer Society: Atlanta, GA, USA, 2012; pp. 248–270.
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: New York, NY, USA, 1989. [Google Scholar]
- Tsujita, Y.; Nose, T.; Hata, T. Thermodynamic Properties of Poly(ethylene glycol) and Poly(tetrahydrofuran). I. P—V—T Relations and Internal Pressure. Polym. J. 1973, 5, 201–207. [Google Scholar] [CrossRef]
- Koizumi, N.; Hanai, T. Dielectric Properties of Polyethylene Glycols: Dielectric Relaxation in Solid State (Special Issue on Polymer Chemistry, I). Bull. Inst. Chem. Res. Kyoto Univ. 1964, 42, 115–127. [Google Scholar]
- Neumann, M. Dipole moment fluctuation formulas in computer simulations of polar systems. Mol. Phys. 1983, 50, 841–858. [Google Scholar] [CrossRef]
- Diddens, D.; Heuer, A. Simulation Study of the Lithium Ion Transport Mechanism in Ternary Polymer Electrolytes: The Critical Role of the Segmental Mobility. J. Phys. Chem. B 2014, 118, 1113–1125. [Google Scholar] [CrossRef]
- Mao, G.; Saboungi, M.L.; Price, D.L.; Armand, M.B.; Howells, W.S. Structure of Liquid PEO-LiTFSI Electrolyte. Phys. Rev. Lett. 2000, 84, 5536. [Google Scholar] [CrossRef]
- Timachova, K.; Watanabe, H.; Balsara, N.P. Effect of molecular weight and salt concentration on ion transport and the transference number in polymer electrolytes. Macromolecules 2015, 48, 7882–7888. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (T > Tm) (K) | (T < Tm) (K) | Dielectric Constant (F/m) | |
|---|---|---|---|
| Experiment [57,58] | 9–11 | ||
| OPLS | 4.36 | ||
| Barbosa | − | 8.08 | |
| OPLSR | 9.94 |
| Unit: cm s | 363 K | 333 K | 298 K | t (333 K) |
|---|---|---|---|---|
| Experiment | − | 0.24 | ||
| OPLS | 0.41 | |||
| OPLSR () | 0.363 | |||
| OPLSR () | 0.215 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, C.-E.; Tsai, Y.-C.; Scheurer, C.; Chiu, C.-C. Revised Atomic Charges for OPLS Force Field Model of Poly(Ethylene Oxide): Benchmarks and Applications in Polymer Electrolyte. Polymers 2021, 13, 1131. https://doi.org/10.3390/polym13071131
Fang C-E, Tsai Y-C, Scheurer C, Chiu C-C. Revised Atomic Charges for OPLS Force Field Model of Poly(Ethylene Oxide): Benchmarks and Applications in Polymer Electrolyte. Polymers. 2021; 13(7):1131. https://doi.org/10.3390/polym13071131
Chicago/Turabian StyleFang, Chan-En, Yi-Chen Tsai, Christoph Scheurer, and Chi-Cheng Chiu. 2021. "Revised Atomic Charges for OPLS Force Field Model of Poly(Ethylene Oxide): Benchmarks and Applications in Polymer Electrolyte" Polymers 13, no. 7: 1131. https://doi.org/10.3390/polym13071131
APA StyleFang, C.-E., Tsai, Y.-C., Scheurer, C., & Chiu, C.-C. (2021). Revised Atomic Charges for OPLS Force Field Model of Poly(Ethylene Oxide): Benchmarks and Applications in Polymer Electrolyte. Polymers, 13(7), 1131. https://doi.org/10.3390/polym13071131