
Influence of the Prepolymer Type and Synthesis Parameters on Self-Healing Anticorrosion Properties of Composite Coatings Containing Isophorone Diisocyanate-Loaded Polyurethane Microcapsules


 , , ,
, , ,  and
and 
Abstract
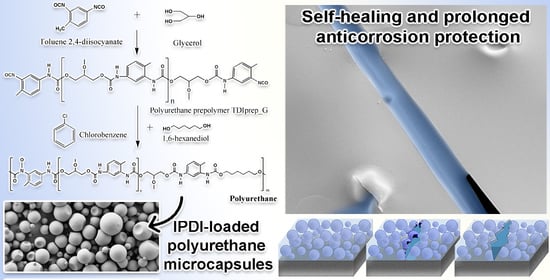
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
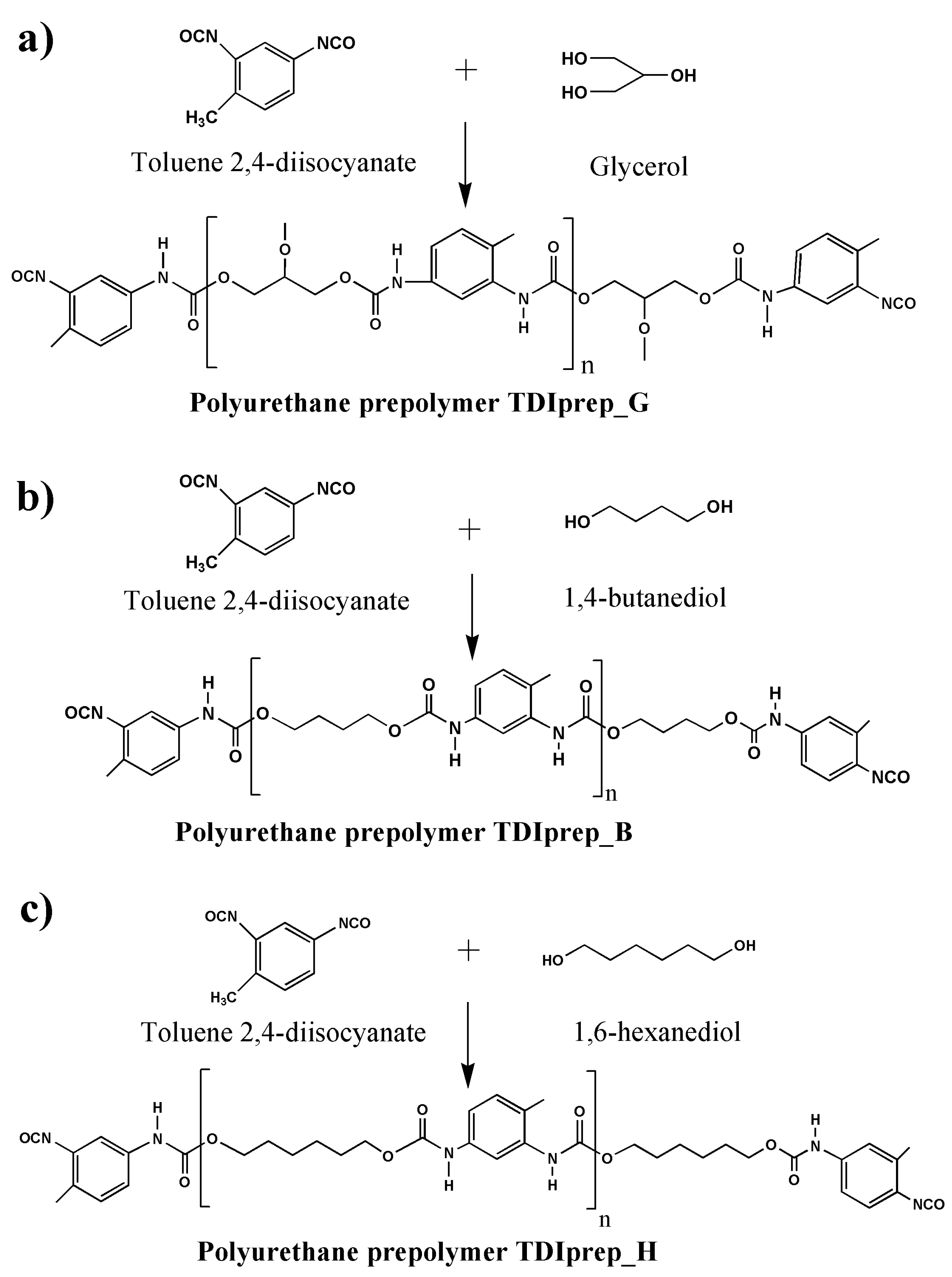
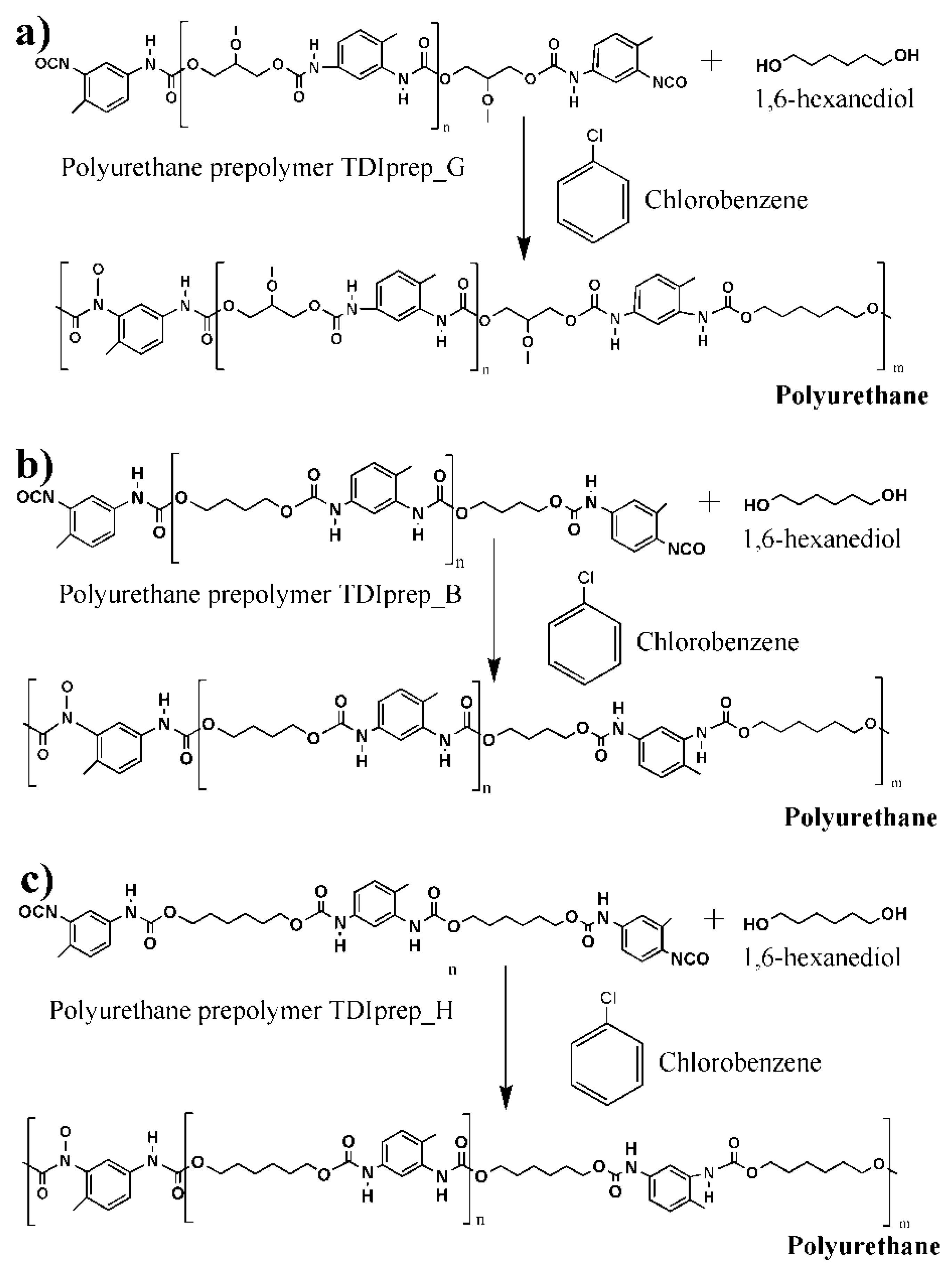
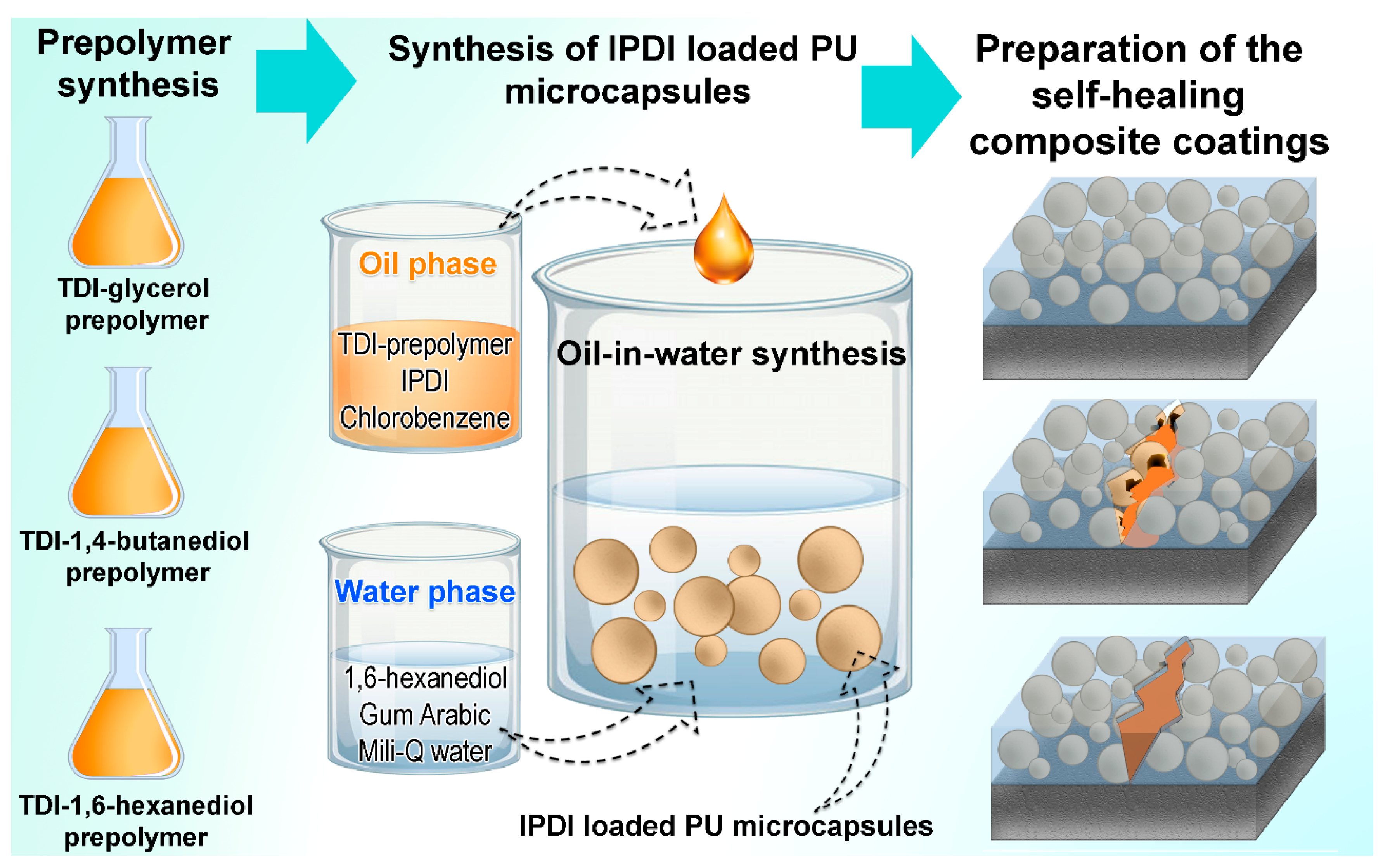
2.2.1. Synthesis of TDI-Prepolymers
2.2.2. Synthesis of IPDI-Loaded Polyurethane Microcapsules
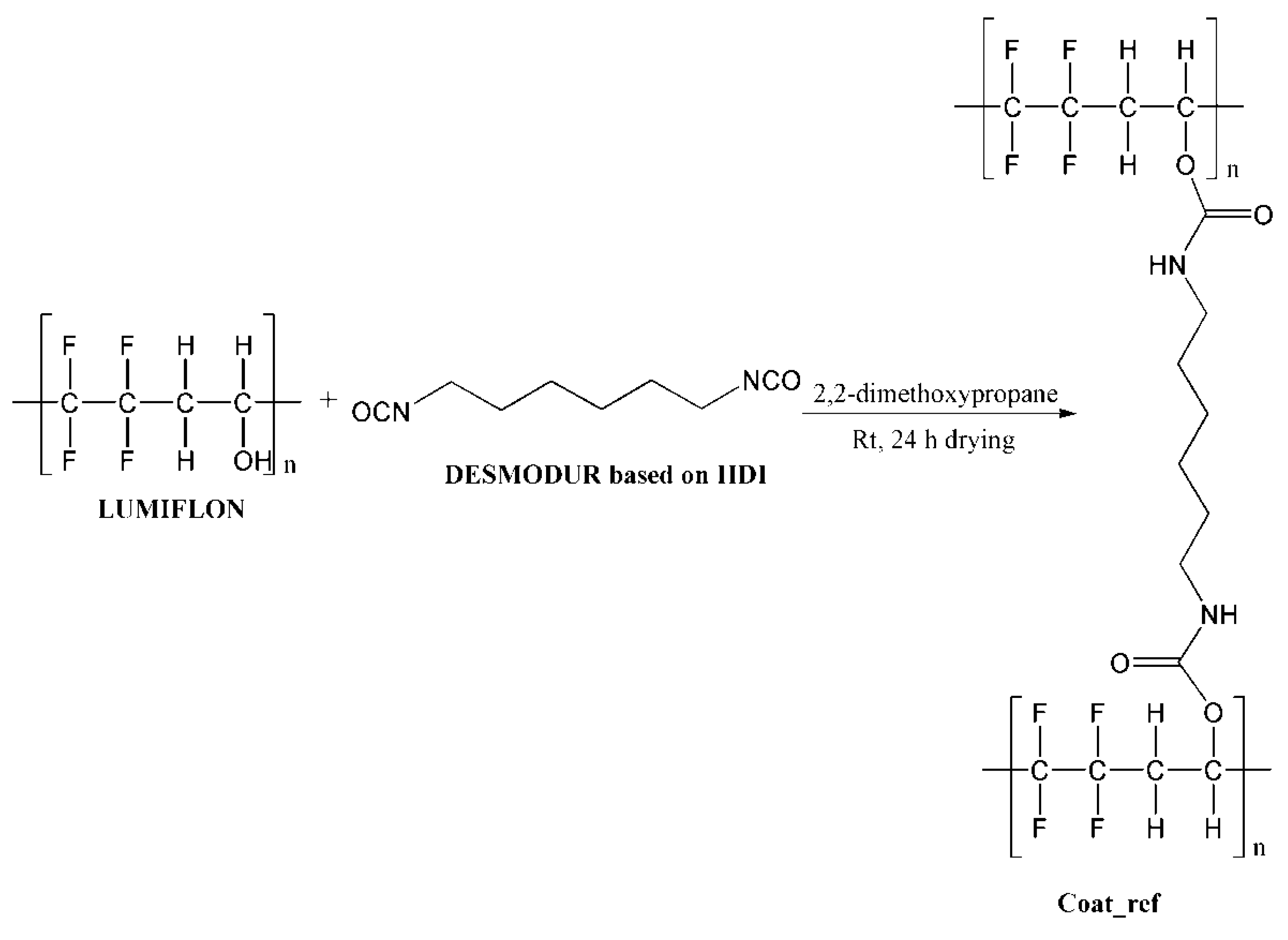
2.2.3. Preparation of the Composite Coatings
2.3. Analysis and Measurements
2.3.1. Chemical Properties of the Synthesized Materials
2.3.2. Thermogravimetric Properties of the Materials
2.3.3. Morphological Properties of the Materials
2.3.4. Determination of Self-Healing and Anticorrosion Properties of the Coatings
3. Results and Discussion
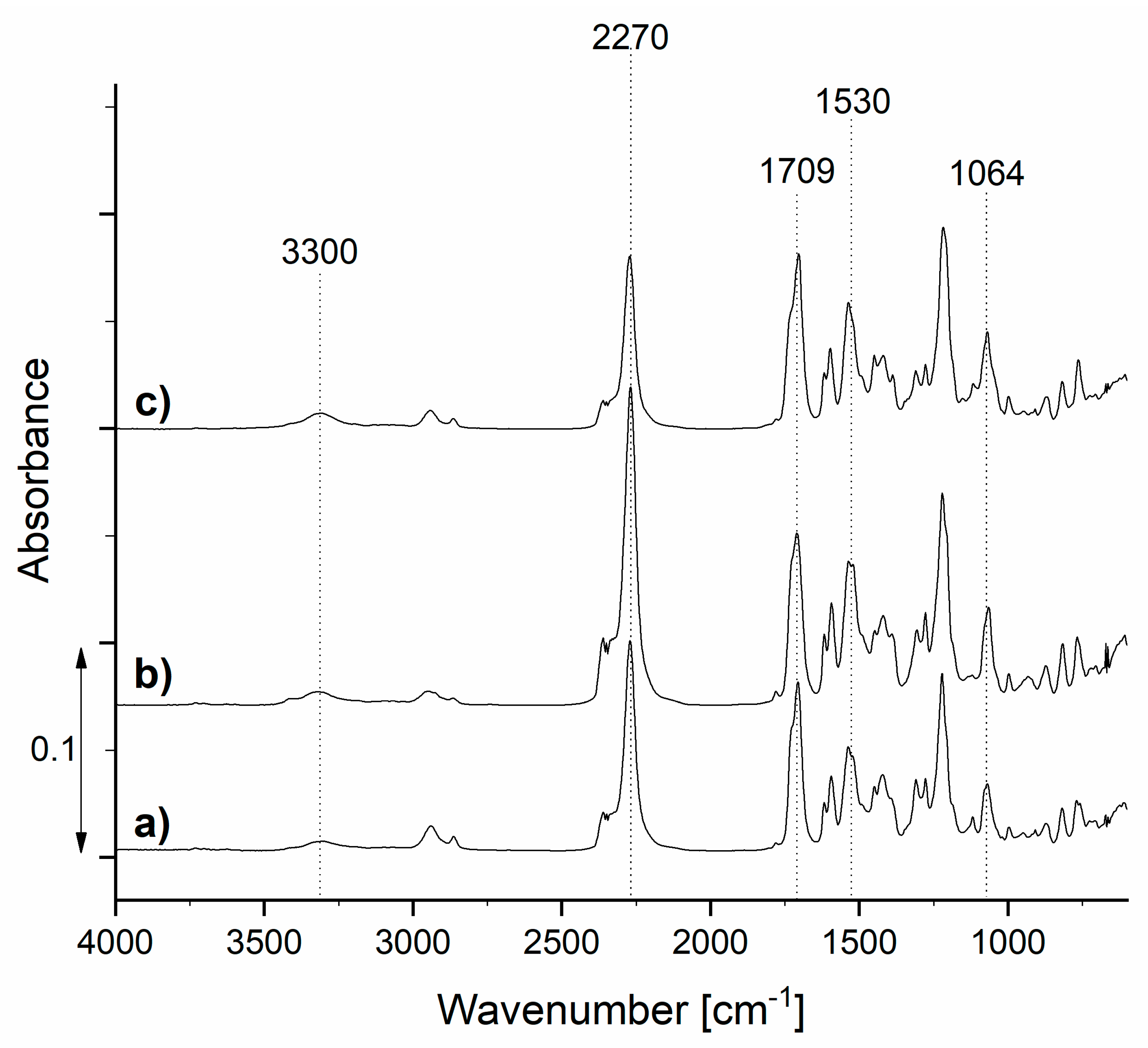
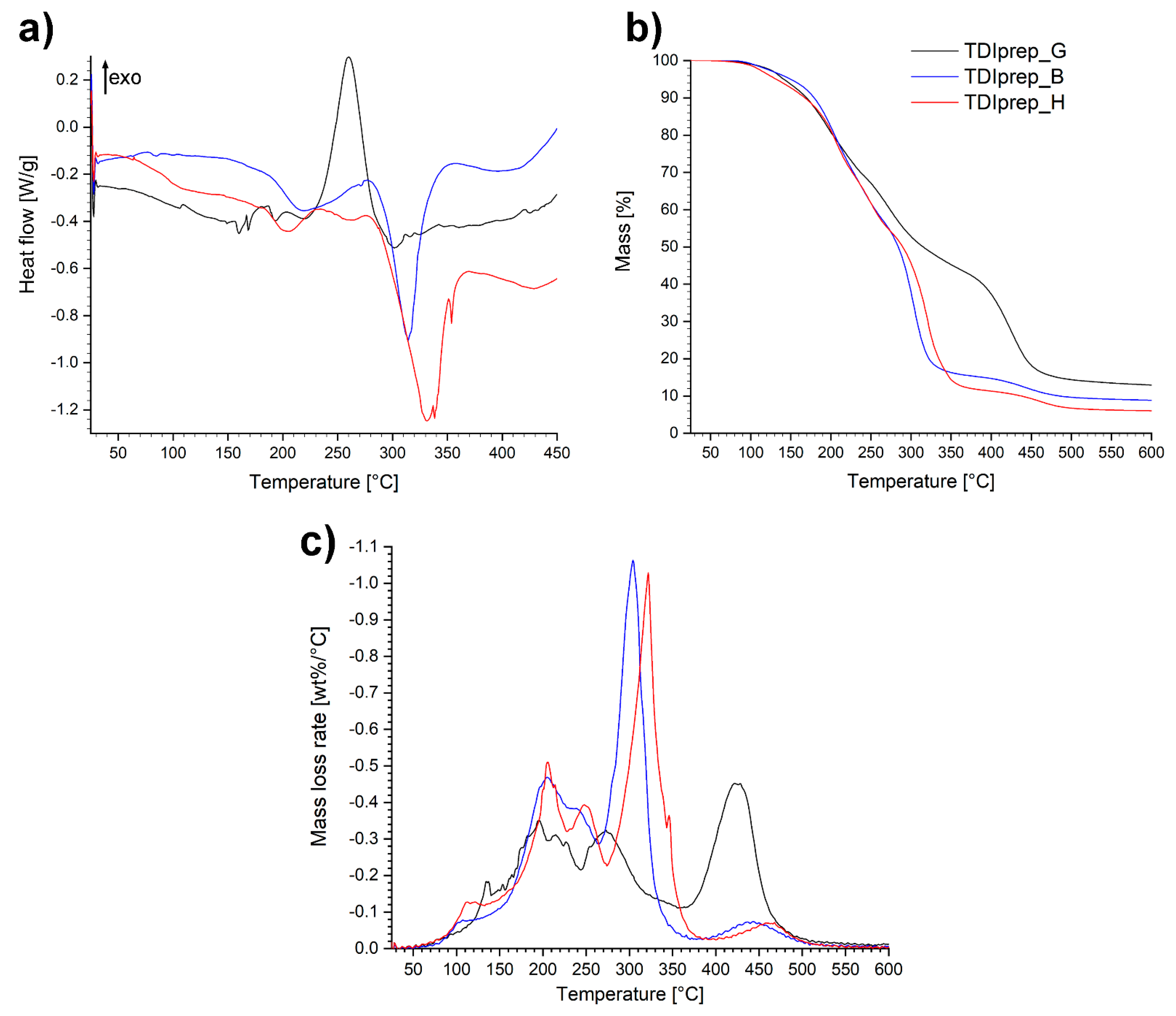
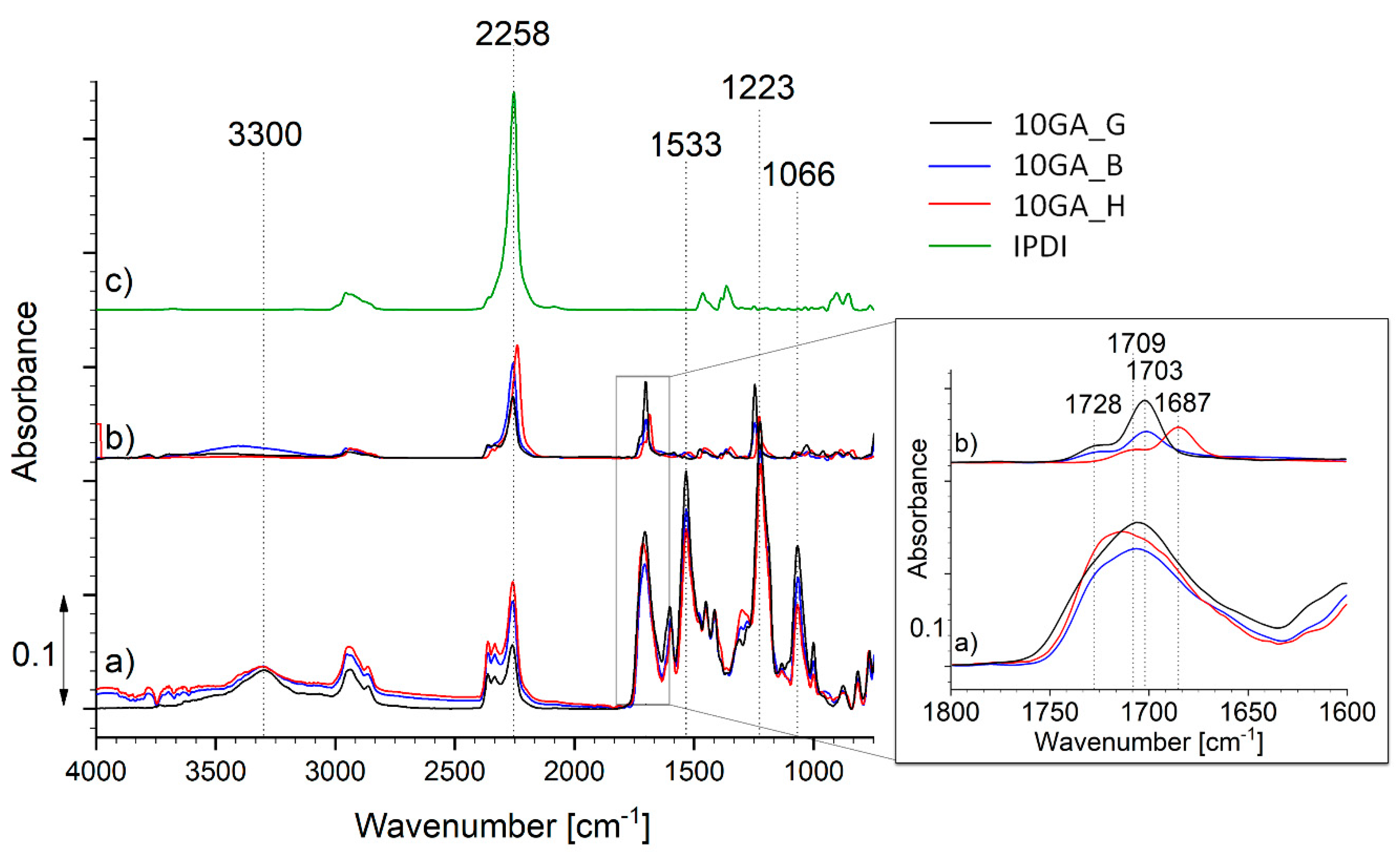
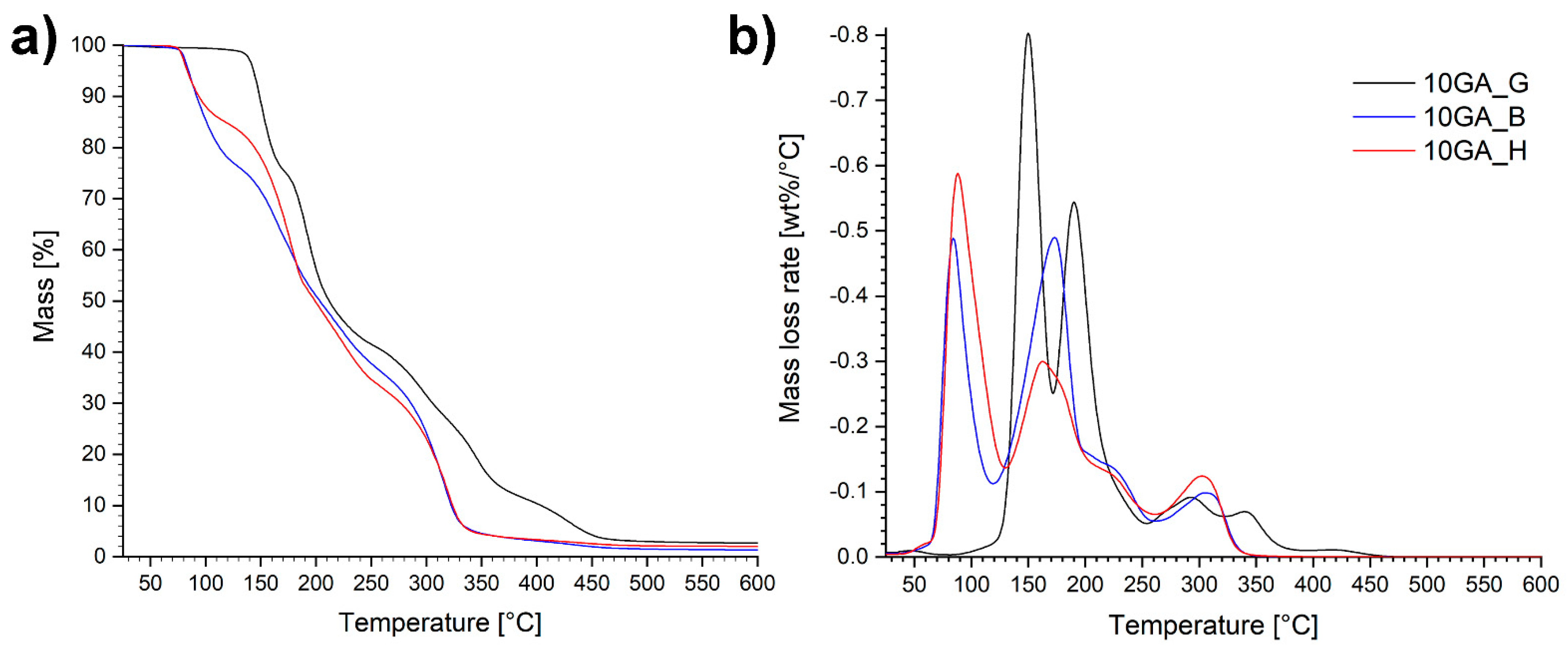
3.1. Characterization of Prepolymers
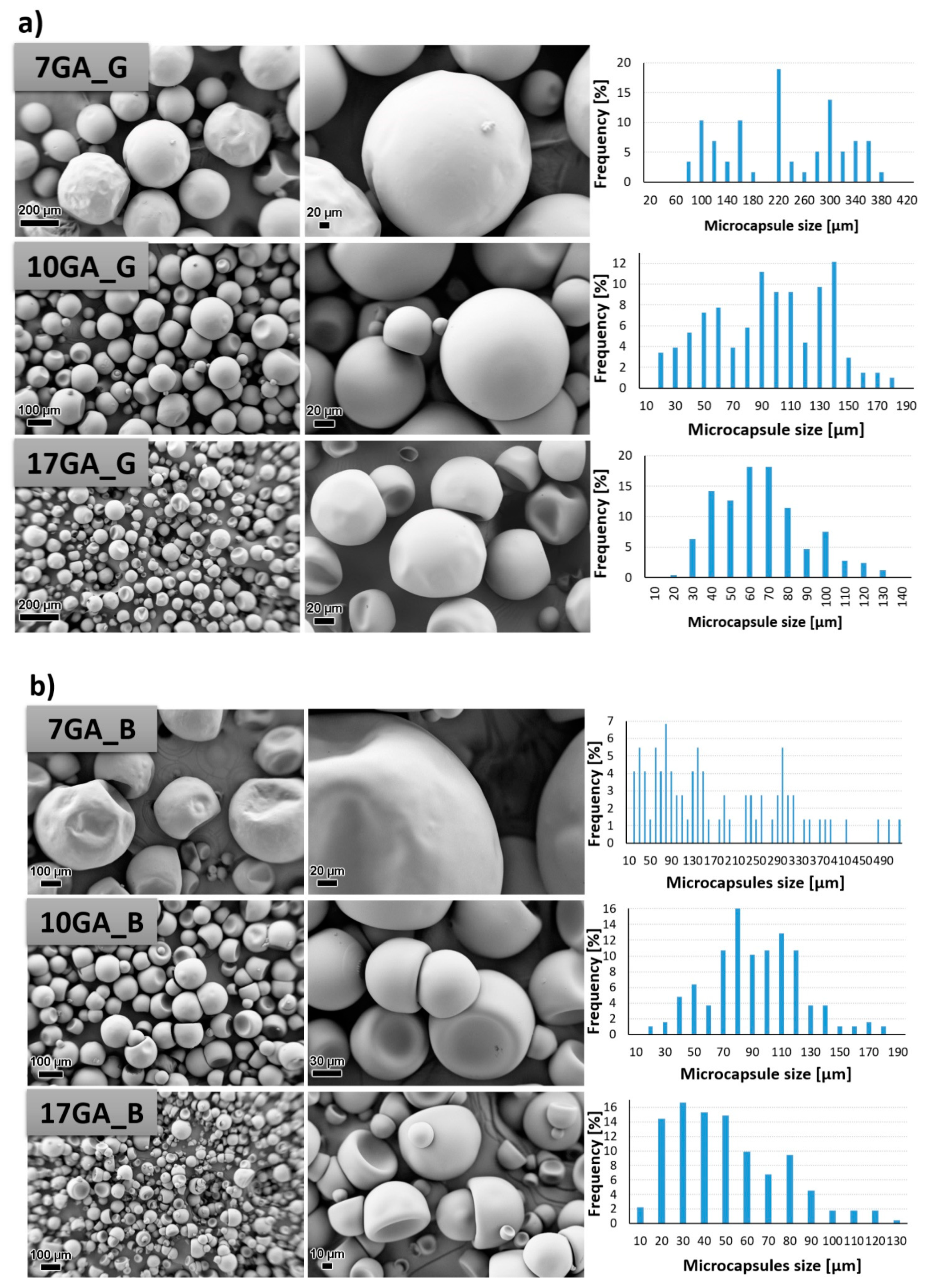
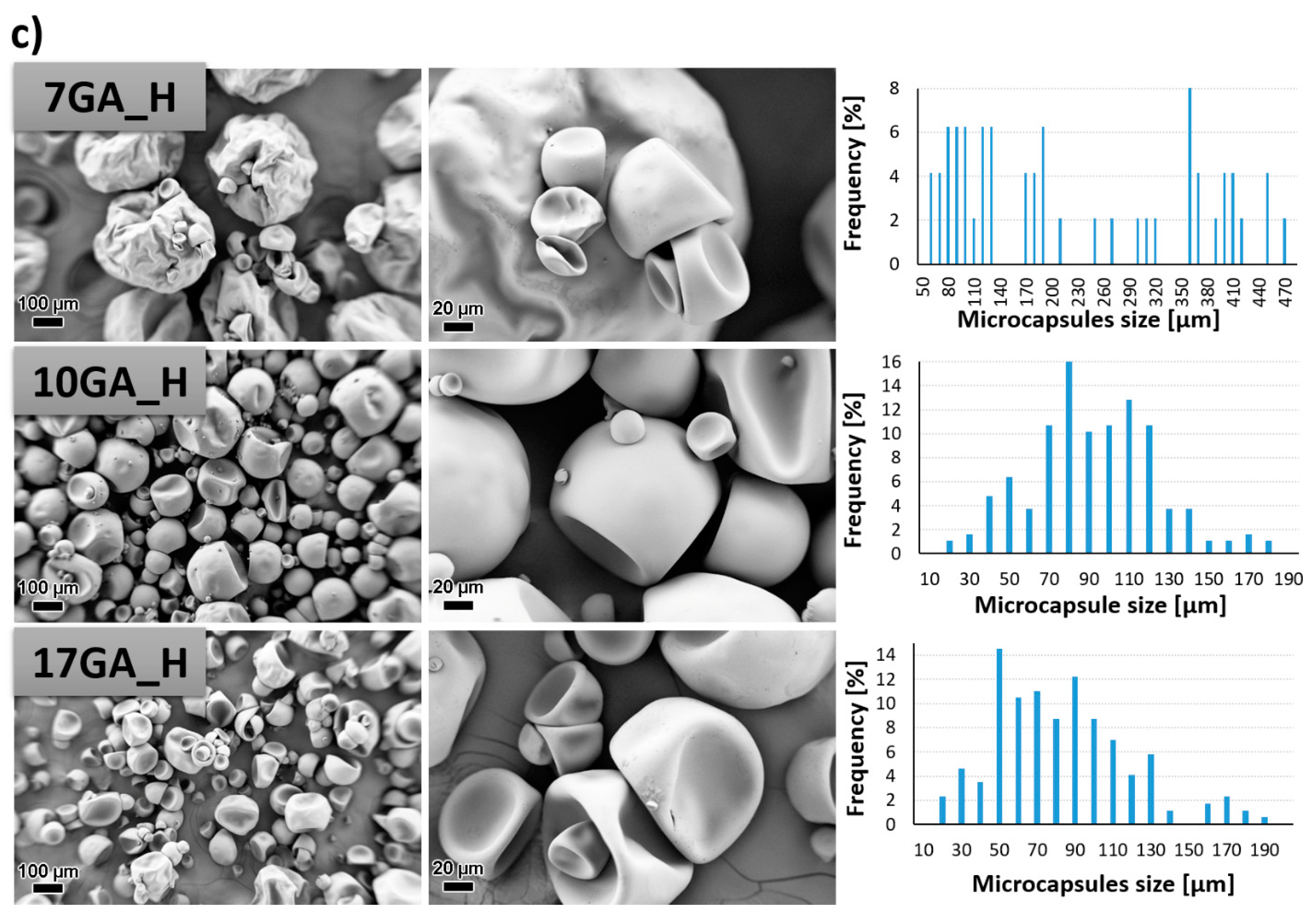
3.2. Characterization of the Microcapsules
3.3. Self-Healing and Anticorrosion Evaluation of the Coatings
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hays, G.F. Now Is the Time. Available online: http://corrosion.org/Corrosion+Resources/Publications/_/nowisthetime.pdf (accessed on 12 February 2021).
- An, S.; Wook Lee, M.; Yarin, A.L.; Yoon, S.S. A review on corrosion-protective extrinsic self-healing: Comparison of microcapsule-based systems and those based on core-shell vascular networks. Chem. Eng. J. 2018, 344, 206–220. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Yu, H.C.; Shen, M.C.; Chern, Y.T.; Li, C.C. Synthesis and application of self-healing microcapsules containing curable glue. Mater. Chem. Phys. 2020, 240, 122161. [Google Scholar] [CrossRef]
- Bekas, D.G.; Tsirka, K.; Baltzis, D.; Paipetis, A.S. Self-healing materials: A review of advances in materials, evaluation, characterization and monitoring techniques. Compos. B Eng. 2016, 87, 92–119. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Wang, J.; Wang, Z.; Wang, S. Preparation of epoxy microcapsule based self-healing coatings and their behavior. Surf. Coat. Technol. 2012, 206, 4976–4980. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, H.; Yang, J. Synthesis of organic silane microcapsules for self-healing corrosion resistant polymer coatings. Corros. Sci. 2012, 65, 561–566. [Google Scholar] [CrossRef]
- Zhang, F.; Ju, P.; Pan, M.; Zhang, D.; Huang, Y.; Li, G.; Li, X. Self-healing mechanisms in smart protective coatings: A review. Corros. Sci. 2018, 144, 74–88. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Li, X.; Yang, Y.; An, E. Self-healing properties of protective coatings containing isophorone diisocyanate microcapsules on carbon steel surfaces. Corros. Sci. 2014, 80, 528–535. [Google Scholar] [CrossRef]
- Lapprand, A.; Boisson, F.; Delolme, F.; Méchin, F.; Pascault, J.-P. Reactivity of isocyanates with urethanes: Conditions for allophanate formation. Polym. Degrad. Stab. 2005, 90, 363–373. [Google Scholar] [CrossRef]
- Attaei, M.M.; Calado, L.; Taryba, M.G.; Morozov, Y.; Shakoor, R.A.; Kahraman, R.; Marques, A.C.; Montemor, M.F. Autonomous self-healing in epoxy coatings provided by high efficiency isophorone diisocyanate (IPDI) microcapsules for protection of carbon steel. Prog. Org. Coat. 2020, 139, 105445. [Google Scholar] [CrossRef]
- Hia, I.L.; Pasbakhsh, P.; Chan, E.S.; Chai, S.P. Electrosprayed multi-core alginate microcapsules as novel self-healing containers. Sci. Rep. 2016, 6, 34674. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-F.; Wang, L.X.; Ren, L.; Huang, Z.; Meng, X.W. Preparation and characterization of polyurethane microcapsules containing n-octadecane with styrene-maleic anhydride as a surfactant by interfacial polycondensation. J. Appl. Polym. Sci. 2006, 102, 34674. [Google Scholar] [CrossRef]
- Yang, H.; Mo, Q.; Li, W.; Gu, F. Preparation and properties of self-healing and self-lubricating epoxy coatings with polyurethane microcapsules containing bifunctional linseed oil. Polymers 2019, 11, 1578. [Google Scholar] [CrossRef]
- Yang, J.; Keller, M.W.; Moore, J.S.; White, S.R.; Sottos, N.R. Microencapsulation of isocyanates for self-healing polymers. Macromolecules 2008, 41, 9650–9655. [Google Scholar] [CrossRef]
- Di Credico, B.; Levi, M.; Turri, S. An efficient method for the output of new self-repairing materials through a reactive isocyanate encapsulation. Eur. Polym. J. 2013, 49, 2467–2476. [Google Scholar] [CrossRef]
- Kardar, P. Preparation of polyurethane microcapsules with different polyols component for encapsulation of isophorone diisocyanate healing agent. Prog. Org. Coat. 2015, 89, 271–276. [Google Scholar] [CrossRef]
- Haghayegh, M.; Mirabedini, S.M.; Yeganeh, H. Microcapsules containing multi-functional reactive isocyanateterminated polyurethane prepolymer as a healing agent. Part 1: Synthesis and optimization of reaction conditions. J. Mater. Sci. 2016, 51, 3056–3068. [Google Scholar] [CrossRef]
- Alizadegan, F.; Pazokifard, S.; Mirabedini, S.M.; Danaei, M.; Farnood, R. Polyurethane-based microcapsules containing reactive isocyanate compounds: Study on preparation procedure and solvent replacement. Colloids Surf. A Physicochem. Eng. Asp. 2017, 529, 750–759. [Google Scholar] [CrossRef]
- Alizadegan, F.; Mirabedini, S.M.; Pazokifard, S.; Moghadam, S.G.; Farnood, R. Improving self-healing performance of polyurethane coatings using PU microcapsules containing bulky-IPDI-BA and nano-clay. Prog. Org. Coat. 2018, 123, 350–361. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Qu, J.; Wen, Y.; Sun, W. Synthesis, characterizations and mechanical properties of microcapsules with dual shell of polyurethane (PU)/melamine formaldehyde (MF): Effect of different chain extenders. Eng. Chem. Res. 2018, 57, 3591–3601. [Google Scholar] [CrossRef]
- De Souza Rodrigues, V.H.; Carrara, A.E.; Rossi, S.S.; Silva, L.M.; de Cássia Lazzarini Dutra, R.; Narciso Dutra, J.C. Synthesis, characterization and qualitative assessment of self-healing capacity of PU microcapsules containing TDI and IPDI as a core agent. Mater. Today Commun. 2019, 21, 100698. [Google Scholar] [CrossRef]
- Attaei, M.; Loureiro, M.V.; do Vale, M.; Condeço, J.A.D.; Pinho, I.; Bordado, J.C.; Marques, A.C. Isophorone diisocyanate (IPDI) microencapsulation for mono-component adhesives: Effect of the active H and NCO sources. Polymers 2018, 10, 825. [Google Scholar] [CrossRef]
- Sondari, D.; Septevani, A.A.; Randy, A.; Triwulandari, E. Polyurethane microcapsule with glycerol as the polyol component for encapsulated self healing agent. Int. J. Eng. Technol. 2010, 2, 466–471. [Google Scholar]
- Loureiro, M.V.; Attaei, M.; Rocha, S.; Vale, M.; Bordado, J.C.; Simões, R.; Pinho, I.; Marques, A.C. The role played by different active hydrogen sources in the microencapsulation of a commercial oligomeric diisocyanate. J. Mater. Sci. 2020, 55, 4607–4623. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.; Kim, N.K.; Shin, J.; Kim, Y.W. Polyurethane microcapsules for self-healing paint coatings. RSC Adv. 2014, 4, 16214–16223. [Google Scholar] [CrossRef]
- Cakic, S.M.; Stamenkovic, J.V.; Djordjevic, D.M.; Ristic, I.S. Synthesis and degradation profile of cast films of PPG-DMPA-IPDI aqueous polyurethane dispersions based on selective catalysts. Polym. Degrad. Stab. 2009, 94, 2015–2022. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, S.; Hao, C.; Verdi, C.; Liu, W.; Liu, H.; Zhang, J. Glycerol induced catalyst-free curing of epoxy and vitrimer preparation. Macromol. Rapid Commun. 2019, 40, 1800889. [Google Scholar] [CrossRef]
- Petrović, Z.S. Polyurethanes from vegetable oils. Polym. Rev. 2008, 48, 109–155. [Google Scholar] [CrossRef]
- Guo, J.; He, Y.; Xie, D.; Zhang, X. Process investigating and modelling for the self-polymerization of toluene diisocyanate (TDI)-based polyurethane prepolymer. J. Mater. Sci. 2015, 50, 5844–5855. [Google Scholar] [CrossRef]
- Mishra, A.; Aswal, V.K.; Maiti, P. Nanostructure to microstructure self-assembly of aliphatic polyurethanes: The effect on mechanical properties. J. Phys. Chem. B 2010, 114, 5292–5300. [Google Scholar] [CrossRef]
- Mattia, J.; Painter, P. A comparison of hydrogen bonding and order in a polyurethane and poly(urethane-urea) and their blends with poly(ethylene glycol). Macromolecules 2007, 40, 1546–1554. [Google Scholar] [CrossRef]
- Lee, D.-W.; Kim, H.-N.; Lee, D.-S. Introduction of reversible urethane bonds based on vanillyl alcohol for efficient self-healing of polyurethane elastomers. Molecules 2019, 24, 2201. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Abbreviations | Description of the Material |
|---|---|
| TDIprep_G | Prepolymer prepared from toluene 2,4-diisocyanate and glycerol |
| TDIprep_B | Prepolymer prepared from toluene 2,4-diisocyanate and 1,4-butanediol |
| TDIprep_H | Prepolymer prepared from toluene 2,4-diisocyanate and 1,6-hexanediol |
| xGA_G | Microcapsules loaded with isophorone diisocyanate, prepared from TDIprep_G, chain extender 1,6-hexanediol and various concentration of the stabilizer, GA, where variable x represents the wt% of GA (7, 10 or 17) |
| xGA_B | Microcapsules loaded with isophorone diisocyanate, prepared from TDIprep_B, chain extender 1,6-hexanediol and various concentration of the stabilizer, GA, where variable x represents the wt% of GA (7, 10 or 17) |
| xGA_H | Microcapsules loaded with isophorone diisocyanate, prepared from TDIprep_H, chain extender 1,6-hexanediol and various concentration of the stabilizer, GA, where variable x represents the wt% of GA (7, 10 or 17) |
| Coat_ref | Metal plates coated with coating reference without the microcapsules |
| Coat_M | Metal plates coated with composite coatings containing 20 wt% of microcapsules 10GA_G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šobak, M.; Štular, D.; Štirn, Ž.; Žitko, G.; Čelan Korošin, N.; Jerman, I. Influence of the Prepolymer Type and Synthesis Parameters on Self-Healing Anticorrosion Properties of Composite Coatings Containing Isophorone Diisocyanate-Loaded Polyurethane Microcapsules. Polymers 2021, 13, 840. https://doi.org/10.3390/polym13050840
Šobak M, Štular D, Štirn Ž, Žitko G, Čelan Korošin N, Jerman I. Influence of the Prepolymer Type and Synthesis Parameters on Self-Healing Anticorrosion Properties of Composite Coatings Containing Isophorone Diisocyanate-Loaded Polyurethane Microcapsules. Polymers. 2021; 13(5):840. https://doi.org/10.3390/polym13050840
Chicago/Turabian StyleŠobak, Matic, Danaja Štular, Žiga Štirn, Gregor Žitko, Nataša Čelan Korošin, and Ivan Jerman. 2021. "Influence of the Prepolymer Type and Synthesis Parameters on Self-Healing Anticorrosion Properties of Composite Coatings Containing Isophorone Diisocyanate-Loaded Polyurethane Microcapsules" Polymers 13, no. 5: 840. https://doi.org/10.3390/polym13050840
APA StyleŠobak, M., Štular, D., Štirn, Ž., Žitko, G., Čelan Korošin, N., & Jerman, I. (2021). Influence of the Prepolymer Type and Synthesis Parameters on Self-Healing Anticorrosion Properties of Composite Coatings Containing Isophorone Diisocyanate-Loaded Polyurethane Microcapsules. Polymers, 13(5), 840. https://doi.org/10.3390/polym13050840