
New Light in Polymer Science: Photoinduced Reversible Addition-Fragmentation Chain Transfer Polymerization (PET-RAFT) as Innovative Strategy for the Synthesis of Advanced Materials



Abstract

1. Introduction
2. Form RAFT to PET-RAFT Polymerization
2.1. Reversible Addition-Fragmentation Chain-Transfer Polymerization (RAFT)
RAFT Agent
2.2. Photoinduced Electron/Energy Transfer (PET)-RAFT Polymerization
2.2.1. PET-RAFT Mechanism
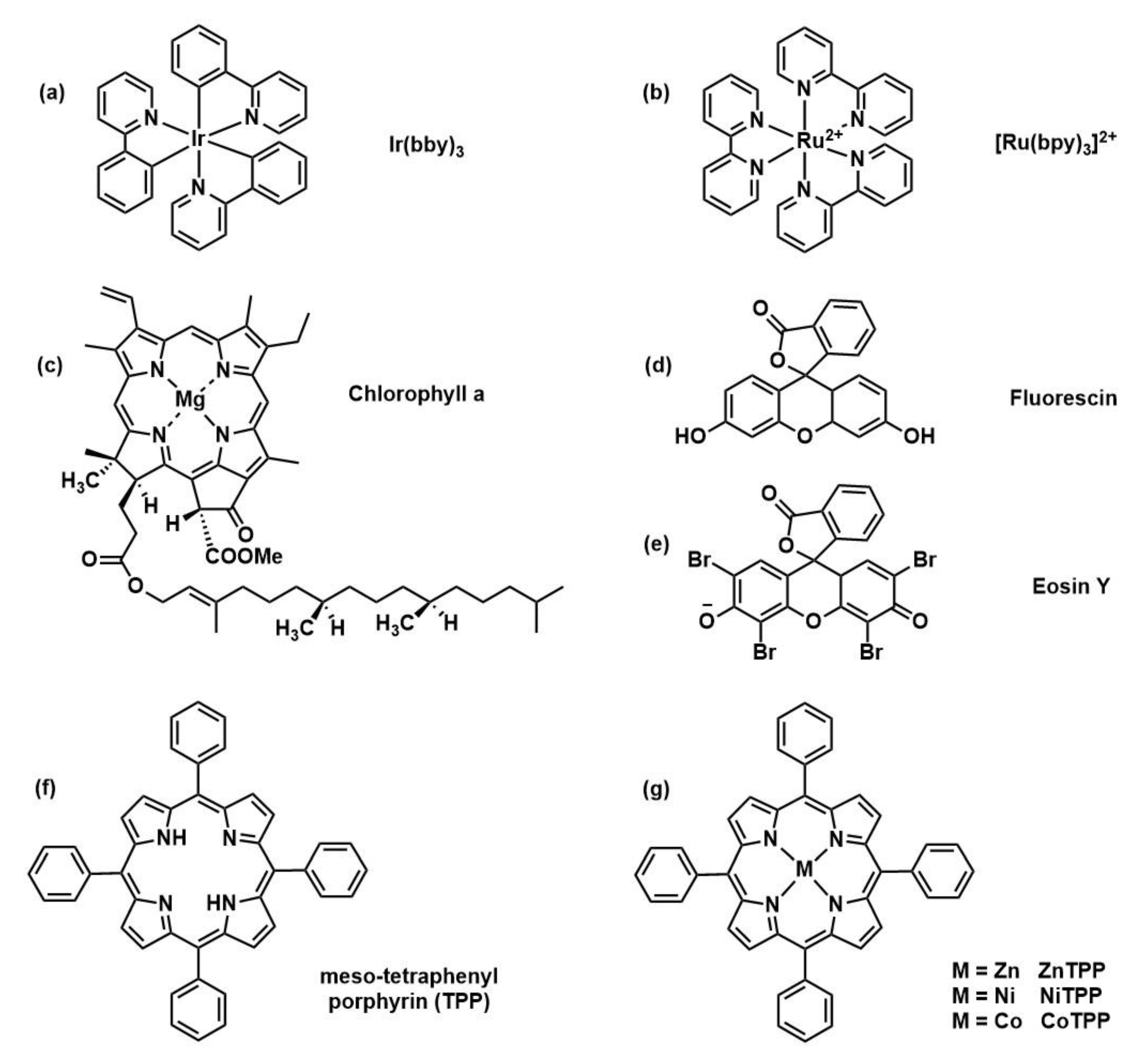
2.2.2. Photoredox Catalysts
2.2.3. Compatibility with Other Controlled Polymerization Techniques
3. PET-RAFT Polymerization: A Greener Approach
3.1. Light Sources
3.2. Recyclable Photocatalysts
3.3. Oxygen Tolerance and High Throughput Polymerization
3.4. Waste Minimization
4. Applications
4.1. Fabrication of Advanced Materials for Commercial Applications
4.1.1. Manufacturing of Nanocomposites
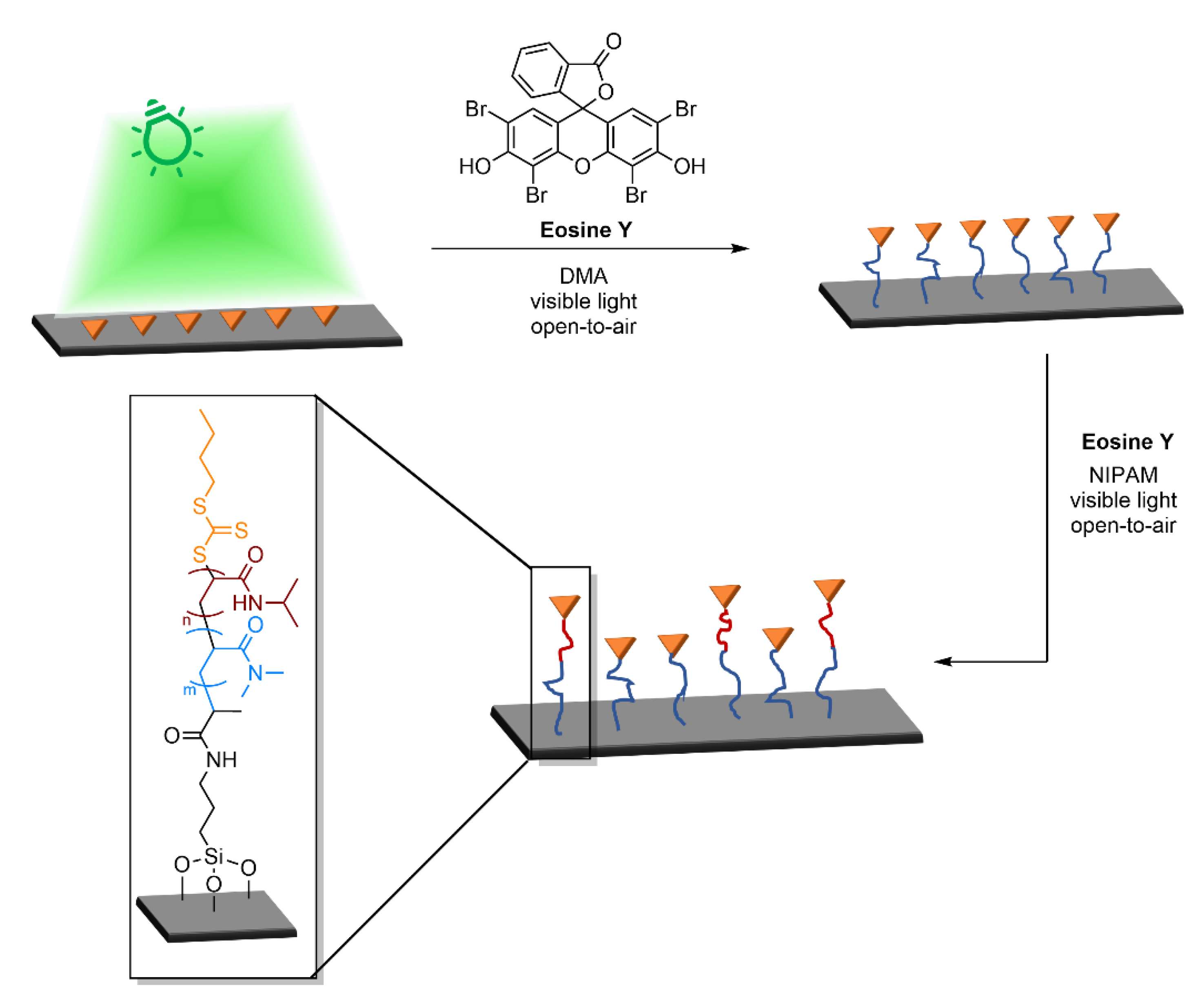
4.1.2. Design of 2D Patterns for Lithography
4.1.3. New Advances in 3D Printing
4.1.4. New Industrial Possibilities
4.2. PET-RAFT for Medical Purposes
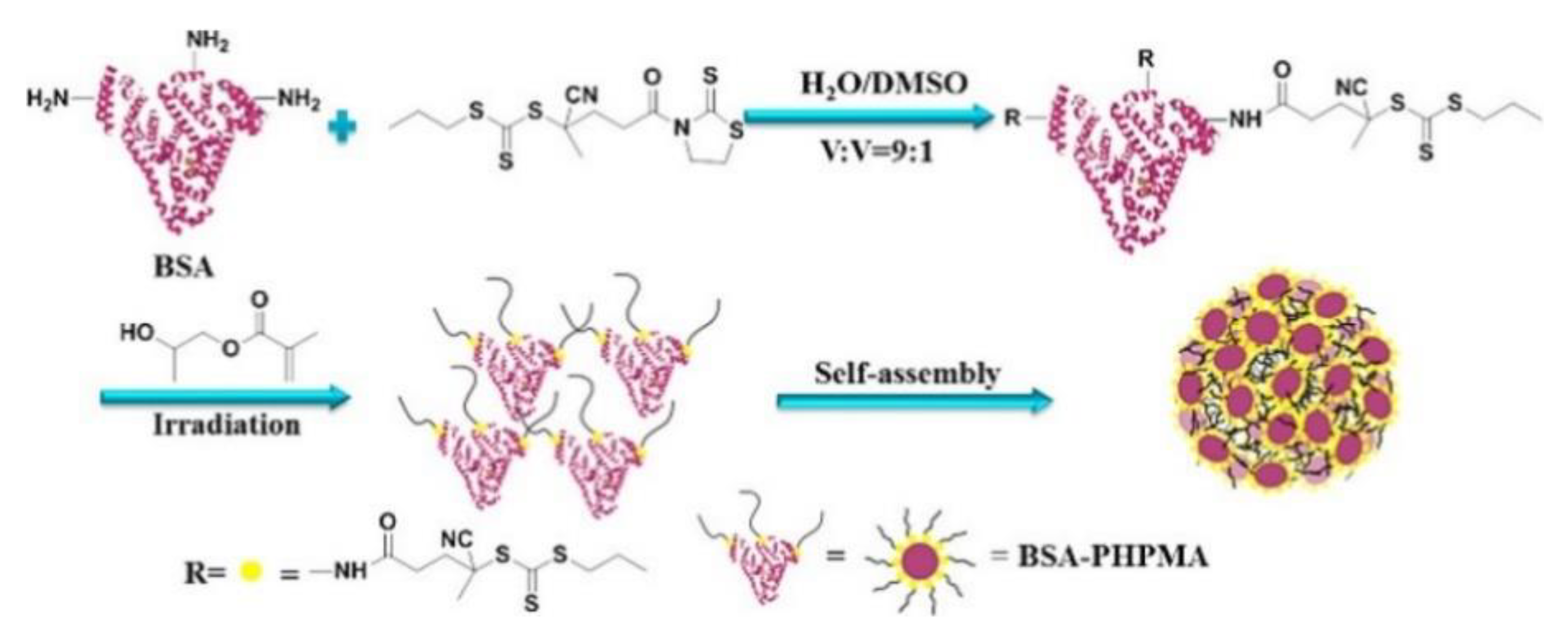
4.2.1. Compatibility with Biological Moieties: Bioconjugation
4.2.2. Antifouling Properties
4.2.3. Antimicrobial Activity

5. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AcFl | Diacryloyl-fluorescein |
| AIBN | Azobisisobutyronitrile |
| BA | Butyl acrylate |
| BSTP | 3-benzylsulfanyl-thiocarbonylthiosulfanyl propionic acid |
| BTPA | 2-(n-butyltrithiocarbonate)-propionic acid |
| CPADB | 4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid |
| DEA | N,N-Diethylacrylamide |
| DEGA | Di(ethylene glycol) methyl ether acrylate |
| DMA | N,N-Dimethylacrylamide |
| DMAEMA | 2-Dimethylaminoethyl methacrylate |
| DMAEA | 2-Dimethylaminoethyl acrylate |
| DPPE | 1,2-Dipalmitoyl-sn-glycero-3-phosphorylethanolamine |
| EGDA | Ethylene glycol diacrylate |
| EGMEMA | Ethylene glycol methyl ether methacrylate |
| EHA | 2-Ethylhexyl acrylate |
| EtBriB | Ethyl α-bromoisobutyrate |
| HEA | 2-Hydroxyethyl Acrylate |
| MMA | Methyl methacrylate |
| NIPAM | N-isopropyl acrylamide |
| NHS | N-Hydroxysuccinimide |
| PEA | Phenoxyethyl acrylate |
| PEGA | Poly(ethylene glycol) methyl ether acrylate |
| PDP | 2-(pyridine-2-yldisulfanyl)ethyl-2-(((dodecylthio)carbonthioyl)thio)propanoate |
| PMMA | Poly(methyl methacrylate) |
| SA | 3-sulfopropyl methacrylate |
| TPP | Tetraphenylporphyrin |
References
- Goto, A.; Fukuda, T. Kinetics of living radical polymerization. Prog. Polym. Sci. 2004, 29, 329–385. [Google Scholar] [CrossRef]
- Gregory, A.; Stenzel, M.H. Complex polymer architectures via RAFT polymerization: From fundamental process to extending the scope using click chemistry and nature’s building blocks. Prog. Polym. Sci. 2012, 37, 38–105. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Proc. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Radical addition-fragmentation chemistry in polymer synthesis. Polymer 2008, 49, 1079–1131. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process a third update. Proc. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Manfredi, N.; Bianchi, A.; Causin, V.; Ruffo, R.; Simonutti, R.; Abbotto, A. Electrolytes for quasi solid-state dye-sensitized solar cells based on block copolymers. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 719–727. [Google Scholar] [CrossRef]
- Vaccaro, G.; Bianchi, A.; Mauri, M.; Bonetti, S.; Meinardi, F.; Sanguineti, A.; Simonutti, R.; Beverina, L. Direct monitoring of self-assembly of copolymeric micelles by a luminescent molecular rotor. Chem. Commun. 2013, 49, 8474–8476. [Google Scholar] [CrossRef] [PubMed]
- Piloni, A.; Simonutti, R.; Stenzel, M.H. The effect of cationic groups on the stability of 19F MRI contrast agents in nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1994–2001. [Google Scholar] [CrossRef]
- Gregori, M.; Bertani, D.; Cazzaniga, E.; Orlando, A.; Mauri, M.; Bianchi, A.; Re, F.; Sesana, S.; Minniti, S.; Francolini, M.; et al. Investigation of Functionalized Poly(N,N-dimethylacrylamide)-block-polystyrene Nanoparticles As Novel Drug Delivery System to Overcome the Blood-Brain Barrier in Vitro. Macromol. Biosci. 2015, 15, 1687–1697. [Google Scholar] [CrossRef]
- Bianchi, A.; Mauri, M.; Bonetti, S.; Koynov, K.; Kappl, M.; Lieberwirth, I.; Butt, H.J.; Simonutti, R. Hierarchical self-assembly of PDMA-b-PS chains into granular nanoparticles: Genesis and fate. Macromol. Rapid Commun. 2014, 35, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT agent design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Moad, G.; Keddie, D.; Guerrero-Sanchez, C.; Rizzardo, E.; Thang, S.H. Advances in switchable RAFT polymerization. Macromol. Symp. 2015, 350, 34–42. [Google Scholar] [CrossRef]
- Otsu, T.; Kuriyama, A. Living Radical Polymerization in Homogeneous System by Using Iniferter: Design of block copolymers. J. Macromol. Sci. Chem. 1984, 21, 961–977. [Google Scholar] [CrossRef]
- Otsu, T. Iniferter concept and living radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2121–2136. [Google Scholar] [CrossRef]
- Quinn, J.F.; Barner, L.; Davis, T.P.; Thang, S.H.; Rizzardo, E. Living free radical polymerisation under a constant source of gamma radiation—An example of reversible addition-fragmentation chain transfer or reversible termination? Macromol. Rapid Commun. 2002, 23, 717–721. [Google Scholar] [CrossRef]
- McKenzie, T.G.; Fu, Q.; Uchiyama, M.; Satoh, K.; Xu, J.; Boyer, C.; Kamigaito, M.; Qiao, G.G. Beyond Traditional RAFT: Alternative Activation of Thiocarbonylthio Compounds for Controlled Polymerization. Adv. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Quinn, J.F.; Rizzardo, E.; Barner, L.; Barner-Kowollik, C.; Davis, T.P. Reversible addition-fragmentation chain transfer polymerization initiated with ultraviolet and γ radiation: A comparison. Am. Chem. Soc. Polym. Prepr. Div. Polym. Chem. 2002, 43, 319–320. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. End-functional polymers, thiocarbonylthio group removal/transformation and reversible addition-fragmentation-chain transfer (RAFT) polymerization. Polym. Int. 2011, 60, 9–25. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. A robust and versatile photoinduced living polymerization of conjugated and unconjugated monomers and its oxygen tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Phommalysack-lovan, J.; Chu, Y.; Boyer, C. PET-RAFT polymerisation: Towards green and precision polymer manufacturing. Chem. Commun. 2018, 6591–6606. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef]
- Corrigan, N.; Xu, J.; Boyer, C.; Allonas, X. Exploration of the PET-RAFT Initiation Mechanism for Two Commonly Used Photocatalysts. ChemPhotoChem 2019, 3, 1193–1199. [Google Scholar] [CrossRef]
- Christmann, J.; Ibrahim, A.; Charlot, V.; Croutxé-Barghorn, C.; Ley, C.; Allonas, X. Elucidation of the Key Role of [Ru(bpy)3]2+ in Photocatalyzed RAFT Polymerization. ChemPhysChem 2016, 2309–2314. [Google Scholar] [CrossRef]
- Seal, P.; Xu, J.; Luca, S.; Boyer, C.; Smith, S.C. Unraveling Photocatalytic Mechanism and Selectivity in PET-RAFT Polymerization. Adv. Theory Simul. 2019, 1900038. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Corrigan, N.A.; Boyer, C. Aqueous photoinduced living/controlled polymerization: Tailoring for bioconjugation. Chem. Sci. 2014, 5, 3568–3575. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Utilizing the electron transfer mechanism of chlorophyll a under light for controlled radical polymerization. Chem. Sci. 2015, 6, 1341–1349. [Google Scholar] [CrossRef]
- Xu, J.; Shanmugam, S.; Duong, H.T.; Boyer, C. Organo-photocatalysts for photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization. Polym. Chem. 2015, 6, 5615–5624. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths. J. Am. Chem. Soc. 2015, 137, 9174–9185. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Xu, J.; Boyer, C. Light-regulated polymerization under near-infrared/far-red irradiation catalyzed by bacteriochlorophyll a. Angew. Chem. Int. Ed. 2016, 55, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shanmugam, S.; Boyer, C. Organic Electron Donor-Acceptor Photoredox Catalysts: Enhanced Catalytic Efficiency toward Controlled Radical Polymerization. ACS Macro Lett. 2015, 4, 926–932. [Google Scholar] [CrossRef]
- Chen, M.; MacLeod, M.J.; Johnson, J.A. Visible-light-controlled living radical polymerization from a trithiocarbonate iniferter mediated by an organic photoredox catalyst. ACS Macro Lett. 2015, 4, 566–569. [Google Scholar] [CrossRef]
- Cheng, B.F.; Wang, L.H.; You, Y.Z. Photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization using titanium dioxide. Macromol. Res. 2016, 24, 811–815. [Google Scholar] [CrossRef]
- Liang, E.; Liu, M.; He, B.; Wang, G.X. ZnO as photocatalyst for photoinduced electron transfer–reversible addition–fragmentation chain transfer of methyl methacrylate. Adv. Polym. Technol. 2018, 37, 2879–2884. [Google Scholar] [CrossRef]
- He, J.Y.; Lu, M. Photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization of acrylonitrile in miniemulsion. J. Macromol. Sci. Part A Pure Appl. Chem. 2019, 56, 443–449. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Y.; Miller, K.A.; Zhu, H.; Egap, E. Lead Halide Perovskite Nanocrystals as Photocatalysts for PET-RAFT Polymerization under Visible and Near-Infrared Irradiation. ACS Macro Lett. 2020, 9, 725–730. [Google Scholar] [CrossRef]
- Liang, Y.; Ma, H.; Zhang, W.; Cui, Z.; Fu, P.; Liu, M.; Qiao, X.; Pang, X. Size effect of semiconductor quantum dots as photocatalysts for PET-RAFT polymerization. Polym. Chem. 2020. [Google Scholar] [CrossRef]
- Zhu, Y.; Egap, E. PET-RAft polymerization catalyzed by cadmium selenide quantum dots (qds): Grafting-from qds photocatalysts to make polymer nanocomposites. Polym. Chem. 2020, 11, 1018–1024. [Google Scholar] [CrossRef]
- Fu, Q.; McKenzie, T.G.; Tan, S.; Nam, E.; Qiao, G.G. Tertiary amine catalyzed photo-induced controlled radical polymerization of methacrylates. Polym. Chem. 2015, 6, 5362–5368. [Google Scholar] [CrossRef]
- Fu, Q.; Ruan, Q.; McKenzie, T.G.; Reyhani, A.; Tang, J.; Qiao, G.G. Development of a Robust PET-RAFT Polymerization Using Graphitic Carbon Nitride (g-C3N4). Macromolecules 2017, 50, 7509–7516. [Google Scholar] [CrossRef]
- Fu, C.; Xu, J.; Kokotovic, M.; Boyer, C. One-Pot Synthesis of Block Copolymers by Orthogonal Ring-Opening Polymerization and PET-RAFT Polymerization at Ambient Temperature. ACS Macro Lett. 2016, 5, 444–449. [Google Scholar] [CrossRef]
- Fu, C.; Xu, J.; Boyer, C. Photoacid-mediated ring opening polymerization driven by visible light. Chem. Commun. 2016, 52, 7126–7129. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, T.Y.; Xia, L.; Hong, C.Y.; Wu, D.C.; You, Y.Z. Synthe sis of polymers with on-demand sequence structures via dually switchable and interconvertible polymerizations. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Theriot, J.C.; Miyake, G.M.; Boyer, C.A. N, N-Diaryl Dihydrophenazines as Photoredox Catalysts for PET-RAFT and Sequential PET-RAFT/O-ATRP. ACS Macro Lett. 2018, 7, 662–666. [Google Scholar] [CrossRef]
- Yeow, J.; Chapman, R.; Xu, J.; Boyer, C. Oxygen tolerant photopolymerization for ultralow volumes. Polym. Chem. 2017, 8, 5012–5022. [Google Scholar] [CrossRef]
- Corrigan, N.; Yeow, J.; Judzewitsch, P.; Xu, J.; Boyer, C. Seeing the Light: Advancing Materials Chemistry through Photopolymerization. Angew. Chem. Int. Ed. 2019, 58, 5170–5189. [Google Scholar] [CrossRef]
- Sender, M.; Ziegenbalg, D. Light Sources for Photochemical Processes—Estimation of Technological Potentials. Chem. Ing. Tech. 2017, 89, 1159–1173. [Google Scholar] [CrossRef]
- Leonard, D.L.; Charlton, D.G.; Roberts, H.W.; Cohen, M.E. Polymerization efficiency of LED curing lights. J. Esthet. Restor. Dent. 2002, 14, 286–295. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization of vinyl acetate and N-vinylpyrrolidinone: Kinetic and oxygen tolerance study. Macromolecules 2014, 47, 4930–4942. [Google Scholar] [CrossRef]
- Chu, Y.; Corrigan, N.; Wu, C.; Boyer, C.; Xu, J. A Process for Well-Defined Polymer Synthesis through Textile Dyeing Inspired Catalyst Immobilization. ACS Sustain. Chem. Eng. 2018, 6, 15245–15253. [Google Scholar] [CrossRef]
- Chu, Y.; Huang, Z.; Liang, K.; Guo, J.; Boyer, C.; Xu, J. A photocatalyst immobilized on fibrous and porous monolithic cellulose for heterogeneous catalysis of controlled radical polymerization. Polym. Chem. 2018, 9, 1666–1673. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, S.; Adnan, N.N.M.; Boyer, C. Heterogeneous Photocatalysis as a Means for Improving Recyclability of Organocatalyst in “living” Radical Polymerization. Macromolecules 2018, 51, 779–790. [Google Scholar] [CrossRef]
- Chen, M.; Deng, S.; Gu, Y.; Lin, J.; MacLeod, M.J.; Johnson, J.A. Logic-Controlled Radical Polymerization with Heat and Light: Multiple-Stimuli Switching of Polymer Chain Growth via a Recyclable, Thermally Responsive Gel Photoredox Catalyst. J. Am. Chem. Soc. 2017, 139, 2257–2266. [Google Scholar] [CrossRef]
- Wang, J.; Rivero, M.; Muñoz Bonilla, A.; Sanchez-Marcos, J.; Xue, W.; Chen, G.; Zhang, W.; Zhu, X. Natural RAFT Polymerization: Recyclable-Catalyst-Aided, Opened-to-Air, and Sunlight-Photolyzed RAFT Polymerizations. ACS Macro Lett. 2016, 5, 1278–1282. [Google Scholar] [CrossRef]
- McClelland, K.P.; Clemons, T.D.; Stupp, S.I.; Weiss, E.A. Semiconductor Quantum Dots Are Efficient and Recyclable Photocatalysts for Aqueous PET-RAFT Polymerization. ACS Macro Lett. 2020, 9, 7–13. [Google Scholar] [CrossRef]
- Yeow, J.; Chapman, R.; Gormley, A.J.; Boyer, C. Up in the air: Oxygen tolerance in controlled/living radical polymerisation. Chem. Soc. Rev. 2018, 47, 4357–4387. [Google Scholar] [CrossRef]
- Ligon, S.C.; Husár, B.; Wutzel, H.; Holman, R.; Liska, R. Strategies to reduce oxygen inhibition in photoinduced polymerization. Chem. Rev. 2014, 114, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.; Yeow, J.; Xu, J.; Boyer, C. Application of oxygen tolerant PET-RAFT to polymerization-induced self-assembly. Polym. Chem. 2017, 8, 2841–2851. [Google Scholar] [CrossRef]
- Chapman, R.; Gormley, A.J.; Herpoldt, K.L.; Stevens, M.M. Highly controlled open vessel RAFT polymerizations by enzyme degassing. Macromolecules 2014, 47, 8541–8547. [Google Scholar] [CrossRef]
- Chapman, R.; Gormley, A.J.; Stenzel, M.H.; Stevens, M.M. Combinatorial Low-Volume Synthesis of Well-Defined Polymers by Enzyme Degassing. Angew. Chem. Int. Ed. 2016, 55, 4500–4503. [Google Scholar] [CrossRef]
- Tan, J.; Liu, D.; Bai, Y.; Huang, C.; Li, X.; He, J.; Xu, Q.; Zhang, L. Enzyme-Assisted Photoinitiated Polymerization-Induced Self-Assembly: An Oxygen-Tolerant Method for Preparing Block Copolymer Nano-Objects in Open Vessels and Multiwell Plates. Macromolecules 2017, 50, 5798–5806. [Google Scholar] [CrossRef]
- Gormley, A.J.; Yeow, J.; Ng, G.; Conway, Ó.; Boyer, C.; Chapman, R. An Oxygen-Tolerant PET-RAFT Polymerization for Screening Structure–Activity Relationships. Angew. Chem. Int. Ed. 2018, 57, 1557–1562. [Google Scholar] [CrossRef]
- Ng, G.; Yeow, J.; Chapman, R.; Isahak, N.; Wolvetang, E.; Cooper-White, J.J.; Boyer, C. Pushing the Limits of High Throughput PET-RAFT Polymerization. Macromolecules 2018, 51, 7600–7607. [Google Scholar] [CrossRef]
- Yeow, J.; Joshi, S.; Chapman, R.; Boyer, C. A Self-Reporting Photocatalyst for Online Fluorescence Monitoring of High Throughput RAFT Polymerization. Angew. Chem. 2018, 130, 10259–10263. [Google Scholar] [CrossRef]
- Kango, S.; Kalia, S.; Celli, A.; Njuguna, J.; Habibi, Y.; Kumar, R. Surface modification of inorganic nanoparticles for development of organic-inorganic nanocomposites—A review. Prog. Polym. Sci. 2013, 38, 1232–1261. [Google Scholar] [CrossRef]
- Müller, K.; Bugnicourt, E.; Latorre, M.; Jorda, M.; Sanz, Y.E.; Lagaron, J.M.; Miesbauer, O.; Bianchin, A.; Hankin, S.; Bölz, U.; et al. Review on the processing and properties of polymer nanocomposites and nanocoatings and their applications in the packaging, automotive and solar energy fields. Nanomaterials 2017, 7, 74. [Google Scholar] [CrossRef]
- Macchione, M.A.; Biglione, C.; Strumia, M. Design, synthesis and architectures of hybrid nanomaterials for therapy and diagnosis applications. Polymers 2018, 10, 527. [Google Scholar] [CrossRef]
- Maity, N.; Dawn, A. Conducting polymer grafting: Recent and key developments. Polymers 2020, 12, 709. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.C.; Radzinski, S.C.; Matson, J.B. Graft polymer synthesis by RAFT transfer-to. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2865–2876. [Google Scholar] [CrossRef]
- Mazloomi-Rezvani, M.; Salami-Kalajahi, M.; Roghani-Mamaqani, H. “Grafting to” approach for surface modification of AuNPs with RAFT-mediated synthesized smart polymers: Stimuli-responsive behaviors of hybrid nanoparticles. J. Phys. Chem. Solids 2018, 123, 183–190. [Google Scholar] [CrossRef]
- Klein, T.; Parkin, J.; de Jongh, P.A.J.M.; Esser, L.; Sepehrizadeh, T.; Zheng, G.; De Veer, M.; Alt, K.; Hagemeyer, C.E.; Haddleton, D.M.; et al. Functional Brush Poly(2-ethyl-2-oxazine)s: Synthesis by CROP and RAFT, Thermoresponsiveness and Grafting onto Iron Oxide Nanoparticles. Macromol. Rapid Commun. 2019, 40, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.M.; Tawfilas, M.; Khani, M.M.; Golian, K.; Marsh, Z.M.; Jhalaria, M.; Simonutti, R.; Stefik, M.; Kumar, S.K.; Benicewicz, B.C. Reinforcement of polychloroprene by grafted silica nanoparticles. Polymer 2019, 171, 96–105. [Google Scholar] [CrossRef]
- Matsumura, S.; Hlil, A.R.; Lepiller, C.; Gaudet, J.; Guay, D.; Shi, Z.; Holdcroft, S.; Hay, A.S. Stability and Utility of Pyridyl Disulfide Functionality in RAFT and Conventional Radical Polymerizations. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7207–7224. [Google Scholar] [CrossRef]
- Crippa, M.; Bianchi, A.; Cristofori, D.; D’Arienzo, M.; Merletti, F.; Morazzoni, F.; Scotti, R.; Simonutti, R. High dielectric constant rutile-polystyrene composite with enhanced percolative threshold. J. Mater. Chem. C 2013, 1, 484–492. [Google Scholar] [CrossRef]
- Chen, J.; Liu, M.; Huang, H.; Deng, F.; Mao, L.; Wen, Y.; Huang, L.; Tian, J.; Zhang, X.; Wei, Y. Facile preparation of thermoresponsive fluorescent silica nanopaprticles based composites through the oxygen tolerance light-induced RAFT polymerization. J. Mol. Liq. 2018, 259, 179–185. [Google Scholar] [CrossRef]
- Li, X.; Ye, S.; Huang, Y.; Li, J.L.; Cai, T. Precise growth of polymer brushes on silica-based nanocomposites via visible-light-regulated controlled radical polymerization. J. Mater. Chem. A 2019, 7, 6173–6179. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, L.; Cui, Z.; Fu, P.; Liu, M.; Qiao, X.; Pang, X. Dual Roles of Amino-Functionalized Silicon Quantum Dots (SiQDs) for Visible-Light-Induced Surface-Initiated PET-RAFT Polymerization on Substrates. ACS Appl. Mater. Interfaces 2020, 12, 42161–42168. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Goldmann, A.S.; Schacher, F.H. Polymer Interfaces: Synthetic Strategies Enabling Functionality, Adaptivity, and Spatial Control. Macromolecules 2016, 49, 5001–5016. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Russell, G.T. Chain-length-dependent termination in radical polymerization: Subtle revolution in tackling a long-standing challenge. Prog. Polym. Sci. 2009, 34, 1211–1259. [Google Scholar] [CrossRef]
- Discekici, E.H.; Pester, C.W.; Treat, N.J.; Lawrence, J.; Mattson, K.M.; Narupai, B.; Toumayan, E.P.; Luo, Y.; McGrath, A.J.; Clark, P.G.; et al. Simple Benchtop Approach to Polymer Brush Nanostructures Using Visible-Light-Mediated Metal-Free Atom Transfer Radical Polymerization. ACS Macro Lett. 2016, 5, 258–262. [Google Scholar] [CrossRef]
- Poelma, J.E.; Fors, B.P.; Meyers, G.F.; Kramer, J.W.; Hawker, C.J. Fabrication of complex three-dimensional polymer brush nanostructures through light-mediated living radical polymerization. Angew. Chem. Int. Ed. 2013, 52, 6844–6848. [Google Scholar] [CrossRef] [PubMed]
- Narupai, B.; Poelma, J.E.; Pester, C.W.; McGrath, A.J.; Toumayan, E.P.; Luo, Y.; Kramer, J.W.; Clark, P.G.; Ray, P.C.; Hawker, C.J. Hierarchical comb brush architectures via sequential light-mediated controlled radical polymerizations. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2276–2284. [Google Scholar] [CrossRef]
- Pester, C.W.; Narupai, B.; Mattson, K.M.; Bothman, D.P.; Klinger, D.; Lee, K.W.; Discekici, E.H.; Hawker, C.J. Engineering Surfaces through Sequential Stop-Flow Photopatterning. Adv. Mater. 2016, 28, 9292–9300. [Google Scholar] [CrossRef]
- Li, M.; Fromel, M.; Ranaweera, D.; Rocha, S.; Boyer, C.; Pester, C.W. SI-PET-RAFT: Surface-Initiated Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer Polymerization. ACS Macro Lett. 2019, 8, 374–380. [Google Scholar] [CrossRef]
- Seo, S.E.; Discekici, E.H.; Zhang, Y.; Bates, C.M.; Hawker, C.J. Surface-initiated PET-RAFT polymerization under metal-free and ambient conditions using enzyme degassing. J. Polym. Sci. 2020, 58, 70–76. [Google Scholar] [CrossRef]
- Jung, K.; Corrigan, N.; Ciftci, M.; Xu, J.; Seo, S.E.; Hawker, C.J.; Boyer, C. Designing with Light: Advanced 2D, 3D, and 4D Materials. Adv. Mater. 2020, 32, 1–21. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, M.; Zhou, Z.; Gou, J.; Hui, D. 3D printing of polymer matrix composites: A review and prospective. Compos. Part B Eng. 2017, 110, 442–458. [Google Scholar] [CrossRef]
- Corrigan, N.; Boyer, C. In the Limelight: 2D and 3D Materials via Photo-Controlled Radical Polymerization. Trends Chem. 2020, 2, 689–706. [Google Scholar] [CrossRef]
- Bagheri, A.; Bainbridge, C.W.A.; Engel, K.E.; Qiao, G.G.; Xu, J.; Boyer, C.; Jin, J. Oxygen Tolerant PET-RAFT Facilitated 3D Printing of Polymeric Materials under Visible LEDs. ACS Appl. Polym. Mater. 2020, 2, 782–790. [Google Scholar] [CrossRef]
- Momeni, F.; Mehdi, M.; Hassani, N.S.; Liu, X.; Ni, J. A review of 4D printing. Mater. Des. 2017, 122, 42–79. [Google Scholar] [CrossRef]
- Choi, J.; Kwon, O.C.; Jo, W.; Lee, H.J.; Moon, M.W. 4D printing technology: A review. 3D Print. Addit. Manuf. 2015, 2, 159–167. [Google Scholar] [CrossRef]
- Zhang, Z.; Corrigan, N.; Bagheri, A.; Jin, J.; Boyer, C. A Versatile 3D and 4D Printing System through Photocontrolled RAFT Polymerization. Angew. Chem. Int. Ed. 2019, 58, 17954–17963. [Google Scholar] [CrossRef]
- Bainbridge, C.W.A.; Engel, K.E.; Jin, J. 3D printing and growth induced bending based on PET-RAFT polymerization. Polym. Chem. 2020, 11, 4084–4093. [Google Scholar] [CrossRef]
- Ko, J.H.; Maynard, H.D. A guide to maximizing the therapeutic potential of protein-polymer conjugates by rational design. Chem. Soc. Rev. 2018, 47, 8998–9014. [Google Scholar] [CrossRef]
- Messina, M.S.; Messina, K.M.M.; Bhattacharya, A.; Montgomery, H.R.; Maynard, H.D. Preparation of biomolecule-polymer conjugates by grafting-from using ATRP, RAFT, or ROMP. Prog. Polym. Sci. 2020, 100, 101186. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Wu, G.; Zhou, P.; Zhou, Y.; Wang, L.; Huang, X. Efficient Way to Generate Protein-Based Nanoparticles by in-Situ Photoinitiated Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6, 689–694. [Google Scholar] [CrossRef]
- Kovaliov, M.; Allegrezza, M.L.; Richter, B.; Konkolewicz, D.; Averick, S. Synthesis of lipase polymer hybrids with retained or enhanced activity using the grafting-from strategy. Polymer 2018, 137, 338–345. [Google Scholar] [CrossRef]
- Tucker, B.S.; Coughlin, M.L.; Figg, C.A.; Sumerlin, B.S. Grafting-From Proteins Using Metal-Free PET-RAFT Polymerizations under Mild Visible-Light Irradiation. ACS Macro Lett. 2017, 6, 452–457. [Google Scholar] [CrossRef]
- Watanabe, A.; Niu, J.; Lunn, D.J.; Lawrence, J.; Knight, A.S.; Zhang, M.; Hawker, C.J. PET-RAFT as a facile strategy for preparing functional lipid–polymer conjugates. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 1259–1268. [Google Scholar] [CrossRef]
- Lueckerath, T.; Strauch, T.; Koynov, K.; Barner-Kowollik, C.; Ng, D.Y.W.; Weil, T. DNA-Polymer Conjugates by Photoinduced RAFT Polymerization. Biomacromolecules 2019, 20, 212–221. [Google Scholar] [CrossRef]
- Niu, J.; Lunn, D.J.; Pusuluri, A.; Yoo, J.I.; O’Malley, M.A.; Mitragotri, S.; Soh, H.T.; Hawker, C.J. Engineering live cell surfaces with functional polymers via cytocompatible controlled radical polymerization. Nat. Chem. 2017, 9, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Bixler, G.D.; Bhushan, B. Review article: Biofouling: Lessons from nature. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 2381–2417. [Google Scholar] [CrossRef]
- De Los Santos Pereira, A.; Rodriguez-Emmenegger, C.; Surman, F.; Riedel, T.; Alles, A.B.; Brynda, E. Use of pooled blood plasmas in the assessment of fouling resistance. RSC Adv. 2014, 4, 2318–2321. [Google Scholar] [CrossRef]
- Baggerman, J.; Smulders, M.M.J.; Zuilhof, H. Romantic Surfaces: A Systematic Overview of Stable, Biospecific, and Antifouling Zwitterionic Surfaces. Langmuir 2019, 35, 1072–1084. [Google Scholar] [CrossRef]
- Dalsin, J.L.; Messersmith, P.B. Bioinspired antifouling polymers. Mater. Today 2005, 8, 38–46. [Google Scholar] [CrossRef]
- Leonardi, A.K.; Ober, C.K. Polymer-based marine antifouling and fouling release surfaces: Strategies for synthesis and modification. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 241–264. [Google Scholar] [CrossRef]
- Kuzmyn, A.R.; Nguyen, A.T.; Teunissen, L.W.; Zuilhof, H.; Baggerman, J. Antifouling Polymer Brushes via Oxygen-Tolerant Surface-Initiated PET-RAFT. Langmuir 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, L.; Zhou, J.; Ye, L.; Hu, H.; Luo, Z.; Zhou, L. Surface modification of PVA hydrogel membranes with carboxybetaine methacrylate via PET-RAFT for anti-fouling. Polymer 2019, 162, 80–90. [Google Scholar] [CrossRef]
- Zhou, J.; Ye, L.; Lin, Y.; Wang, L.; Zhou, L.; Hu, H.; Zhang, Q.; Yang, H.; Luo, Z. Surface modification PVA hydrogel with zwitterionic via PET-RAFT to improve the antifouling property. J. Appl. Polym. Sci. 2019, 136, 2–10. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, Y.; Ye, L.; Wang, L.; Zhou, L.; Hu, H.; Zhang, Q.; Yang, H.; Luo, Z. PVA Hydrogel Functionalization via PET-RAFT Grafting with Glycidyl Methacrylate and Immobilization with 2-Hydroxypropyltrimethyl Ammonium Chloride Chitosan via Ring-Open Reaction. Macromol. Res. 2019, 27, 1144–1154. [Google Scholar] [CrossRef]
- Timofeeva, L.; Kleshcheva, N. Antimicrobial polymers: Mechanism of action, factors of activity, and applications. Appl. Microbiol. Biotechnol. 2011, 89, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; Wong, E.H.H.; Boyer, C.; Qiao, G.G. Antimicrobial polymeric nanoparticles. Prog. Polym. Sci. 2018, 76, 40–64. [Google Scholar] [CrossRef]
- Tew, G.N.; Scott, R.W.; Klein, M.L.; Degrado, W.F. De Novo Design of Antimicrobial Polymers, Foldamers, and Small Molecules: From Discovery to Practical Applications. Acc. Chem. Res. 2011, 43, 30–39. [Google Scholar] [CrossRef]
- Siedenbiedel, F.; Tiller, J.C. Antimicrobial polymers in solution and on surfaces: Overview and functional principles. Polymers 2012, 4, 46–71. [Google Scholar] [CrossRef]
- Zheng, Y.; Luo, Y.; Feng, K.; Zhang, W.; Chen, G. High Throughput Screening of Glycopolymers: Balance between Cytotoxicity and Antibacterial Property. ACS Macro Lett. 2019, 8, 326–330. [Google Scholar] [CrossRef]
- Richards, S.J.; Jones, A.; Tomás, R.M.F.; Gibson, M.I. Photochemical “In-Air” Combinatorial Discovery of Antimicrobial Co-polymers. Chem. A Eur. J. 2018, 24, 13758–13761. [Google Scholar] [CrossRef]
- Judzewitsch, P.R.; Zhao, L.; Wong, E.H.H.; Boyer, C. High-Throughput Synthesis of Antimicrobial Copolymers and Rapid Evaluation of Their Bioactivity. Macromolecules 2019, 52, 3975–3986. [Google Scholar] [CrossRef]
- Judzewitsch, P.R.; Nguyen, T.-K.; Shanmugam, S.; Wong, E.H.H.; Boyer, C. Towards Sequence-Controlled Antimicrobial Polymers: Effect of Polymer Block Order on Antimicrobial Activity. Angew. Chem. 2018, 130, 4649–4654. [Google Scholar] [CrossRef]
- Judzewitsch, P.R.; Corrigan, N.; Trujillo, F.; Xu, J.; Moad, G.; Hawker, C.J.; Wong, E.H.H.; Boyer, C. High-Throughput Process for the Discovery of Antimicrobial Polymers and Their Upscaled Production via Flow Polymerization. Macromolecules 2020, 53, 631–639. [Google Scholar] [CrossRef]
- Sherwood, J.; De bruyn, M.; Constantinou, A.; Moity, L.; McElroy, C.R.; Farmer, T.J.; Duncan, T.; Raverty, W.; Hunt, A.J.; Clark, J.H. Dihydrolevoglucosenone (Cyrene) as a bio-based alternative for dipolar aprotic solvents. Chem. Commun. 2014, 50, 9650–9652. [Google Scholar] [CrossRef] [PubMed]
- Cseri, L.; Szekely, G. Towards cleaner PolarClean: Efficient synthesis and extended applications of the polar aprotic solvent methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate. Green Chem. 2019, 21, 4178–4188. [Google Scholar] [CrossRef]
- Kubisa, P. Application of ionic liquids as solvents for polymerization processes. Prog. Polym. Sci. 2004, 29, 3–12. [Google Scholar] [CrossRef]
- Kubisa, P. Ionic liquids as solvents for polymerization processes-Progress and challenges. Prog. Polym. Sci. 2009, 34, 1333–1347. [Google Scholar] [CrossRef]
- Vijayakrishna, K.; Jewrajka, S.K.; Ruiz, A.; Marcilla, R.; Pomposo, J.A.; Mecerreyes, D.; Taton, D.; Gnanou, Y. Synthesis by RAFT and ionic responsiveness of double hydrophilic block copolymers based on ionic liquid monomer units. Macromolecules 2008, 41, 6299–6308. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PC | Ref | Advantages | Disadvantages |
|---|---|---|---|
| (Ir(ppy)3) | [23] | Low energy absorption | Precious metal, expensive, soluble in few organic solvents |
| [Ru(bpy)3Cl2] | [30] | Solubility in wide variety of solvents (also polar) | Precious metal, expensive |
| Porphyrin-based | [31,33,34] | Non precious metal, low-cost, low-toxicity, multiple absorption peaks | Presence of impurity, effective only with trithiocarbonates |
| Eosine Y | [32] | Metal free, low cost | Sacrificial electron donor required, slow kinetics |
| Metal oxides (ZnO, TiO2) | [37,38] | Easy to recycle low-cost, low-toxicity | High energy irradiation |
| QDs | [41,42] | Easy to recycle, modulable absorption wavelength | Toxic compound |
| Tertiary amines | [43] | Metal free | High energy irradiation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellotti, V.; Simonutti, R. New Light in Polymer Science: Photoinduced Reversible Addition-Fragmentation Chain Transfer Polymerization (PET-RAFT) as Innovative Strategy for the Synthesis of Advanced Materials. Polymers 2021, 13, 1119. https://doi.org/10.3390/polym13071119
Bellotti V, Simonutti R. New Light in Polymer Science: Photoinduced Reversible Addition-Fragmentation Chain Transfer Polymerization (PET-RAFT) as Innovative Strategy for the Synthesis of Advanced Materials. Polymers. 2021; 13(7):1119. https://doi.org/10.3390/polym13071119
Chicago/Turabian StyleBellotti, Valentina, and Roberto Simonutti. 2021. "New Light in Polymer Science: Photoinduced Reversible Addition-Fragmentation Chain Transfer Polymerization (PET-RAFT) as Innovative Strategy for the Synthesis of Advanced Materials" Polymers 13, no. 7: 1119. https://doi.org/10.3390/polym13071119
APA StyleBellotti, V., & Simonutti, R. (2021). New Light in Polymer Science: Photoinduced Reversible Addition-Fragmentation Chain Transfer Polymerization (PET-RAFT) as Innovative Strategy for the Synthesis of Advanced Materials. Polymers, 13(7), 1119. https://doi.org/10.3390/polym13071119