
A New Era in Engineering Plastics: Compatibility and Perspectives of Sustainable Alipharomatic Poly(ethylene terephthalate)/Poly(ethylene 2,5-furandicarboxylate) Blends

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Synthesis of 2,5-Dimethylfuran-Dicarboxylate (DMF)
2.3. Polyester Synthesis
2.4. Preparation of Blends
2.5. Polyester Characterization
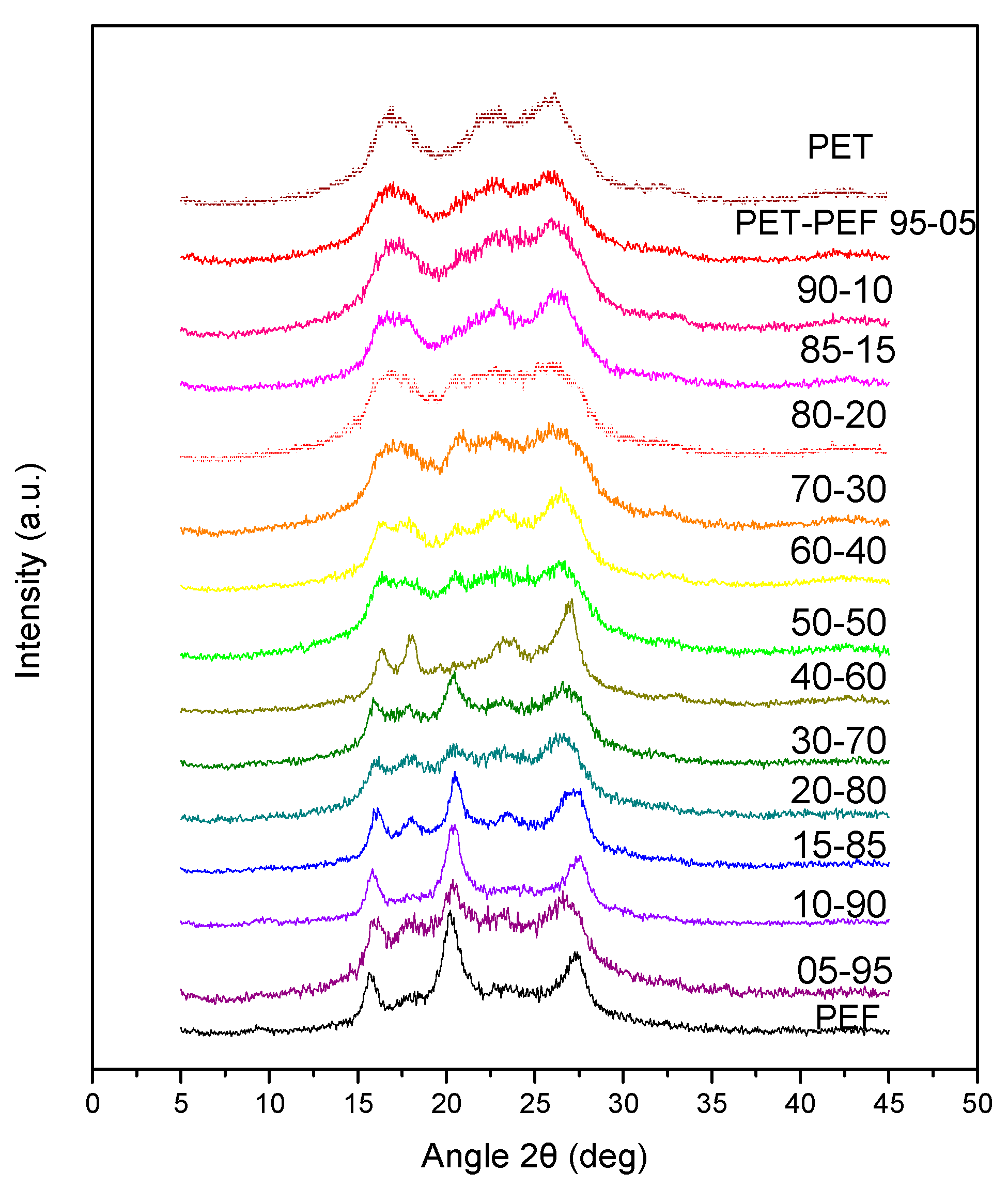
2.5.1. Wide Angle X-Ray Diffraction Patterns (WAXD)
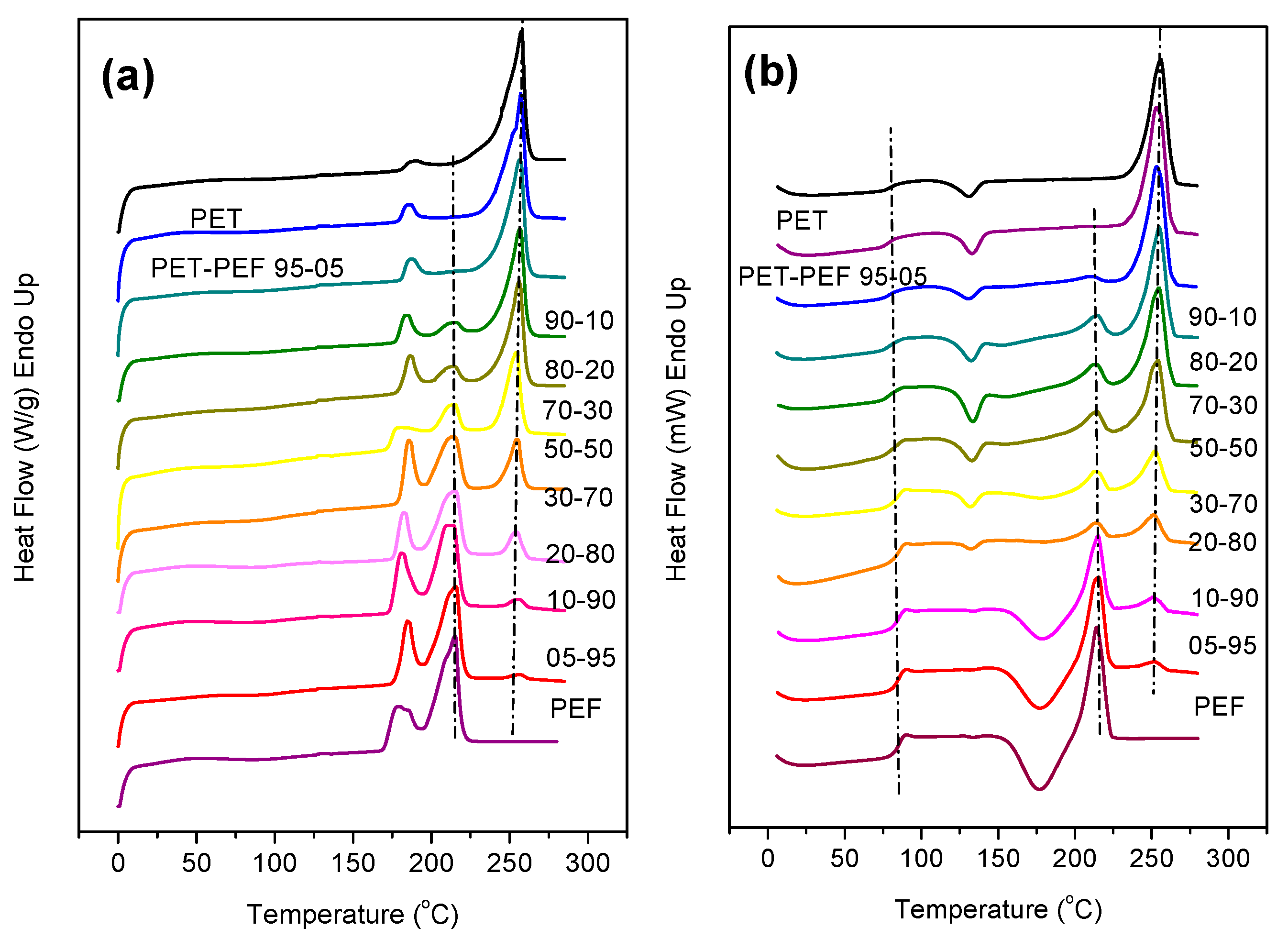
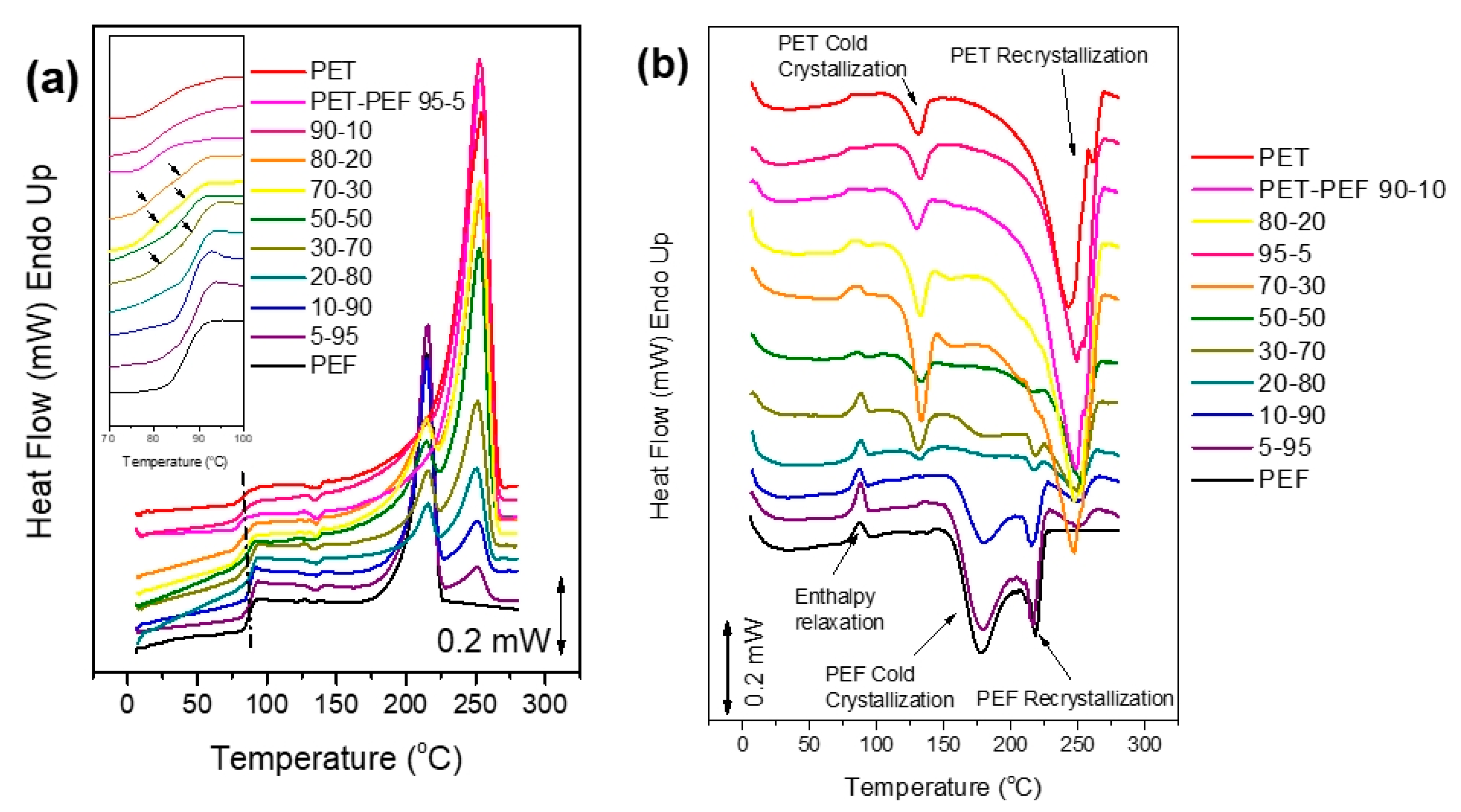
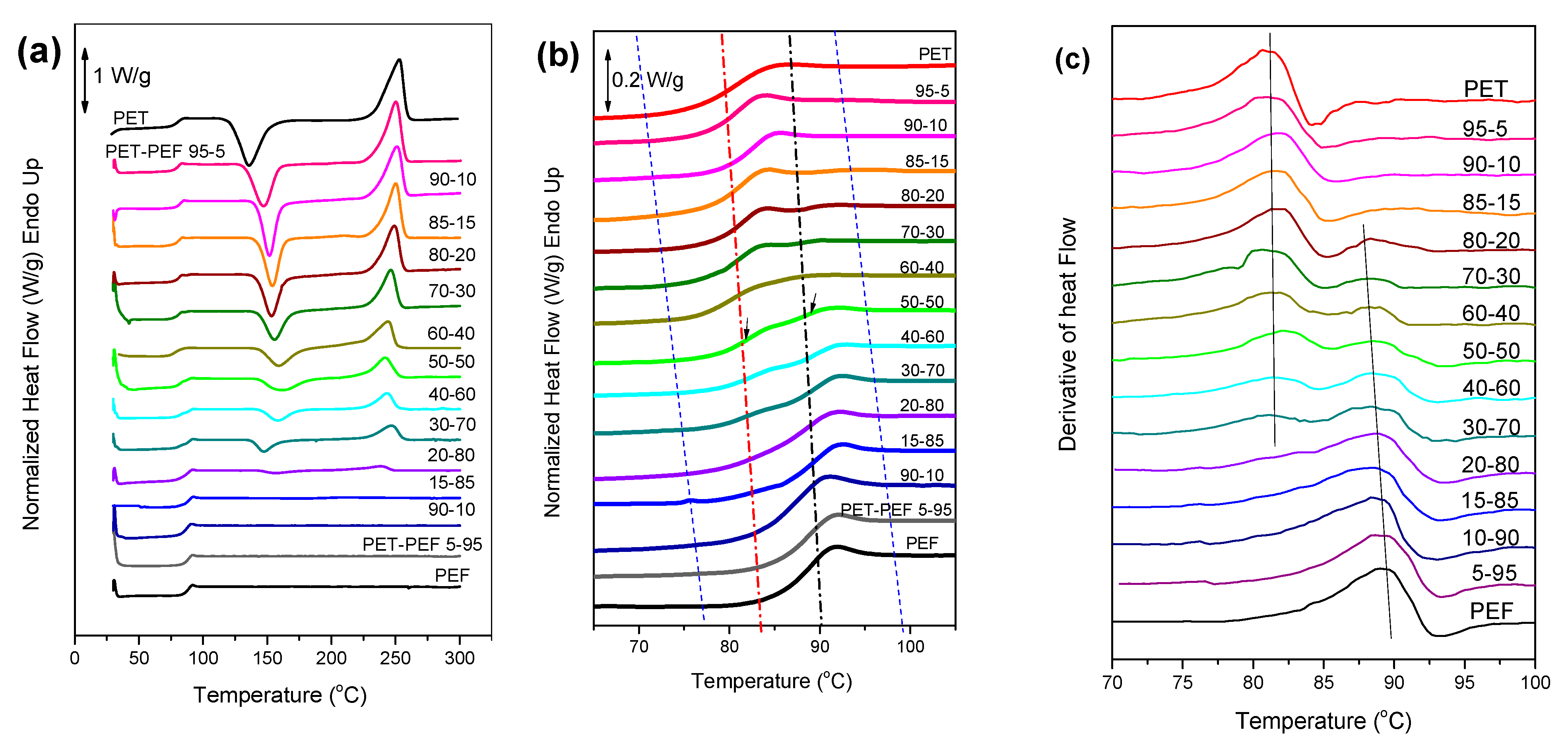
2.5.2. Differential Scanning Calorimetry (DSC)
2.5.3. Polarizing Light Microscopy (PLM)
3. Results and Discussion
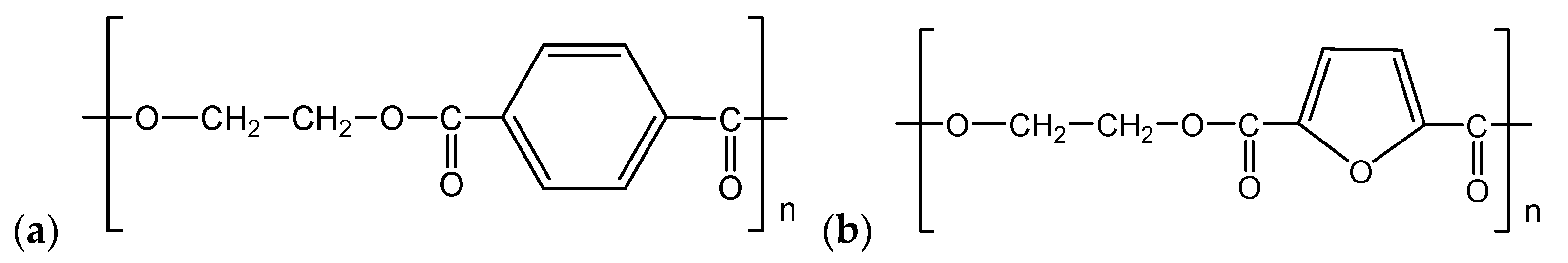
3.1. Primary Characterisation
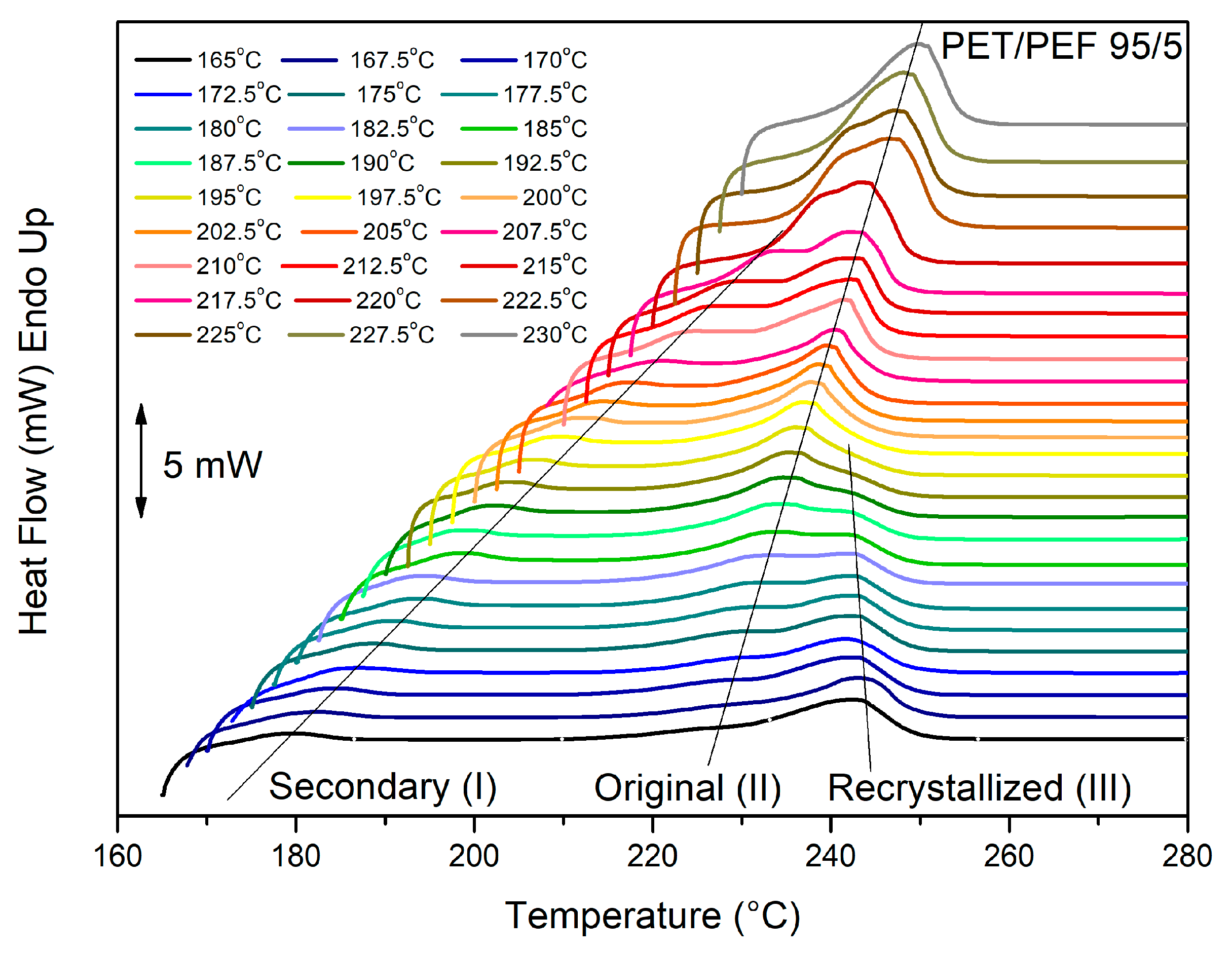
3.2. Melting Behavior of the Blends
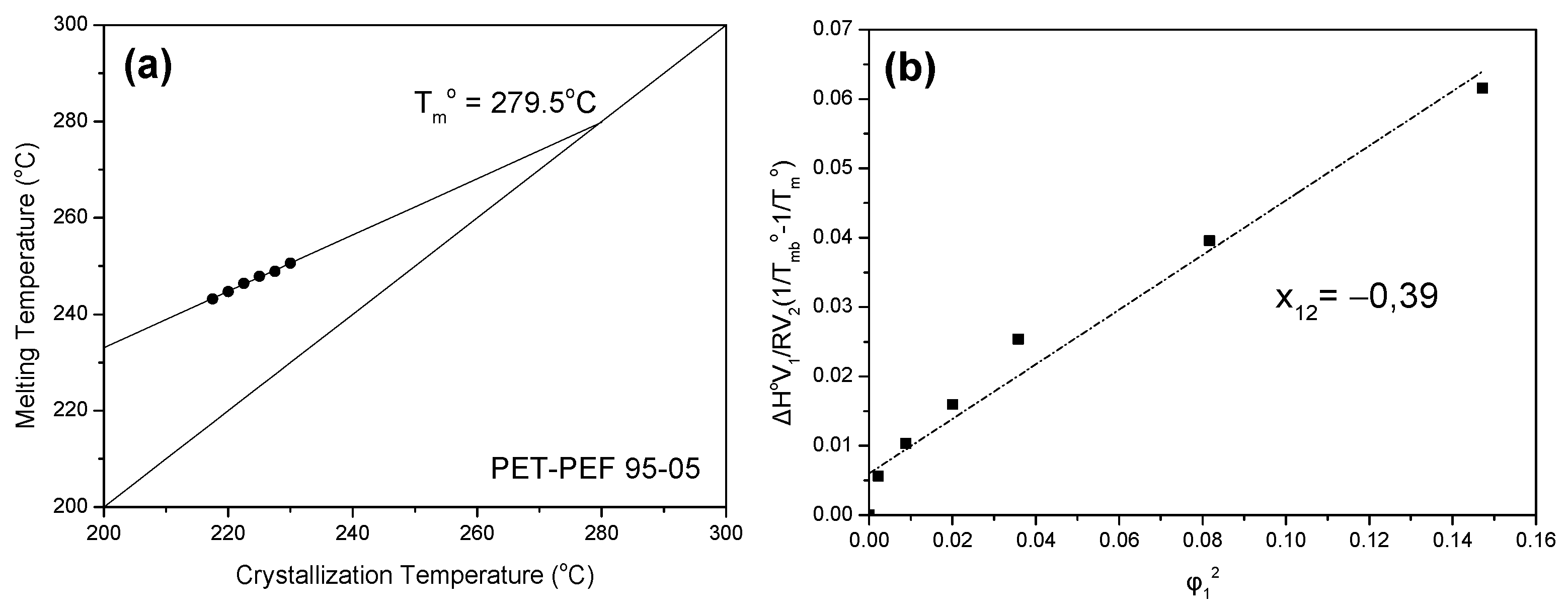
3.3. Equilibrium Melting Point
3.4. Crystallization of the Blends
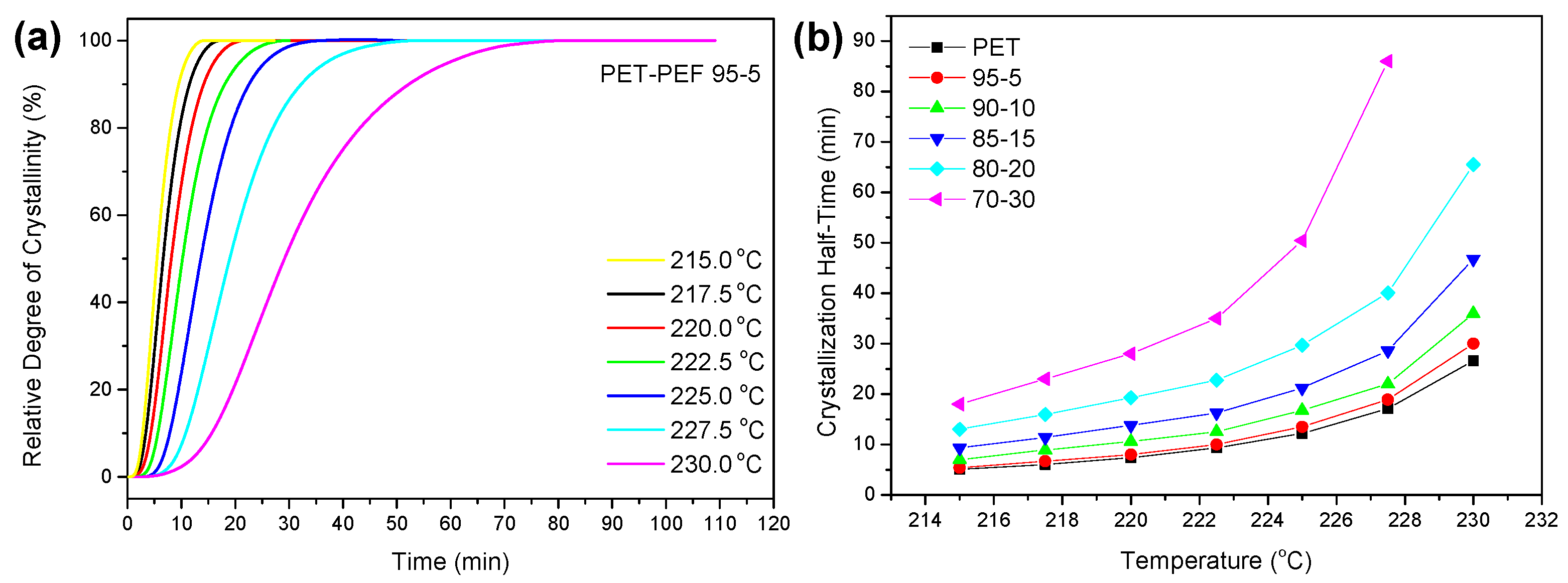
3.4.1. Isothermal Crystallization
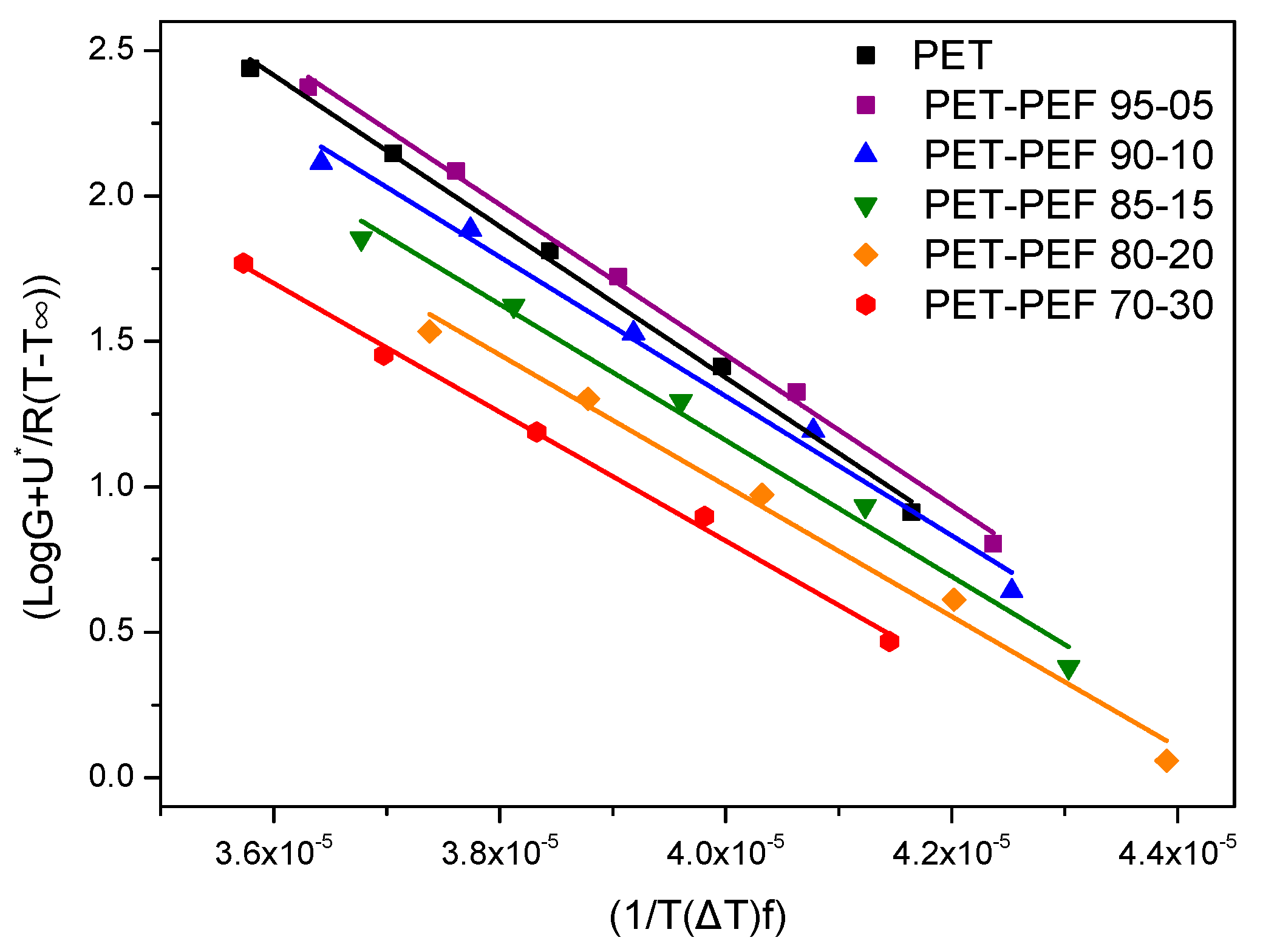
3.4.2. Application of Secondary Nucleation Theory
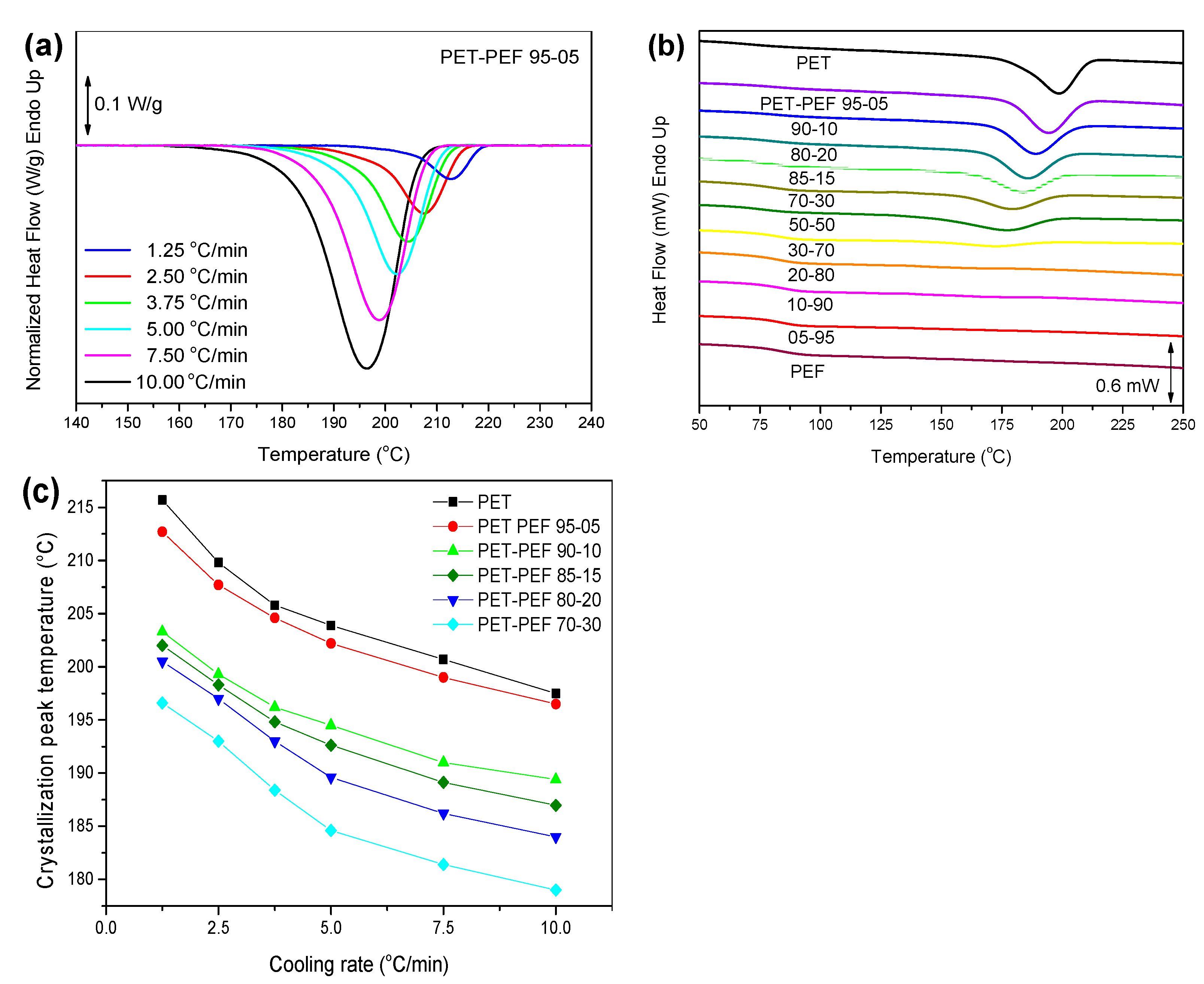
3.4.3. Non-Isothermal Crystallization
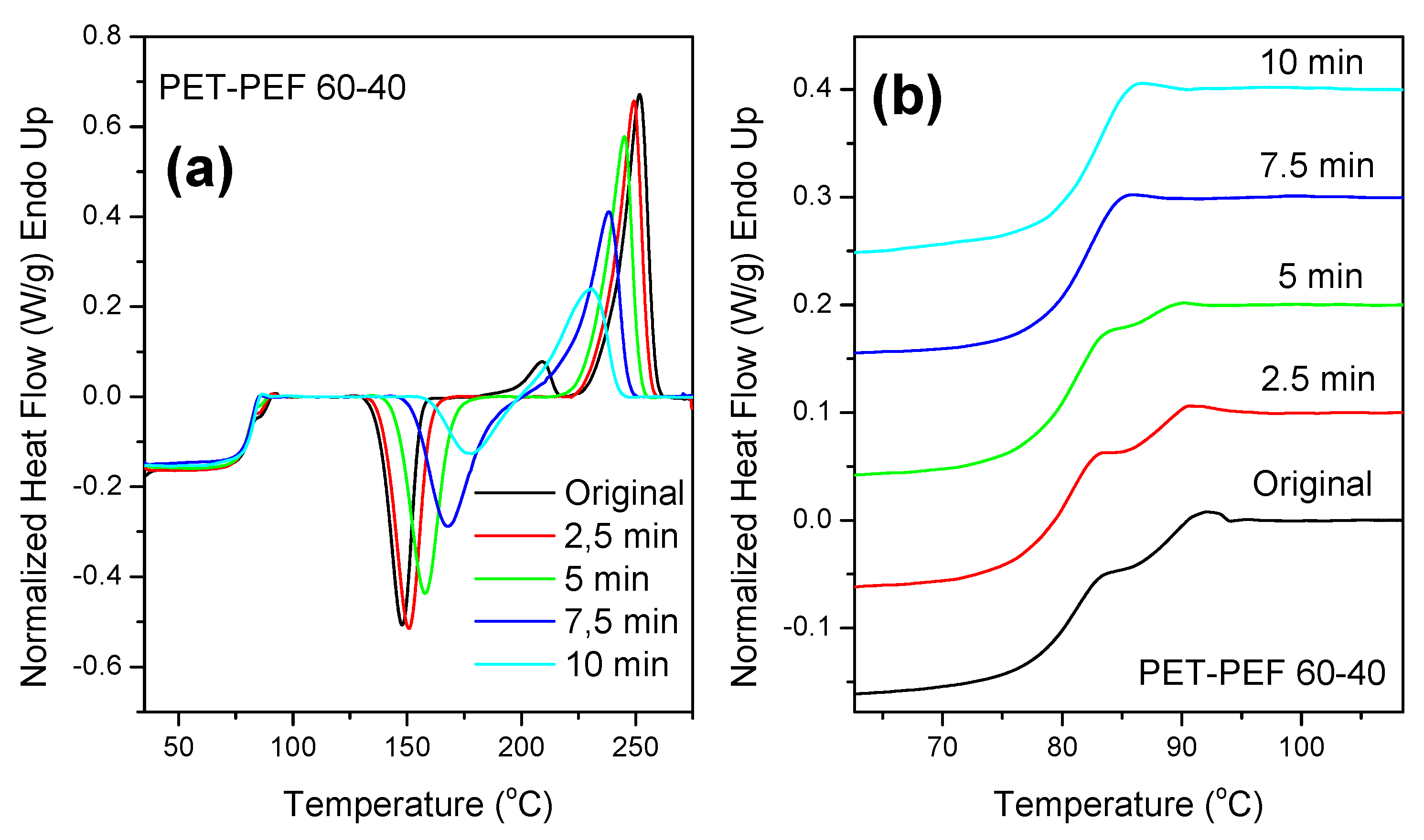
3.5. Reactive Blending
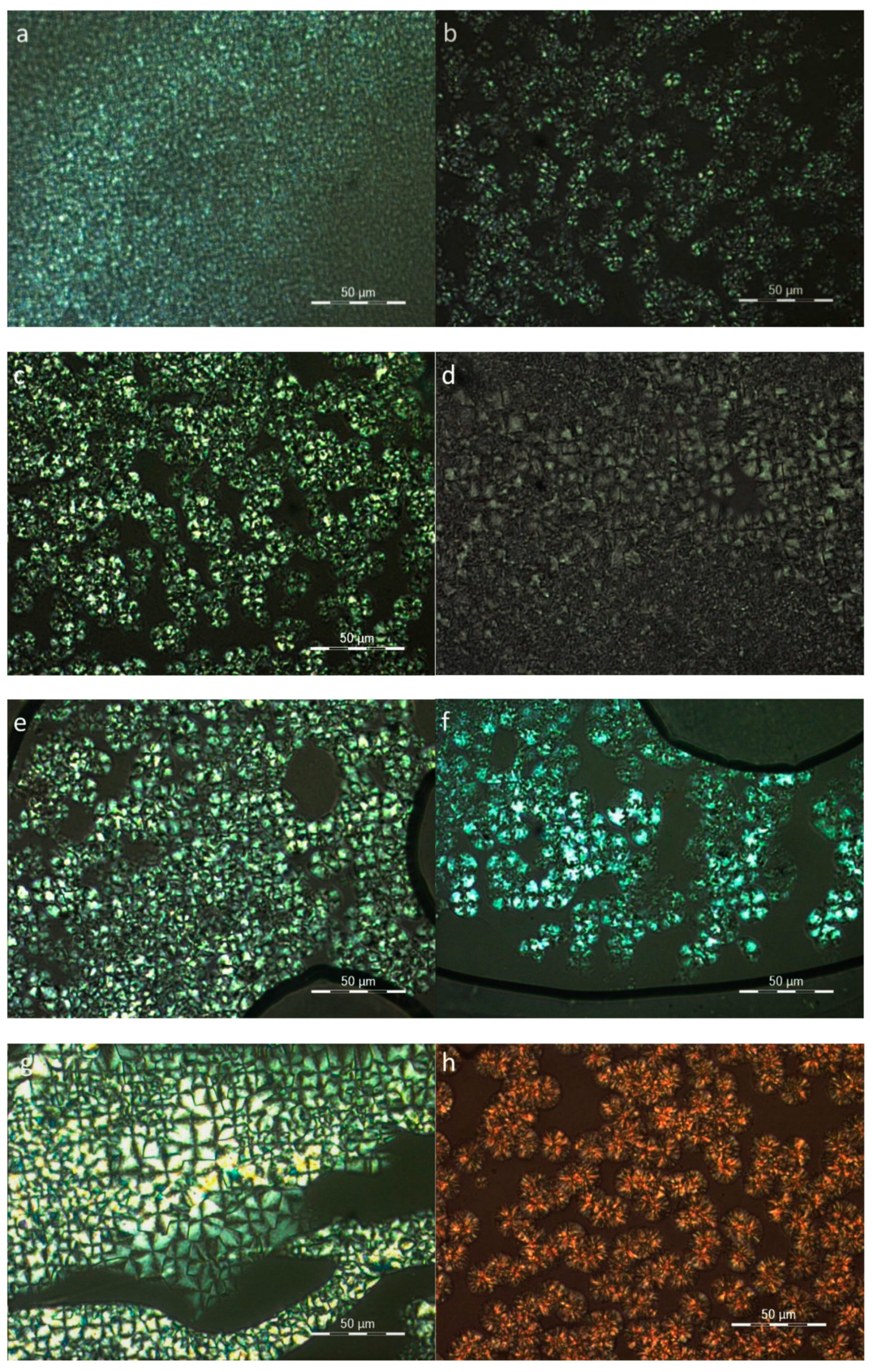
3.6. Morphological Study of the Blends
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanders, J.P.M.; Clark, J.H.; Harmsen, G.J.; Heeres, H.J.; Heijnen, J.J.; Kersten, S.R.A.; Swaaij, W.P.M.; Moulijin, J.A. Process intensification in the future production of base chemicals from biomass. Chem. Eng. Process. 2012, 51, 117–136. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Y.; Xiong, Z.; Liu, X.; Na, H.; Zhang, R.; Zhu, J. Highly recoverable rosin-based shape memory polyurethanes. J. Mater. Chem. A 2013, 1, 3263–3267. [Google Scholar] [CrossRef]
- Storz, H.; Vorlop, K.D. Bio-based plastics: Status, challenges and trends. Appl. Agric. For. Res. 2013, 63, 321–332. [Google Scholar]
- Mülhaupt, R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2012, 214, 159–174. [Google Scholar] [CrossRef]
- Schneiderman, D.K.; Hillmyer, M.A. 50th Anniversary Perspective: There Is a Great Future in Sustainable Polymers. Macromolecules 2017, 50, 3733–3749. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, L.; Li, B.-G. High Molecular Weight Polyesters Derived from Biobased 1,5-Pentanediol and a Variety of Aliphatic Diacids: Synthesis, Characterization, and Thermo-Mechanical Properties. ACS Sustain. Chem. Eng. 2017, 5, 6159–6166. [Google Scholar] [CrossRef]
- Sousa, A.F.; Vilela, C.; Fonseca, A.C.; Matos, M.; Freire, C.S.R.; Gruter, G.-J.M.; Coelho, J.F.J.; Silvestre, A.J.D. Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: A tribute to furan excellency. Polym. Chem. 2015, 6, 5961–5983. [Google Scholar] [CrossRef]
- Morales-Huerta, J.C.; Ciulik, C.B.; De Ilarduya, A.M.; Muñoz-Guerra, S. Fully bio-based aromatic–aliphatic copolyesters: Poly(butylene furandicarboxylate-co-succinate)s obtained by ring opening polymerization. Polym. Chem. 2017, 8, 748–760. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Papageorgiou, D.G.; Exarhopoulos, S.; Papageorgiou, M.; Bikiaris, D.N. Evaluation of polyesters from renewable resources as alternatives to the current fossil-based polymers. Phase transitions of poly(butylene 2,5-furan-dicarboxylate). Polymer 2014, 55, 3846–3858. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Papageorgiou, D.G.; Chrissafis, K.; Exarhopoulos, S.; Bikiaris, D.N. Furan-based polyesters from renewable resources: Crystallization and thermal degradation behavior of poly(hexamethylene 2,5-furan-dicarboxylate). Eur. Polym. J. 2015, 67, 383–396. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, K. Recent Advances in the Catalytic Synthesis of 2,5-Furandicarboxylic Acid and Its Derivatives. ACS Catal. 2015, 5, 6529–6544. [Google Scholar] [CrossRef]
- Rosenboom, J.-G.; Hohl, D.K.; Fleckenstein, P.; Storti, G.; Morbidelli, M. Bottle-grade polyethylene furanoate from ring-opening polymerisation of cyclic oligomers. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.K.; Wenz, G.B.; Kriegel, R.M.; Koros, W.J. Penetrant transport in semicrystalline poly(ethylene furanoate). Polymer 2016, 98, 305–310. [Google Scholar] [CrossRef]
- Burgess, S.K.; Leisen, J.E.; Kraftschik, B.E.; Mubarak, C.R.; Kriegel, R.M.; Koros, W.J. Chain Mobility, Thermal, and Mechanical Properties of Poly(ethylene furanoate) Compared to Poly(ethylene terephthalate). Macromolecules 2014, 47, 1383–1391. [Google Scholar] [CrossRef]
- Van Berkel, J.G.; Guigo, N.; Kolstad, J.J.; Sipos, L.; Wang, B.; Dam, M.A.; Sbirrazzuoli, N. Isothermal Crystallization Kinetics of Poly (Ethylene 2,5-Furandicarboxylate). Macromol. Mater. Eng. 2015, 300, 466–474. [Google Scholar] [CrossRef]
- Dimitriadis, T.; Bikiaris, D.N.; Papageorgiou, G.Z.; Floudas, G. Molecular Dynamics of Poly(ethylene-2,5-furanoate) (PEF) as a Function of the Degree of Crystallinity by Dielectric Spectroscopy and Calorimetry. Macromol. Chem. Phys. 2016, 217, 2056–2062. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Terzopoulou, Z.; Bikiaris, D.N. Production of bio-based 2,5-furan dicarboxylate polyesters: Recent progress and critical aspects in their synthesis and thermal properties. Eur. Polym. J. 2016, 83, 202–229. [Google Scholar] [CrossRef]
- Tomisawa, R.; Ikaga, T.; Kim, K.; Ohkoshi, Y.; Okada, K.; Masunaga, H.; Kanaya, T.; Masuda, M.; Maeda, Y. Effect of draw ratio on fiber structure development of polyethylene terephthalate. Polymer 2017, 116, 357–366. [Google Scholar] [CrossRef]
- Van Uytvanck, P.; Haire, G.; Marshall, P.; Dennis, J. Impact on the Polyester Value Chain of Using p-Xylene Derived from Biomass. ACS Sustain. Chem. Eng. 2017, 5, 4119–4126. [Google Scholar] [CrossRef]
- Galbis, J.A.; García-Martín, M.D.G.; De Paz, M.V.; Galbis, E. Synthetic Polymers from Sugar-Based Monomers. Chem. Rev. 2016, 116, 1600–1636. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Guerra, S.; Lavilla, C.; Japu, C.; De Ilarduya, A.M. Renewable terephthalate polyesters from carbohydrate-based bicyclic monomers. Green Chem. 2014, 16, 1716–1739. [Google Scholar] [CrossRef]
- Kim, G.S.; Son, J.M.; Lee, J.K.; Lee, K.H. Morphology development and crystallization behavior of poly(ethylene terephthalate)/poly(trimethylene terephthalate) blends. Eur. Polym. J. 2010, 46, 1696–1704. [Google Scholar] [CrossRef]
- Aoyama, S.; Park, Y.T.; Ougizawa, T.; Macosko, C.W. Melt crystallization of poly(ethylene terephthalate): Comparing addition of graphene vs. carbon nanotubes. Polymer 2014, 55, 2077–2085. [Google Scholar] [CrossRef]
- Docampo, P.; Ball, J.M.; Darwich, M.; Eperon, G.E.; Snaith, H.J. Efficient organometal trihalide perovskite planar-heterojunction solar cells on flexible polymer substrates. Nat. Commun. 2013, 4, 2761. [Google Scholar] [CrossRef] [PubMed]
- Kawai, F.; Kawabata, T.; Oda, M. Current State and Perspectives Related to the Polyethylene Terephthalate Hydrolases Available for Biorecycling. ACS Sustain. Chem. Eng. 2020, 8, 8894–8908. [Google Scholar] [CrossRef]
- Kim, H.J.; Peng, X.; Shin, Y.; Hillmyer, M.A.; Ellison, C.J. Blend Miscibility of Poly(ethylene terephthalate) and Aromatic Polyesters from Salicylic Acid. J. Phys. Chem. B 2021, 125, 450–460. [Google Scholar] [CrossRef]
- Bai, H.; Xiu, H.; Gao, J.; Deng, H.; Zhang, Q.; Yang, M.; Fu, Q. Tailoring impact toughness of poly (L-lactide)/poly (ε-caprolactone)(PLLA/PCL) blends by controlling crystallization of PLLA matrix. ACS Appl. Mater. Interfaces 2012, 4, 897–905. [Google Scholar] [CrossRef]
- Doroshenko, M.; Gonzales, M.; Best, A.; Butt, H.-J.; Koynov, K.; Floudas, G. Monitoring the Dynamics of Phase Separation in a Polymer Blend by Confocal Imaging and Fluorescence Correlation Spectroscopy. Macromol. Rapid Commun. 2012, 33, 1568–1573. [Google Scholar] [CrossRef]
- Blochowiak, M.; Pakula, T.; Butt, H.-J.; Floudas, G. Miscibility of binary blends of ethylene/norbornene copolymers: Comparison to a lattice cluster theory. Polymer 2007, 48, 6010–6017. [Google Scholar] [CrossRef]
- Suzuki, T.; Tanaka, H.; Nishi, T. Miscibility and transesterification in bisphenol A polycarbonate/poly(ethylene terephthalate) blends. Polymer 1989, 30, 1287–1297. [Google Scholar] [CrossRef]
- Daubeny, R.D.P.; Bunn, C. The crystal structure of polyethylene terephthalate. Proc. Royal 1954, 226, 531–542. [Google Scholar]
- Mao, Y.; Kriegel, R.M.; Bucknall, D.G. The crystal structure of poly(ethylene furanoate). Polymer 2016, 102, 308–314. [Google Scholar] [CrossRef]
- Tsanaktsis, V.; Papageorgiou, D.G.; Exarhopoulos, S.; Bikiaris, D.N.; Papageorgiou, G.Z. Crystallization and Polymorphism of Poly(ethylene furanoate). Cryst. Growth Des. 2015, 15, 5505–5512. [Google Scholar] [CrossRef]
- Lotti, N.; Munari, A.; Gigli, M.; Gazzano, M.; Tsanaktsis, V.; Bikiaris, D.N.; Papageorgiou, G.Z. Thermal and structural response of in situ prepared biobased poly(ethylene 2,5-furan dicarboxylate) nanocomposites. Polymer 2016, 103, 288–298. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Chrissafis, K.; Bikiaris, D.; Will, J.; Hoppe, A.; Roether, J.A.; Boccaccini, A.R. Crystallization and Melting Behavior of Poly(Butylene Succinate) Nanocomposites Containing Silica-Nanotubes and Strontium Hydroxyapatite Nanorods. Ind. Eng. Chem. Res. 2013, 53, 678–692. [Google Scholar] [CrossRef]
- Stoclet, G.; du Sart, G.G.; Yeniad, B.; de Vos, S.; Lefebvre, J. Isothermal crystallization and structural characterization of poly(ethylene-2,5-furanoate). Polymer 2015, 72, 165–176. [Google Scholar] [CrossRef]
- Martino, L.; Guigo, N.; Van Berkel, J.G.; Sbirrazzuoli, N. Influence of organically modified montmorillonite and sepiolite clays on the physical properties of bio-based poly(ethylene 2,5-furandicarboxylate). Compos. Part B Eng. 2017, 110, 96–105. [Google Scholar] [CrossRef]
- Xie, H.; Meng, H.; Wu, L.; Li, B.G.; Dubois, P. In-situ synthesis, thermal and mechanical properties of biobased poly (ethylene 2, 5-furandicarboxylate)/montmorillonite (PEF/MMT) nanocomposites. Eur. Polym. J. 2019, 121, 109266. [Google Scholar] [CrossRef]
- Woo, E.M.; Kuo, Y.-H. Complete miscibility of ternary aryl polyesters demonstrating a new criterion and horizon for miscibility characterization. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 2394–2404. [Google Scholar] [CrossRef]
- Lodge, T.P.; McLeish, T.C.B. Self-Concentrations and Effective Glass Transition Temperatures in Polymer Blends. Macromolecules 2000, 33, 5278–5284. [Google Scholar] [CrossRef]
- Lodge, T.P.; Wood, E.R.; Haley, J.C. Two calorimetric glass transitions do not necessarily indicate immiscibility: The case of PEO/PMMA. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 756–763. [Google Scholar] [CrossRef]
- Kordjazi, Z.; Ajji, A. Partially miscible polymer blends of ethyl cellulose and hydroxyl terminated polybutadiene. Polymer 2020, 211, 123067. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Bikiaris, D.N. Synthesis of poly(ethylene furandicarboxylate) polyester using monomers derived from renewable resources: Thermal behavior comparison with PET and PEN. Phys. Chem. Chem. Phys. 2014, 16, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-J.; Shi, Q.-S. Transforming Sugarcane Bagasse into Bioplastics via Homogeneous Modification with Phthalic Anhydride in Ionic Liquid. ACS Sustain. Chem. Eng. 2015, 3, 2510–2515. [Google Scholar] [CrossRef]
- Minakov, A.A.; Mordvintsev, D.A.; Schick, C. Melting and reorganization of poly(ethylene terephthalate) on fast heating (1000 K/s). Polymer 2004, 45, 3755–3763. [Google Scholar] [CrossRef]
- Zhou, C.; Clough, S.B. Multiple melting endotherms of poly(ethylene terephthalate). Polym. Eng. Sci. 1988, 28, 65–68. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Weeks, J.J. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1962, 66A, 13. [Google Scholar] [CrossRef]
- Runt, J.; Miley, D.M.; Zhang, X.; Gallagher, K.P.; McFeaters, K.; Fishburn, J. Crystallization of poly(butylene terephthalate) and its blends with polyarylate. Macromolecules 1992, 25, 1929–1934. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of Crystallization in High Polymers. IV. A Theory of Crystalline States and Fusion in Polymers, Copolymers, and Their Mixtures with Diluents. J. Chem. Phys. 1949, 17, 223–240. [Google Scholar] [CrossRef]
- Flory, P.J. Theory of crystallization in copolymers. Trans. Faraday Soc. 1955, 51, 848–857. [Google Scholar] [CrossRef]
- Konstantopoulou, M.; Terzopoulou, Z.; Nerantzaki, M.; Tsagkalias, J.; Achilias, D.S.; Bikiaris, D.N.; Exarhopoulos, S.; Papageorgiou, D.G.; Papageorgiou, G.Z. Poly(ethylene furanoate-co-ethylene terephthalate) biobased copolymers: Synthesis, thermal properties and cocrystallization behavior. Eur. Polym. J. 2017, 89, 349–366. [Google Scholar] [CrossRef]
- Meaurio, E.; Sanchez-Rexach, E.; Hernandez, E.Z.; Lejardi, A.; Sanchez-Camargo, A.D.P.; Sarasua, J.-R. Predicting miscibility in polymer blends using the Bagley plot: Blends with poly(ethylene oxide). Polymer 2017, 113, 295–309. [Google Scholar] [CrossRef]
- Qiu, Z.; Komura, M.; Ikehara, T.; Nishi, T. Poly (butylene succinate)/poly (vinyl phenol) blends. Part 1. Miscibility and crystallization. Polymer 2003, 44, 8111–8117. [Google Scholar] [CrossRef]
- Penning, J.P.; Manley, R.S.J. Miscible Blends of Two Crystalline Polymers. 1. Phase Behavior and Miscibility in Blends of Poly(vinylidene fluoride) and Poly(1,4-butylene adipate). Macromolecules 1996, 29, 77–83. [Google Scholar] [CrossRef]
- Lauritzen, J.I., Jr.; Hoffman, J.D. Formation of polymer crystals with folded chains from dilute solution. J. Chem. Phys. 1959, 31, 1680–1681. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Medellín-Rodríguez, F.J.; Phillips, P.J.; Lin, J.S. Application of Secondary Nucleation Theory to Semirigid Macromolecules: PEEK, PET, and PEN. Macromolecules 1995, 28, 7744–7755. [Google Scholar] [CrossRef]
- Rahman, M.; Nandi, A.K. On the crystallization mechanism of poly(ethylene terepthalate) in its blends with poly(vinylidene fluoride). Polymer 2002, 43, 6863–6870. [Google Scholar] [CrossRef]
- Lu, X.; Hay, J. Isothermal crystallization kinetics and melting behaviour of poly(ethylene terephthalate). Polymer 2001, 42, 9423–9431. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Karandrea, E.; Giliopoulos, D.J.; Papageorgiou, D.G.; Ladavos, A.; Katerinopoulou, A.; Achilias, D.S.; Triantafyllidis, K.S.; Bikiaris, D.N. Effect of clay structure and type of organomodifier on the thermal properties of poly(ethylene terephthalate) based nanocomposites. Thermochim. Acta 2014, 576, 84–96. [Google Scholar] [CrossRef]
- Thomas, D.G.; Staveley, L.A.K. 889. A study of the supercooling of drops of some molecular liquids. J. Chem. Soc. 1952, 4569–4577. [Google Scholar] [CrossRef]
- Jeziorny, A. Parameters characterizing the kinetics of the non-isothermal crystallization of poly(ethylene terephthalate) determined by d.s.c. Polymer 1978, 19, 1142–1144. [Google Scholar] [CrossRef]
- Di Lorenzo, M.; Silvestre, C. Non-isothermal crystallization of polymers. Prog. Polym. Sci. 1999, 24, 917–950. [Google Scholar] [CrossRef]
- Guigo, N.; van Berkel, J.; de Jong, E.; Sbirrazzuoli, N. Modelling the non-isothermal crystallization of polymers: Application to poly (ethylene 2, 5-furandicarboxylate). Thermochim. Acta 2017, 650, 66–75. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Formela, K.; Zedler, Ł.; Hejna, A.; Tercjak, A. Reactive extrusion of bio-based polymer blends and composites—Current trends and future developments. Express Polym. Lett. 2018, 12, 24–57. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blend | KgII × 10−5 (K2) | σσe × 104 (J2/m4) | σ × 102 (J/m2) | σe × 102 (J/m2) | q (kJ/mol) |
|---|---|---|---|---|---|
| PET | 2.60 | 11.9 | 1.09 | 10.9 | 35.7 |
| PET-PEF 95-05 | 2.58 | 11.8 | 1.09 | 10.8 | 35.4 |
| PET-PEF 90-10 | 2.40 | 11.0 | 1.09 | 10.1 | 33.1 |
| PET-PEF 85-15 | 2.34 | 10.7 | 1.09 | 9.85 | 32.3 |
| PET-PEF 80-20 | 2.25 | 10.3 | 1.09 | 9.47 | 31.0 |
| PET-PEF 70-30 | 2.21 | 10.2 | 1.09 | 9.37 | 30.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papageorgiou, D.G.; Tsetsou, I.; Ioannidis, R.O.; Nikolaidis, G.N.; Exarhopoulos, S.; Kasmi, N.; Bikiaris, D.N.; Achilias, D.S.; Papageorgiou, G.Z. A New Era in Engineering Plastics: Compatibility and Perspectives of Sustainable Alipharomatic Poly(ethylene terephthalate)/Poly(ethylene 2,5-furandicarboxylate) Blends. Polymers 2021, 13, 1070. https://doi.org/10.3390/polym13071070
Papageorgiou DG, Tsetsou I, Ioannidis RO, Nikolaidis GN, Exarhopoulos S, Kasmi N, Bikiaris DN, Achilias DS, Papageorgiou GZ. A New Era in Engineering Plastics: Compatibility and Perspectives of Sustainable Alipharomatic Poly(ethylene terephthalate)/Poly(ethylene 2,5-furandicarboxylate) Blends. Polymers. 2021; 13(7):1070. https://doi.org/10.3390/polym13071070
Chicago/Turabian StylePapageorgiou, Dimitrios G., Irini Tsetsou, Raphael O. Ioannidis, George N. Nikolaidis, Stylianos Exarhopoulos, Nejib Kasmi, Dimitrios N. Bikiaris, Dimitris S. Achilias, and George Z. Papageorgiou. 2021. "A New Era in Engineering Plastics: Compatibility and Perspectives of Sustainable Alipharomatic Poly(ethylene terephthalate)/Poly(ethylene 2,5-furandicarboxylate) Blends" Polymers 13, no. 7: 1070. https://doi.org/10.3390/polym13071070
APA StylePapageorgiou, D. G., Tsetsou, I., Ioannidis, R. O., Nikolaidis, G. N., Exarhopoulos, S., Kasmi, N., Bikiaris, D. N., Achilias, D. S., & Papageorgiou, G. Z. (2021). A New Era in Engineering Plastics: Compatibility and Perspectives of Sustainable Alipharomatic Poly(ethylene terephthalate)/Poly(ethylene 2,5-furandicarboxylate) Blends. Polymers, 13(7), 1070. https://doi.org/10.3390/polym13071070