
Spatially Controlled Highly Branched Vinylsilicones



Abstract
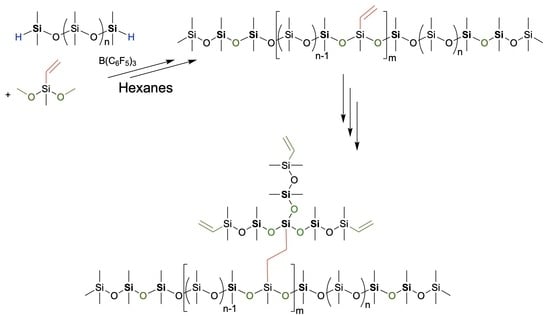
1. Introduction

2. Materials and Methods
2.1. Materials
2.2. Methods
2.3. Synthesis
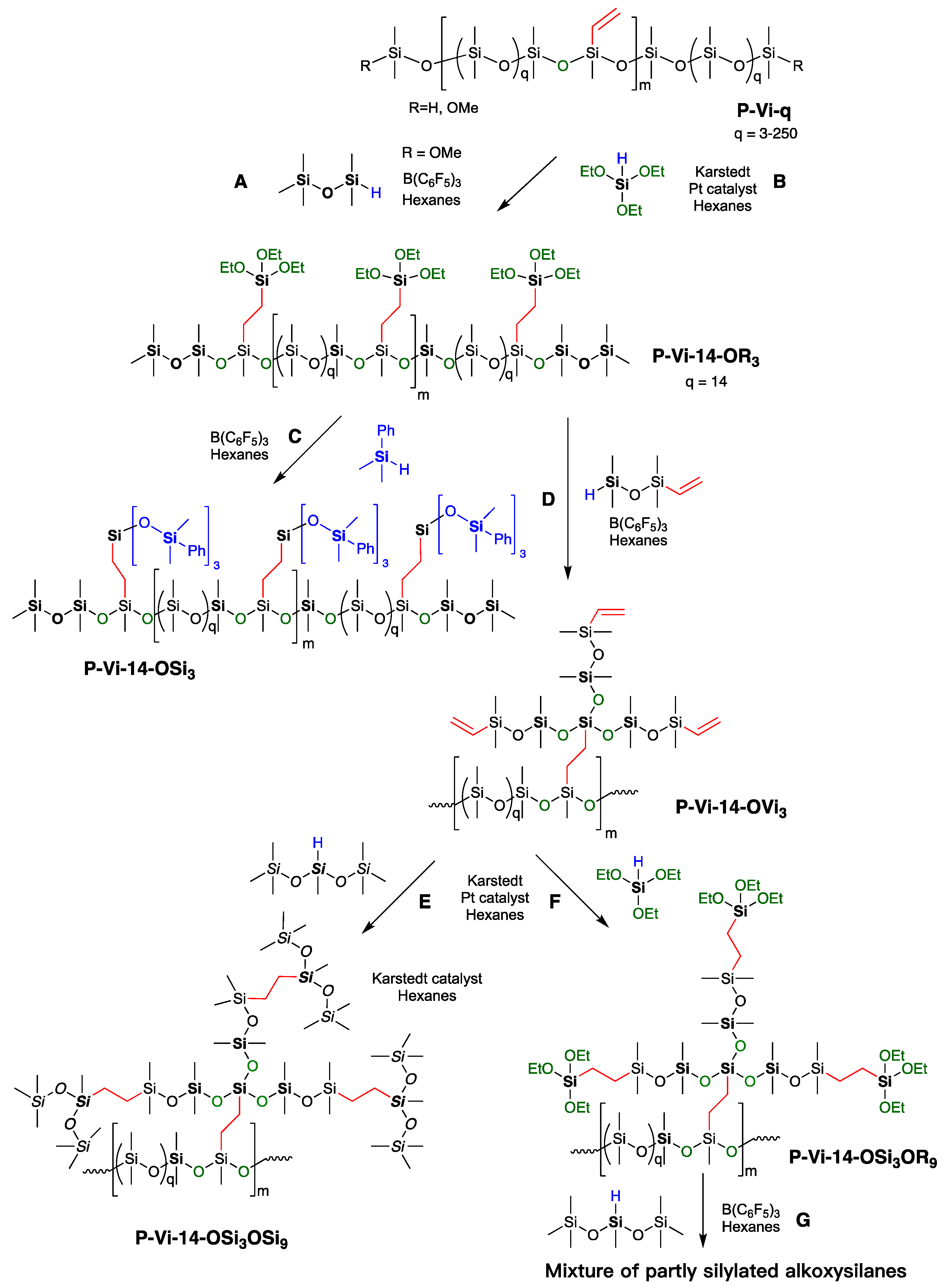
2.3.1. General Procedures for the Preparation of Vinylsilicones with Controlled Spacing, Shown for P-Vi-14

2.3.2. Capping OMe Groups in P-Vi-14
2.3.3. Synthesis of Compound P-Vi-14-OR3

2.3.4. Synthesis of Compound P-Vi-14-OSi3

2.3.5. Synthesis of Compound P-Vi-14-OVi3

2.3.6. Synthesis of Compound P-Vi-14-OSi3OSi9

2.3.7. Attempted Synthesis of Compound P-Vi-14-OVi3OR9, and then Branching

2.3.8. Attempted Branching of Compound P-Vi-14-OVi3OR9
2.3.9. Synthesis of Silicone Elastomers Using Controlled Spacing Hydride-Terminated Vinyl-Containing Linear Silicone
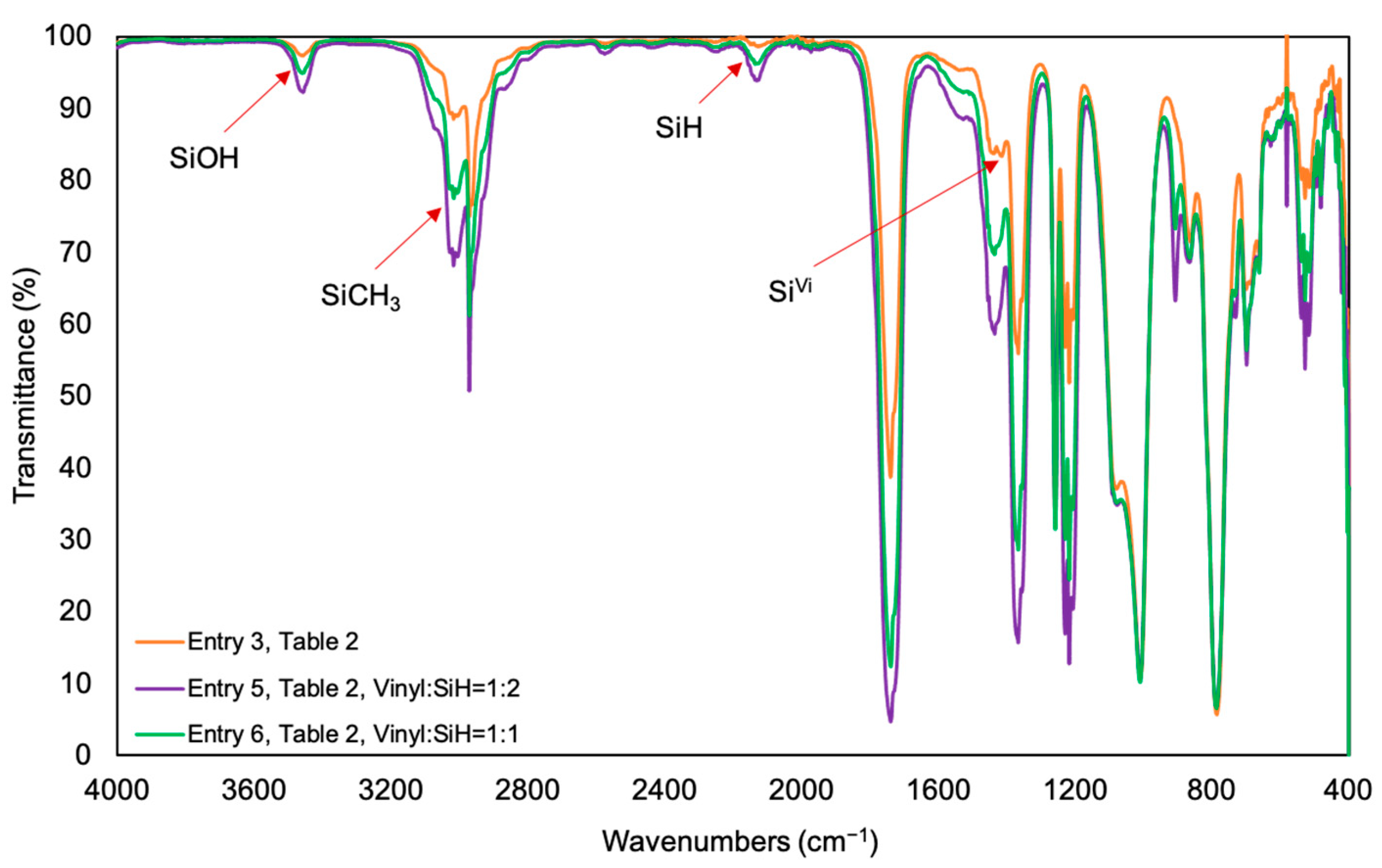
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, D.; Jin, Y.; Zhu, X.; Yan, D. Synthesis and applications of stimuli-responsive hyperbranched polymers. Prog. Polym. Sci. 2017, 64, 114–153. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Monteiro, M.J.; Perrier, S. Dendritic and Hyperbranched Polymers from Macromolecular Units: Elegant Approaches to the Synthesis of Functional Polymers. Macromolecules 2011, 44, 7067–7087. [Google Scholar] [CrossRef]
- Abad, M.; Martínez-Bueno, A.; Mendoza, G.; Arruebo, M.; Oriol, L.; Sebastián, V.; Piñol, M. Supramolecular Functionalizable Linear–Dendritic Block Copolymers for the Preparation of Nanocarriers by Microfluidics. Polymers 2021, 13, 684. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Z.; Zhang, D. Synthesis and application of epoxy-ended hyperbranched polymers. Chem. Eng. J. 2018, 343, 283–302. [Google Scholar] [CrossRef]
- Caminade, A.-M.; Yan, D.; Smith, D.K. Dendrimers and hyperbranched polymers. Chem. Soc. Rev. 2015, 44, 3870–3873. [Google Scholar] [CrossRef] [PubMed]
- Mu, B.; Liu, T.; Tian, W. Long-Chain Hyperbranched Polymers: Synthesis, Properties, and Applications. Macromol. Rapid Commun. 2019, 40, 1800471. [Google Scholar] [CrossRef]
- Yates, C.R.; Hayes, W. Synthesis and applications of hyperbranched polymers. Eur. Polym. J. 2004, 40, 1257–1281. [Google Scholar] [CrossRef]
- Gurunathan, T.; Mohanty, S.; Nayak, S.K. Hyperbranched Polymers for Coating Applications: A Review. Polym. Plast. Technol. Eng. 2016, 55, 92–117. [Google Scholar] [CrossRef]
- Emrick, T.; Chang, H.-T.; Fréchet, J.M.J.; Woods, J.; Baccei, L. Hyperbranched aromatic epoxies in the design of adhesive materials. Polym. Bull. 2000, 45, 1–7. [Google Scholar] [CrossRef]
- Stumpe, K.; Eichhorn, K.-J.; Voit, B. Characterisation of Thin Composite Films from Hyperbranched Polyphenylene and Thermolabile Hyperbranched Polycarbonate. Macromol. Chem. Phys. 2008, 209, 1787–1796. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, W.; Liu, J.; Zhu, X.; Yan, D. Self-Assembly of Hyperbranched Polymers and Its Biomedical Applications. Adv. Mater. 2010, 22, 4567–4590. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Liu, J.; Hao, S.; Song, H.; Zhang, J. 3D Printable, Highly Stretchable, Superior Stable Ionogels Based on Poly(ionic liquid) with Hyperbranched Polymers as Macro-cross-linkers for High-Performance Strain Sensors. ACS Appl. Mater. Interfaces 2021, 13, 5614–5624. [Google Scholar] [CrossRef] [PubMed]
- Voit, B.I.; Lederer, A. Hyperbranched and Highly Branched Polymer Architectures—Synthetic Strategies and Major Characterization Aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar] [CrossRef] [PubMed]
- Kamino, B.A.; Bender, T.P. The use of siloxanes, silsesquioxanes, and silicones in organic semiconducting materials. Chem. Soc. Rev. 2013, 42, 5119–5130. [Google Scholar] [CrossRef]
- Zhang, H.; Cloud, A. The Permeability Characteristics of Silicone Rubber. In Proceedings of the SAMPE—Society for the Advancement of Material and Process Engineering, Fall Technical Conference, Dallas, TX, USA, 6–9 November 2006. [Google Scholar]
- Owen, M.J. Siloxane Surface Activity. In Silicon-Based Polymer Science: A Comprehensive Resource; Zeigler, J.M., Fearon, F.W.G., Eds.; American Chemical Society: Washington, DC, USA, 1990; pp. 705–739. [Google Scholar]
- Kazakova, V.V.; Rebrov, E.A.; Myakushev, V.B.; Strelkova, T.V.; Ozerin, A.N.; Ozerina, L.A.; Chenskaya, T.B.; Sheiko, S.S.; Sharipov, E.Y.; Muzafarov, A.M. From a Hyperbranched Polyethoxysiloxane Toward Molecular Forms of Silica: A Polymer-Based Approach to the Monitoring of Silica Properties. In Silicones and Silicone-Modified Materials; American Chemical Society: Washington, DC, USA, 2000; Volume 729, pp. 503–515. [Google Scholar]
- Jaumann, M.; Rebrov, E.A.; Kazakova, V.V.; Muzafarov, A.M.; Goedel, W.A.; Möller, M. Hyperbranched Polyalkoxysiloxanes via AB3-Type Monomers. Macromol. Chem. Phys. 2003, 204, 1014–1026. [Google Scholar] [CrossRef]
- Paulasaari, J.K.; Weber, W.P. Synthesis of Hyperbranched Polysiloxanes by Base-Catalyzed Proton-Transfer Polymerization. Comparison of Hyperbranched Polymer Microstructure and Properties to Those of Linear Analogues Prepared by Cationic or Anionic Ring-Opening Polymerization. Macromolecules 2000, 33, 2005–2010. [Google Scholar] [CrossRef]
- Paulasaari, J.K.; Weber, W.P. Base catalyzed proton transfer polymerization of 1-hydroxypentamethylcyclotrisiloxane. Comparison of hyperbranched polymer microstructure and properties to those of highly regular linear analogs. Macromol. Chem. Phys. 2000, 201, 1585–1592. [Google Scholar] [CrossRef]
- Chojnowski, J.; Cypryk, M.; Fortuniak, W.; Ścibiorek, M.; Rózga-Wijas, K. Synthesis of Branched Polysiloxanes with Controlled Branching and Functionalization by Anionic Ring-Opening Polymerization. Macromolecules 2003, 36, 3890–3897. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis using SiH Chemistry: The Piers–Rubinsztajn Reaction. Chem. Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Grande, J.B.; Urlich, T.; Dickie, T.; Brook, M.A. Silicone dendrons and dendrimers from orthogonal SiH coupling reactions. Polym. Chem. 2014, 5, 6728–6739. [Google Scholar] [CrossRef]
- Zheng, S.; Liang, S.; Chen, Y.; Brook, M.A. Hyperbranched Silicone MDTQ Tack Promoters. Molecules 2019, 24, 4133. [Google Scholar] [CrossRef]
- Morgan, J.; Chen, T.; Hayes, R.; Dickie, T.; Urlich, T.; Brook, M.A. Facile synthesis of dendron-branched silicone polymers. Polym. Chem. 2017, 8, 2743–2746. [Google Scholar] [CrossRef]
- Madsen, F.B.; Javakhishvili, I.; Jensen, R.E.; Daugaard, A.E.; Hvilsted, S.; Skov, A.L. Synthesis of telechelic vinyl/allyl functional siloxane copolymers with structural control. Polym. Chem. 2014, 5, 7054–7061. [Google Scholar] [CrossRef]
- van Genabeek, B.; Lamers, B.A.G.; Hawker, C.J.; Meijer, E.W.; Gutekunst, W.R.; Schmidt, B.V.K.J. Properties and applications of precision oligomer materials; where organic and polymer chemistry join forces. J. Polym. Sci. 2021, 59, 373–403. [Google Scholar] [CrossRef]
- Chojnowski, J.; Fortuniak, W.; Kurjata, J.; Rubinsztajn, S.; Cella, J.A. Oligomerization of Hydrosiloxanes in the Presence of Tris(pentafluorophenyl)borane. Macromolecules 2006, 39, 3802–3807. [Google Scholar] [CrossRef]
- Liao, M.; Schneider, A.F.; Laengert, S.E.; Gale, C.B.; Chen, Y.; Brook, M.A. Living synthesis of silicone polymers controlled by humidity. Eur. Polym. J. 2018, 107, 287–293. [Google Scholar] [CrossRef]
- Fawcett, A.S.; Grande, J.B.; Brook, M.A. Rapid, metal-free room temperature vulcanization produces silicone elastomers. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 644–652. [Google Scholar] [CrossRef]
- Marciniec, B.; Gulinski, J.; Urbaniak, W.; Kornetka, Z.W. Comprehensive Handbook on Hydrosilylation Chemistry; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Wang, D.; Klein, J.; Mejía, E. Catalytic Systems for the Cross-Linking of Organosilicon Polymers. Chem. Asian J. 2017, 12, 1180–1197. [Google Scholar] [CrossRef] [PubMed]
- Mrozek, R.A.; Cole, P.J.; Otim, K.J.; Shull, K.R.; Lenhart, J.L. Influence of solvent size on the mechanical properties and rheology of polydimethylsiloxane-based polymeric gels. Polymer 2011, 52, 3422–3430. [Google Scholar] [CrossRef]
- Stricher, A.M.; Rinaldi, R.G.; Barrès, C.; Ganachaud, F.; Chazeau, L. How I Met Your Elastomers: From Network Topology to Mechanical Behaviours of Conventional Silicone Materials. RSC Adv. 2015, 5, 53713–53725. [Google Scholar] [CrossRef]
- Laengert, S.E.; Schneider, A.F.; Lovinger, E.; Chen, Y.; Brook, M.A. Sequential Functionalization of a Natural Crosslinker Leads to Designer Silicone Networks. Chem. Asian J. 2017, 12, 1208–1212. [Google Scholar] [CrossRef]
- Rogers, M.G. The structure of epoxy resins using NMR and GPC techniques. J. Appl. Polym. Sci. 1972, 16, 1953–1958. [Google Scholar] [CrossRef]
- Liao, M.; Chen, Y.; Brook, M.A. When Attempting Chain Extension, Even Without Solvent, It Is Not Possible to Avoid Chojnowski Metathesis Giving D3. Molecules 2021, 26, 231. [Google Scholar] [CrossRef]
- Piers, W.E. The chemistry of perfluoroaryl boranes. Adv. Organomet. Chem. 2005, 52, 1–77. [Google Scholar] [CrossRef]
- Chojnowski, J.; Rubinsztajn, S.; Cella, J.A.; Fortuniak, W.; Cypryk, M.; Kurjata, J.; Kazmierski, K. Mechanism of the B(C6F5)3-catalyzed reaction of silyl hydrides with alkoxysilanes. Kinetic and spectroscopic studies. Organometallics 2005, 24, 6077–6084. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Cella, J.A. A new polycondensation process for the preparation of polysiloxane copolymers. Macromolecules 2005, 38, 1061–1063. [Google Scholar] [CrossRef]
- Chojnowski, J.; Kurjata, J.; Fortuniak, W.; Rubinsztajn, S.; Trzebicka, B. Hydride Transfer Ring-Opening Polymerization of a Cyclic Oligomethylhydrosiloxane. Route to a Polymer of Closed Multicyclic Structure. Macromolecules 2012, 45, 2654–2661. [Google Scholar] [CrossRef]
- van Dongen, M.A.; Desai, A.; Orr, B.G.; Baker, J.R.; Banaszak Holl, M.M. Quantitative analysis of generation and branch defects in G5 poly(amidoamine) dendrimer. Polymer 2013, 54, 4126–4133. [Google Scholar] [CrossRef]
- Charlesby, A. Viscosity measurements in branched silicones. J. Polym. Sci. 1955, 17, 379–390. [Google Scholar] [CrossRef]






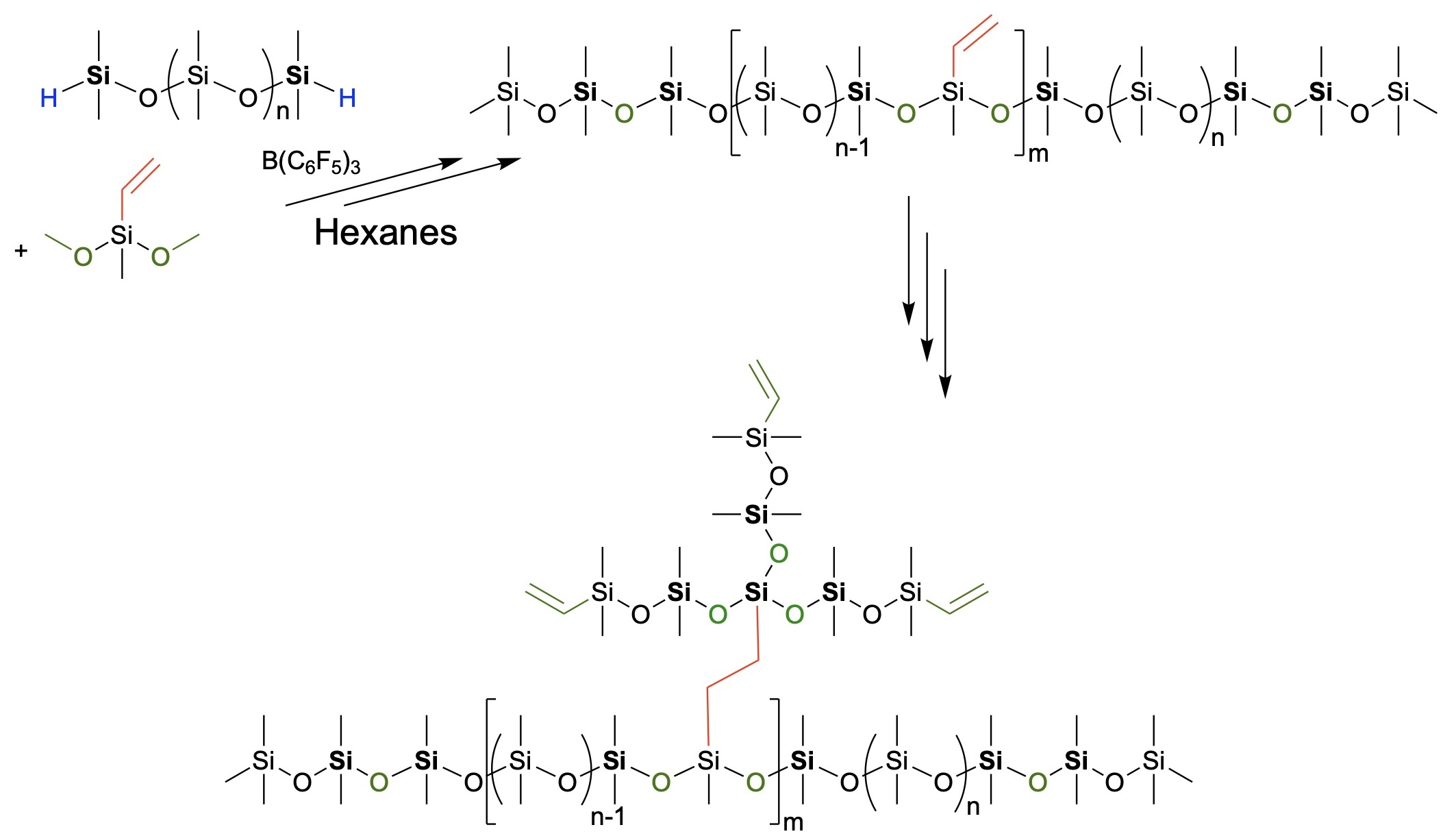{kind=link}
{kind=link}
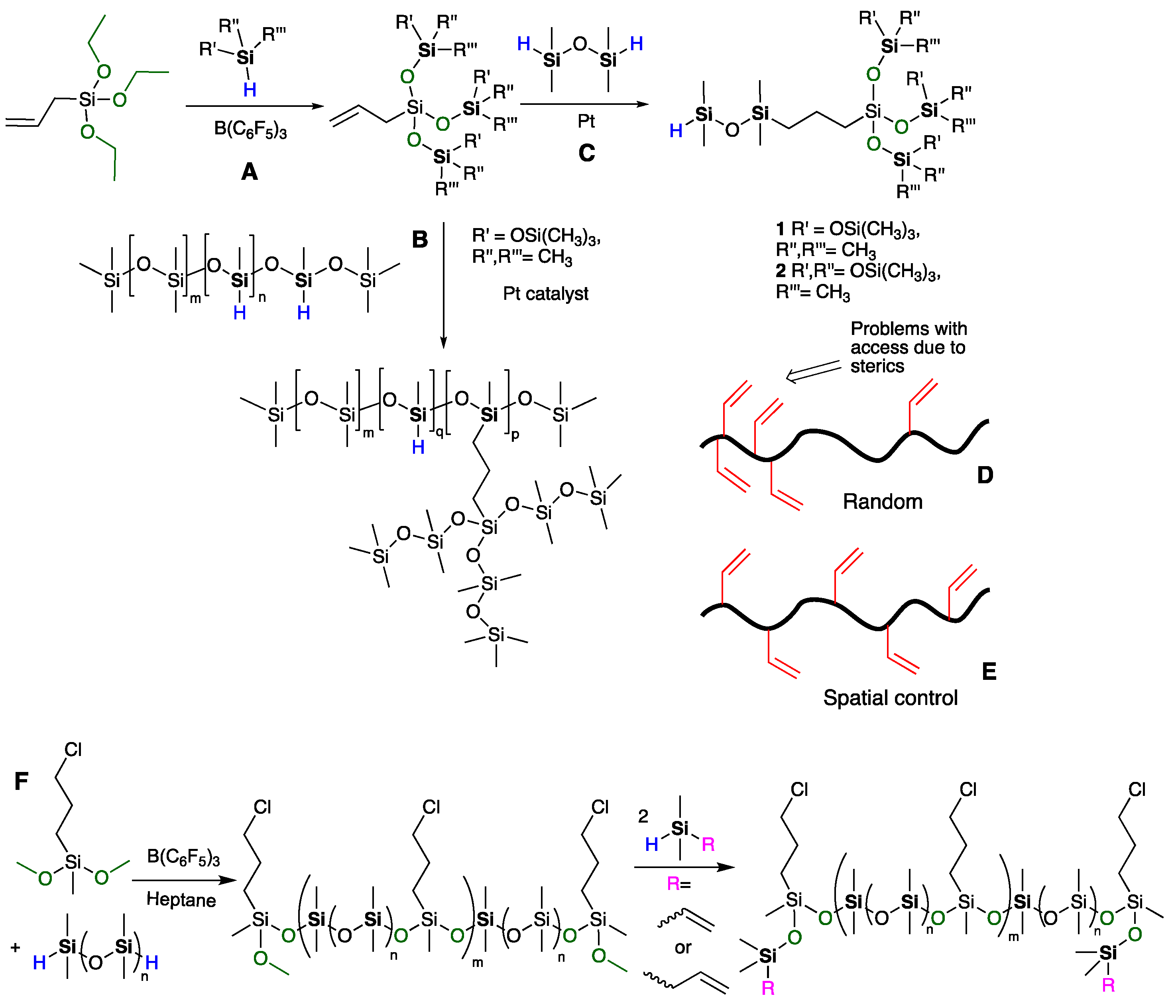{kind=link}
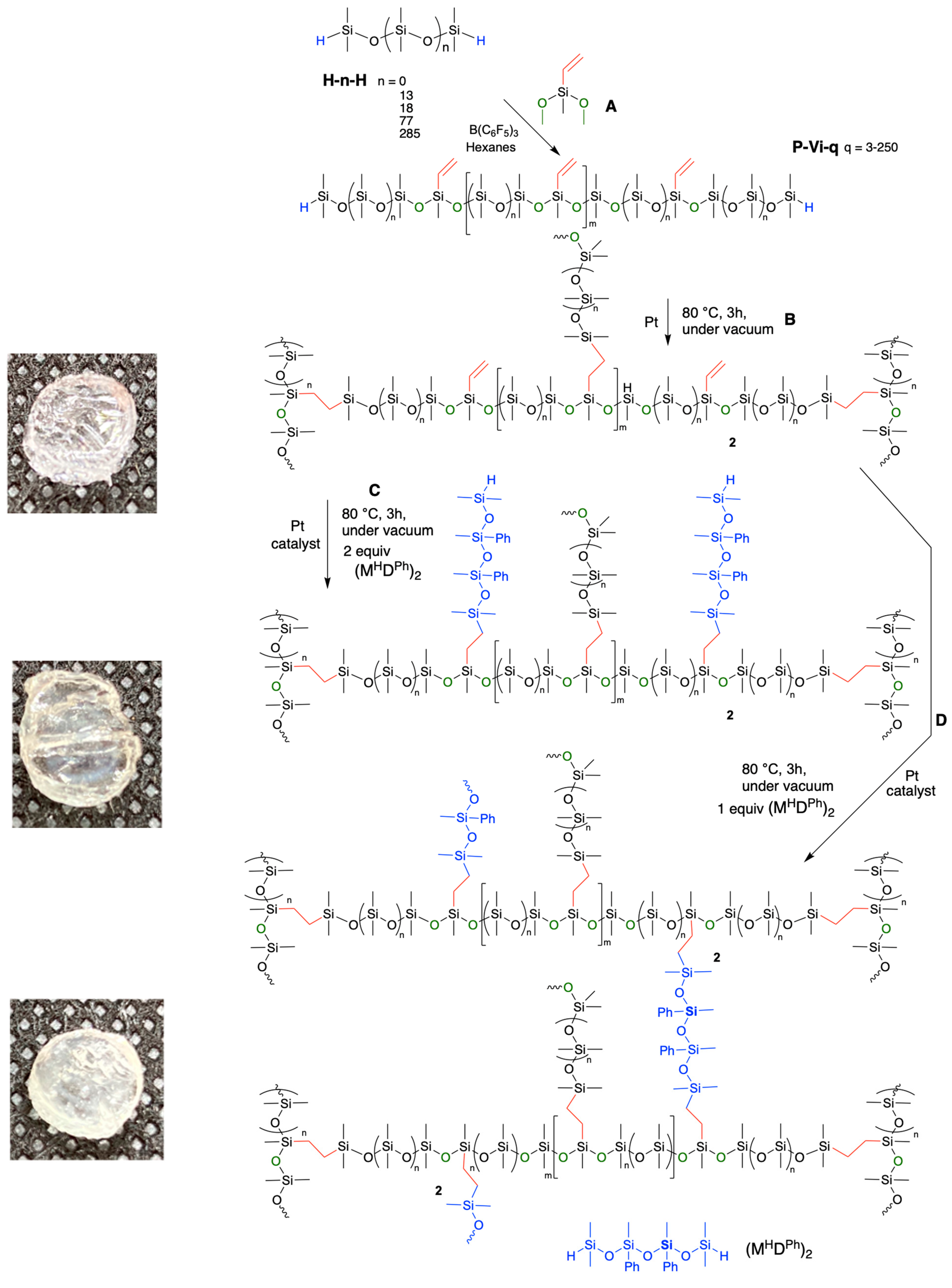{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | H-Dn-H a | Mn (g mol−1) b | [SiH]/[SiOMe] | Mn (g mol−1) c | ĐM | DP d | Product Name P-Vi-q | Vinyl Conc. (%) | Yield (%) | Terminal Groups |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H-0-H | 134 | 1 | N/A e | N/A e | - | P-Vi-3 | 31.5 | 66 | OMe |
| 2 | H-13-H | 1100 | 1 | 85,800 | 1.65 | 78 | P-Vi-16 | 6.1 | 86 | OMe |
| 3 | H-18-H | 1500 | 1 | 102,400 | 1.11 | 68 | P-Vi-26 | 3.9 | 75 | OMe |
| 4 | H-77-H | 5800 | 1 | 87,000 | 1.20 | 15 | P-Vi-200 | 0.5 | 84 | OMe |
| 5 | H-0-H | 134 | 1.2 | 25,200 | 1.68 | 188 | P-Vi-4 | 28.8 | 54 | SiH |
| 6 | H-13-H | 1100 | 1.2 | 26,900 | 1.68 | 24 | P-Vi-29 | 3.4 | 86 | SiH |
| 7 | H-18-H | 1500 | 1.2 | 65,700 | 1.39 | 44 | P-Vi-28 | 3.6 | 84 | SiH |
| 8 | H-77-H | 5800 | 1.2 | 13,300 | 2.09 | 2 | P-Vi-111 | 0.9 | 89 | SiH |
| 9 | H-0-H | 134 | 0.83 | N/A e | N/A e | - | P-Vi-3 | 35.4 | 63 | OMe |
| 10 | H-13-H | 1100 | 0.83 | 37,400 | 1.85 | 34 | P-Vi-14 | 6.9 | 84 | OMe |
| 11 | H-18-H | 1500 | 0.83 | 23,600 | 2.15 | 16 | P-Vi-17 | 5.7 | 99 | OMe |
| 12 | H-77-H | 5800 | 0.83 | 23,400 | 1.18 | 41 | P-Vi-83 | 1.2 | 99 | OMe |
| 13 | H-285-H | 21,200 | 0.83 | 143,200 | 1.02 | 7 | P-Vi-250 | 0.4 | 99 | OMe |
| Self-Crosslinked Starting Polymer a | Elastomers SVinyl Conc. (%) a | MW (g mol−1) a | Product Shore OO | |
|---|---|---|---|---|
| 1 | P-Vi-4 | 28.8 | 25,200 | 46 ± 1 |
| 2 | P-Vi-29 | 3.4 | 26,900 | 73 ± 2 |
| 3 | P-Vi-28 | 3.6 | 65,700 | 64 ± 2 |
| 4 | P-Vi-111 | 0.9 | 13,300 | 52 ± 2 |
| Grafted copolymer networks Starting elastomers | Equiv a MH versus SiVi | |||
| # | ||||
| 5 | entry 3 | 2 | 78 ± 2 b | |
| 6 | entry 3 | 1 | 82 ± 1 b | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, M.; Chen, Y.; Brook, M.A. Spatially Controlled Highly Branched Vinylsilicones. Polymers 2021, 13, 859. https://doi.org/10.3390/polym13060859
Liao M, Chen Y, Brook MA. Spatially Controlled Highly Branched Vinylsilicones. Polymers. 2021; 13(6):859. https://doi.org/10.3390/polym13060859
Chicago/Turabian StyleLiao, Mengchen, Yang Chen, and Michael A. Brook. 2021. "Spatially Controlled Highly Branched Vinylsilicones" Polymers 13, no. 6: 859. https://doi.org/10.3390/polym13060859
APA StyleLiao, M., Chen, Y., & Brook, M. A. (2021). Spatially Controlled Highly Branched Vinylsilicones. Polymers, 13(6), 859. https://doi.org/10.3390/polym13060859